Abstract
Two ruthenium olefin metathesis initiators featuring electronically modified quinoline-based chelating carbene ligands are introduced. Their reactivity in RCM and ROMP reactions was tested and the results were compared to those obtained with the parent unsubstituted compound. The studied complexes are very stable at high temperatures up to 140 °C. The placement of an electron-withdrawing functionality translates into an enhanced activity in RCM. While electronically modified precatalysts, which exist predominantly in the trans-dichloro configuration, gave mostly the RCM and a minor amount of the cycloisomerization product, the unmodified congener, which preferentially exists as its cis-dichloro isomer, shows a switched reactivity. The position of the equilibrium between the cis- and the trans-dichloro species was found to be the crucial factor governing the reactivity of the complexes.
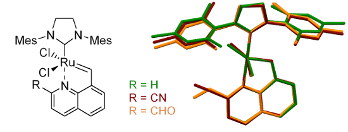
Graphical Abstract
Introduction
Olefin metathesis is a catalytic process during which C–C double bonds are exchanged [1]. Since the first examples were published in the 1950s, many stunning accomplishments have been made in the field resulting in ever increasing interests in the method. Establishment of well-defined molybdenum- and ruthenium-based complexes lead to multitude of applications [2-4]. Especially, the latter class of compounds have gained attention due to their user-friendly character caused by a wide functional group-tolerance and high oxygen and moisture stability. Although a great progress has been made, the unsuitability of ruthenium complexes for high temperature applications remains one of the greatest challenges in the field.
Modifications in the basic structure of ruthenium-based olefin metathesis catalysts led to a diversification of catalytic profiles (Figure 1) [5,6]. Perhaps the most important one was the introduction of bidentate benzylidene ligands instead of simple alkylidenes, thus giving rise to the class of Hoveyda-type complexes with the parent compound 2 [7]. Further modifications of such systems followed. One of the most common is tuning of the properties of the benzylidene ligand so that modified reactivity of the resulting complex is achieved [8]. Various examples of such approaches have been published over the years. Particularly interesting results were obtained by substituting the oxygen atom with sulfur or nitrogen leading to a group of structurally diverse ruthenium chelates [9-11].
![[1860-5397-11-158-1]](/bjoc/content/figures/1860-5397-11-158-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected ruthenium-based complexes.
Figure 1: Selected ruthenium-based complexes.
N-Based chelating complexes offer the advantage of a great availability of precursors for amine ligands making the number of possible structures virtually unlimited. Complexes incorporating alkylidene ligands based on aromatic [12-15] or aliphatic amines [16,17] and Schiff base patterns [18-22] have been prepared so far, exhibiting diverse activities ranging from very fast to very slow initiation. Furthermore, in those compounds, a trans–cis isomerization of the chloride ligands was observed in many cases (Scheme 1) [16]. This phenomenon has been widely discussed in literature with multiple reports of superior activity of trans-configured complexes. The general hypothesis is that the trans-dichloro form of the complex promotes metathesis whereas the cis-dichloro form is postulated to be metathetically inactive [23,24]. Thus, the trans–cis isomerization can be exploited for slowly releasing the olefin metathesis active species [25,26].
![[1860-5397-11-158-i1]](/bjoc/content/inline/1860-5397-11-158-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
Aside from the interesting conformational behavior, nitrogen-chelated complexes possess some practical properties, namely they tend to be thermally stable, enabling applications at elevated temperatures. Being aware of the various advantages of such systems, we set out to study substituted quinolone–ruthenium chelates in view of their trans–cis susceptibility and its consequences.
Results and Discussion
Synthesis and characterization
The shortest pathway to obtain the desired ligands was chosen to provide access to starting materials. It was envisioned as a two-step sequence of triflation of commercially available substituted 8-hydroxyquinolines 8 and 9 followed by a Suzuki coupling as shown in Scheme 2 [14].
![[1860-5397-11-158-i2]](/bjoc/content/inline/1860-5397-11-158-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the ligand precursors.
Scheme 2: Synthesis of the ligand precursors.
The cyano-substituted compound 10 was obtained without difficulty, but esterification of compound 9 was problematic because of purification issues (cf. Supporting Information File 1) so an alternative pathway was established. Starting from 2-bromoaniline upon Doebner–Miller reaction and oxidation, we obtained the corresponding bromide derivative which was subsequently converted via Suzuki coupling into the carbene precursor 13. Both compounds 12 and 13 were then used in a carbene exchange reaction with compound 1 conducted in toluene at 80 °C (see Scheme 3), releasing the desired complexes 14 and 15 as trans-dichloro isomers in good yields.
![[1860-5397-11-158-i3]](/bjoc/content/inline/1860-5397-11-158-i3.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of the ruthenium complexes.
Scheme 3: Synthesis of the ruthenium complexes.
Based on the previously reported trans–cis isomerization of the parent quinoline-based complex 5a, a potential isomerization of 14 and 15 was investigated. Reaction conditions for the isomerization of 5a (complete isomerization after 6 days at 23 °C in CD2Cl2) [14] were proven to be ineffective for the isomerization of 14 and 15, so that more forcing conditions were applied: Compounds 5a, 14 or 15 were dissolved in CDCl3 and heated to 140 °C for 1 h in a microwave reactor.
When the reactions were performed in air, only decomposition of the complexes was observed. Upon using oxygen-free conditions a complete rearrangement of trans-5a to cis-5a in 30 min was obtained. Attempts, to prepare the cis-isomers of electronically modified complexes 14 and 15 in a similar fashion, however, turned out to be difficult. Using methods, such as starting from a pyridine containing the ruthenium complex, which is known to increase the cis-content, or increasing the exposure time of the catalyst up to 8 h in microwave at 140 °C in CDCl3 resulted in limited success.
The formyl-substituted complex 15 underwent isomerization, but only a mixture of 11% cis-15 and 89% trans-15 was obtained (cf. Supporting Information File 1). In the case of the CN-substituted compound 14, on the other hand, no evidence for any isomerization could have been retrieved. These results suggest, that the introduction of the electron withdrawing substituent in 2-position changes the thermodynamic equilibrium favoring the trans- over the cis-isomers. In any case, a remarkable thermal stability of all studied compounds was found.
Intrigued by the observed phenomenon, we turned to structural studies. Structures of 14 and 15 have been determined using single-crystal X-ray diffraction. Each of them includes two molecules of the studied compound in the trans-conformation and one molecule of the solvent (dichloromethane) in the asymmetric part of the unit cell (Figure 2). Both crystals are isotypic, so the crystal packing is identical and lattice dimensions are very similar (Figure 3).
![[1860-5397-11-158-2]](/bjoc/content/figures/1860-5397-11-158-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ADPs (atomic displacement parameters) and atoms labeling of the first molecule in the asymmetric part for 14 (left) and 15 (right). Thermal ellipsoids at 50% level of probability. Hydrogen atoms were omitted for clarity.
Figure 2: ADPs (atomic displacement parameters) and atoms labeling of the first molecule in the asymmetric pa...
![[1860-5397-11-158-3]](/bjoc/content/figures/1860-5397-11-158-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Superposition of the asymmetric parts of units’ cells in both investigated structures: an example of isostructural packing (left). Superposition (right side) trans forms of all studied molecules 5a (green), 14 (ruby) and 15 (orange).
Figure 3: Superposition of the asymmetric parts of units’ cells in both investigated structures: an example o...
Bond lengths and angles of 5a, 14 and 15 are very similar, so their different tendencies to form the corresponding cis-complexes could not be rationalized based on this dataset. The only feature worth mentioning is a slightly decreased Ru(1)-C(22) bond length in compound 15 compared to 5a or 14 (1.829, 1.845 and 1.842 Å, respectively).
DFT calculations
Density functional theory (DFT) calculations were performed with the aim to learn more about the compounds under investigation especially in view of their peculiar trans–cis isomerization behavior. Geometry optimizations were conducted by BP86//SVP, solvent effects were added by single point calculations using M06//TZVP functional (cf. Supporting Information File 1). The first question tackled concerned the anticipated coordination of the carbonyl group in compound 15 to form the corresponding 18-electron complex. Formation of the 18-electron compound is in principle feasible although it is endothermic by 2.6 kcal/mol. Thus, it can be concluded that preferably the corresponding 16-electron species is present in solution. Next, we investigated the relative stabilities of the trans-dichloro versus the cis-dichloro isomers. As already stated in literature, DFT calculations suggested a more preferential arrangement of 5a in the cis-dichloro configuration [23,24]. Here, the equilibrium was investigated assuming solvation in CH2Cl2 and results revealed a preference for the cis-dichloro isomer in the case of 5a (cis-5a is 4.1 kcal/mol more stable than trans-5a) and 15 (cis-15 is 2.4 kcal/mol more stable than trans-15). In contrast, the cyano-group substituted compound 14 exists preferably in the trans configuration of the chloride ligands (trans-14 is 1.4 kcal/mol more stable than cis-14). Because the cis isomer is better stabilized in solvents with high dielectric constants (such as CH2Cl2) than in solvents with low dielectric constants [27], the trans–cis energies were calculated in toluene (as an example for a solvent with low dielectric constant). In this case, 5a is still preferably in the cis configuration (cis-5a is 1.3 kcal/mol more stable than trans-5a), while in 14 and 15 the trans configuration is favored (by 0.7 kcal/mol in case of 15 and 4.2 kcal/mol in case of 14). Further, the energy profile for the isomerization reaction was investigated taking a dissociative and a concerted pathway into consideration (cf. Figure 4).
![[1860-5397-11-158-4]](/bjoc/content/figures/1860-5397-11-158-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Energy profile of trans–cis isomerization, modelled in CH2Cl2, (ΔE in kcal/mol). Geometry optimizations BP86//SVP, solvent effects included by single point calculations, using M06//TZVP.
Figure 4: Energy profile of trans–cis isomerization, modelled in CH2Cl2, (ΔE in kcal/mol). Geometry optimizat...
As disclosed earlier [23,24], the concerted pathway is the most likely operative for the isomerization of 5a. The transition state TS2 (which is the transition state for the concerted pathway) is 5.3 kcal lower in energy than the transition state for closing the chelating ligand towards the cis-dichloro configured form (TS1cis). The energetic preference of the concerted over the dissociative pathway is decreasing when substituents in 2 position of the quinoline ligand are present. For 14 and 15 the transition state for the generation of the catalytically active 14-electron species (TS1trans) is considerably lower compared to 5a. These results go in hand with studies on electronically modified Hoveyda-type catalysts [28,29] and electronically modified ester-chelating benzylidene complexes [30]. Moreover, as already discussed, TS2 becomes energetically more demanding so that for 14 and 15 the pathways for the isomerization (leading to the olefin metathesis inactive cis-dichloro form) becomes less important. Consequently, complex 15 and in particular 14 should be metathetically more active than their unsubsituted version 5a.
Activity in RCM
The consecutive step of the research was devoted to exploring the activity of the obtained complexes in metathesis reactions. The preliminary choice was to conduct ring-closing metathesis (RCM) of diethyl diallylmalonate (16, Scheme 4).
![[1860-5397-11-158-i4]](/bjoc/content/inline/1860-5397-11-158-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Possible reaction pathways of 16.
Scheme 4: Possible reaction pathways of 16.
Initially examined conditions (DCM, rt) originate from the papers previously published by Grela’s group on the subject [14,31]. Unfortunately, along with F. Hoffmann-La Roche AG researchers [32] we were unable to fully reproduce the aforementioned results. The complex was inert at ambient conditions and only tests at elevated temperatures (80 or 140 °C) revealed catalytic activity of 5a. When substrate 16 was subjected to RCM in high temperature conditions, a complex mixture of products was obtained (Figure 5). Upon GC and GC–MS analysis, structures of compounds 17–19 were determined (cf. Supporting Information File 1).
![[1860-5397-11-158-5]](/bjoc/content/figures/1860-5397-11-158-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Time/conversion plots for the transformation of 16 catalyzed by 5 mol % of the trans isomers of trans-5a, 14 and 15. Lines are intended as visual aids.
Figure 5: Time/conversion plots for the transformation of 16 catalyzed by 5 mol % of the trans isomers of tra...
The substituted complexes were inert in ambient conditions similarly to the parent complex. The experiments at elevated temperature (toluene, 80 °C) revealed that precatalysts 14 and 15 led to higher conversions than the unsubstituted version 5a. What is more interesting, both complexes exhibit prolonged activity and similar overall activity under the studied conditions. An interesting observation is that reactions catalyzed with 14 and 15 gave predominantly the RCM product (compound 17) accompanied by a minor amount of a cycloisomerization-derived compound 19. It is worth noting, that even after heating at 80 °C for 100 h the catalysts were still active, as can be assessed from the time/conversion plots (cf. Figure 5a and b). In contrast, the precatalyst 5a promoted predominantly cycloisomerization albeit conversion was poor (cf. Figure 5c). Because all transformations were very slow at 80 °C the next series of test reactions was conducted at 140 °C in xylenes as the solvent (cf. Figure 5d–f). Electronically modified derivatives 14 and 15 promoted a faster transformation of 16. In the first 10 min. about 90% of 16 was converted into about 80% cycloisomerization product 19 and about 12–16% RCM-product 17, thus the selectivity changed upon raising the temperature. In addition, a minor amount of the isomerized RCM-product 18 and isomerized diethyl diallylmalonate 20 were detected in the reaction mixture. After about 1 h reaction time, the composition of the reaction mixture remained virtually unchanged upon prolonged heating.
At this high temperature, the precatalyst trans-5a resulted in a complete conversion of 16 which gave more than 90% cycloisomerization product 19 in 8 h. A small amount of isomerized diethyl diallylmalonate 20 and RCM-product 17 were also observed (cf. Figure 5f). Using cis-5a as the precatalyst resulted in similar results as can be seen in Figure 6. This is not especially peculiar as isomerization is conducted in the very same temperature range within less than 30 minutes meaning that the corresponding equilibrium is reached quickly.
![[1860-5397-11-158-6]](/bjoc/content/figures/1860-5397-11-158-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Composition of a reaction mixture after subjecting 16 to 5 mol % cis-5a in xylene, 140 °C. Lines are intended as visual aids.
Figure 6: Composition of a reaction mixture after subjecting 16 to 5 mol % cis-5a in xylene, 140 °C. Lines ar...
Basing on the described experiments, it can be stated that precatalysts are thermally stable in the absence of oxygen and diethyl diallylmalonate at temperatures as high as 140 °C. Electronically modified precatalysts 14 and 15 initiate significantly faster than the parent precatalyst 5a and when employed in RCM of diethyl diallylmalonate at 80 °C, those complexes gave predominantly the RCM product 17 accompanied with minor amounts of the cycloisomerization product 19, while 5a released predominantly the cycloisomerization product 19.
Switching to 140 °C reaction temperature, all precatalysts released the cycloisomerization product 19 as the main product. These observations make again clear that the thermal stability of the precatalyst becomes irrelevant once it is in the presence of the substrate [33], because it is the thermal stability of the actual active species in the reaction mixture that governs the reaction outcome. In the present case, the methylidene complex formed during metathesis with the terminal olefin diethyl diallylmalonate is probably the most fragile species [32,34-36]. A recent work provides evidence that the catalytic species responsible for (cyclo)isomerization originates from decomposition of the methylidene [37]. Generally, the ability of olefin metathesis precatalysts to promote cycloisomerization [38] is known and has been widely researched both theoretically [39,40] and experimentally [41-43]. Moreover, the methylidene species alone are characterized by a certain degree of stability that is dramatically reduced in the presence of olefins, in particular, ethylene [38]. Accordingly, RCM reactions can be improved in terms of efficiency when ethylene is removed [44-46]. Based on these facts, the catalytic performance of the precatalysts 14 and 15 can be explained as follows: At 80 °C the actual active species is slowly released and performs mainly metathesis with diethyl diallylmalonate (16) leading, amongst other species, to the methylidene complex [47]. The latter is moderately stable under these conditions and participates in the catalytic RCM cycle. A concurring decomposition reaction of the methylidene or another Ru-species is responsible for the cycloisomerization side reaction. Further, the latter species is not able to isomerize the educt or the RCM product. Upon increasing the temperature to 140 °C, the said decomposition reaction is faster leading to the observed switch of reactivity in favor of the cycloisomerization pathway and isomerization of the educt as well as that of the product is observed. However, the results for 5a as the precatalyst make clear that the thermal stability of the methylidene is not the only factor governing the outcome of the studied reactions. If the stability of the methylidene were the only crucial factor in all cases, the product distributions from reactions with 5a would be similar to those from 14 and 15. This is definitely not the case. Therefore, it can be assumed that in case of 5a another, yet unknown decomposition reaction is responsible for the occurrence of the cycloisomerization reaction.
Use as initiators in ROMP
In the next step, compounds 5a, 14 and 15 were tested as initiators in ring-opening metathesis polymerization (ROMP). The active species in ROMP (i.e., the propagating species) can be considered more stable than the methylidene intermediate in RCM, particularly when norbornenes such as endo,exo-bicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic acid dimethyl ester (21) are polymerized (Figure 7) [48].
![[1860-5397-11-158-7]](/bjoc/content/figures/1860-5397-11-158-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Monomers utilized in model ROMP reactions.
Figure 7: Monomers utilized in model ROMP reactions.
First, 300 equivalents of monomer 21 were polymerized with 1 equivalent initiator (5a, 14 or 15) at 110 °C in toluene ([21] = 0.1 M) for 2 days. Initiator 5a polymerized 90% of 21 and the corresponding polymer was characterized by a number average molecular weight (Mn) of 557.0 kg·mol−1 (polydispersity index, PDI = 1.9) as examined by size exclusion chromatography (SEC) in THF against poly(styrene) standards. Initiator 15 gave 93% monomer-conversion and the resulting polymer exhibited a Mn of 516.0 kg·mol−1 (PDI = 2.2) and 14 gave the highest conversion (98%) and the shortest polymer strands (Mn = 326.7 kg·mol−1; PDI = 2.2).
The Mn values allow for an indirect relative assessment of the initiation efficacy [49-54], because they are proportional to the ratio of the propagation rate (kp) and the initiation rate constant (ki), provided that no secondary metathesis occurs. In this case, Mn is only dependent on ki, because in all cases the same propagating species occurs and kp is the same. Accordingly, initiator 14 exhibits the highest initiation efficacy and initiator 5a the lowest. Analyzing these data as disclosed previously, a linear correlation between the Mn values and the difference between the calculated thermodynamic stabilities of the trans- and the cis-dichloro configured isomers (ΔEtrans−cis) can be established (Figure 8). This correlation suggests that the initiation efficacy is above all determined by the position of the trans–cis equilibrium which can be quickly reached at 110 °C [29,55].
![[1860-5397-11-158-8]](/bjoc/content/figures/1860-5397-11-158-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Number-average molecular weight (Mn) of poly-21 prepared with initiators 5a, 14 and 15 plotted against the theoretically determined difference of trans–cis energies (solvation model PCM, solvent toluene).
Figure 8: Number-average molecular weight (Mn) of poly-21 prepared with initiators 5a, 14 and 15 plotted agai...
In the second step, the initiators were tested in neat monomer using simultaneous thermal analysis (STA) for monitoring the polymerizations. As the monomer, either endo,exo-bicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic acid diethyl ester (22) or dicylcopentadiene (23) were used (cf. Figure 7). A distinctly changed initiation trend was observed under these reaction conditions. Initiator 5a started the polymerization at the lowest temperature (onset of the polymerization exotherm at approx. 60 °C; cf. Figure 9, left) while the highest latency was found for initiator 14 (onset at approx. 75 °C). While initiators 14 and 15 exhibited rather sharp exotherms for the polymerization of monomer 22, a much broader shape was found in the case of 5a. This shape can be explained by assuming, that the isomerization of trans-5a to cis-5a is a concurring reaction, thus slowing down the polymerization reaction. The reason for the unexpectedly, at first sight, delayed initiation of 14 and 15 can most probably be attributed to steric effects. It is known that a coordination at the free coordination site (trans to the carbene ligand) can activate the (pre-)catalyst by lowering the energy barrier TS1trans to reach the active 14-electron species [14,56,57]. As both substituents, the CHO and the CN group, sterically shield the vacant coordination site, it is easily conceivable that such substrate-induced activation is impeded.
![[1860-5397-11-158-9]](/bjoc/content/figures/1860-5397-11-158-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: STA analysis of polymerization of 22 (left) and 23 (right), initiated by 5a, 14 and 15. Reaction conditions: [22]:[initiator] = 500 and [23]:[initiator] = 10000:1. Heating rate: 3 K/min. Big symbols: thermogravimetric analysis (TGA); no symbols: differential scanning calorimetry (DSC).
Figure 9: STA analysis of polymerization of 22 (left) and 23 (right), initiated by 5a, 14 and 15. Reaction co...
This effect turned out to be of particular relevance when the polymerization of dicyclopentadiene is regarded. Because polymerizations were carried out in open reaction vessels, the retro-Diels–Alder reaction of 23, releasing volatile cyclopentadiene, is a concurring reaction and responsible for the low(er) polymer yields and pronounced endothermic signals in the DSC traces (cf. Figure 9 right) [58]. While initiator 5a shows an appealing performance in polymerizing 23, the electronically modified congeners 14 and 15 are not particularly interesting for this application.
Conclusion
The present work introduced two ruthenium-based olefin metathesis catalysts/initiators featuring electronically modified quinoline-based chelating carbene ligands. Their reactivity in RCM and ROMP reactions was tested and results were set in comparison to those obtained with the parent compound, bearing the unsubstituted quinoline-based chelating carbene. The entire set of compounds is very stable at high temperatures up to 140 °C in the absence of oxygen and metathesis substrates. Electronic modification of the quinoline moiety changes the position of the trans–cis equilibrium as shown experimentally and theoretically. At the same time, electronic modification lowers the transition state energy for the generation of the catalytically active 14-electron species and increases the energy barrier for the transformation into the corresponding cis-dichloro isomers. Both effects translate into an enhanced activity in RCM at 80 °C when compared to the unmodified catalyst. In particular, the position of the trans–cis equilibrium is the most crucial factor governing the reactivity of the complexes. While electronically modified precatalysts which exist predominantly in trans-dichloro configuration gave mostly RCM and minor amounts of cycloisomerization product, the unmodified congener which preferentially exists in its cis-dichloro isomer, shows a switched reactivity. The reactivity switch is most probably caused by different substrate-induced decomposition reactions being responsible for the occurrence of the cycloisomerization reaction, which are more important at higher temperatures of 140 °C. In ROMP, again the position of the trans–cis equilibrium is the most crucial factor governing the initiation efficacy. Additionally, it has been shown, that steric effects of the substitution are responsible for an altered order of initiation behavior when polymerizations are conducted in bulk.
Experimental
Preparation of 14 and 15. Precursor complex 1 (0.5 mmol, 475 mg) and the respective styrene derivative (0.55 mmol) were put in a Schlenk tube under argon. Reagents were dissolved in anhydrous toluene (25 mL) and the reaction was heated at 80 °C for about an hour. Then the solvent was evaporated and the mixture was purified by flash chromatography using eluents c-hexane/ethyl acetate 10:1 to 1:1 v/v. The solvent was evaporated. The product was then re-dissolved in dichloromethane and cold n-heptane was added to yield the product 14 as dark brown crystals (0.37 mmol, 242 mg, 75%). 1H NMR (CD2Cl2) δ 2.41 (s, 6H), 2.49 (s, 12H), 4.16 (s, 4H), 7.07 (s, 4H), 7.48–7.53 (m, 2H), 7.72 (dd, J = 0.9, 7.2 Hz, 1H), 8.24 (dd, J = 0.9, 8.3 Hz, 1H), 8.33 (d, J = 8.5 Hz, 1H), 16.95 (s, 1H) ppm; 13C NMR (CD2Cl2) δ 19.0, 20.9, 51.8, 116.1, 117.5, 122.6, 128.1, 129.4, 129.7, 133.9, 134.0, 134.6, 136.2, 138.3, 138.9, 146.8, 155.6, 209.4, 285.5 ppm; IR (KBr) ν: 3320, 3042, 3004, 2949, 2912, 2855, 2837, 2810, 2237, 1959, 1704, 1682, 1601, 1586, 1556, 1478, 1454, 1445, 1427, 1418, 1398, 1378, 1326, 1315, 1289, 1256, 1217, 1199, 1176, 1148, 1133, 1102, 1061, 1036, 1014, 985, 966, 930, 911, 879, 848, 822, 813, 792, 777, 752, 734, 721, 701, 653, 643, 622, 580, 533, 498, 428, 415 cm−1; HRMS (ESI) (m/z): [M] calcd, 644.1048; found, 644.1041.
Complex 15 was prepared analogously yielding dark brown crystals (0.43 mmol, 280 mg, 86%). 1H NMR (CD2Cl2) δ 2.43–2.55 (m, 16H), 4.19 (s, 4H), 7.11 (s, 4H), 7.48–7.55 (m, 1H), 7.72 (dd, J = 0.9, 7.2 Hz, 1H), 7.78 (d, J = 8.5 Hz, 1H), 8.28 (dd, J = 0.9, 8.2 Hz, 1H), 8.35 (d, J = 8.3 Hz, 1H), 8.96 (d, J = 0.6, Hz, 1H), 17.11 (s, 1H) ppm; 13C NMR (CD2Cl2) δ 19.1, 20.9, 51.6, 117.0, 122.8, 129.2, 130.8, 133.4, 134.3, 136.0, 138.9, 139.0, 145.9, 152.2, 156.3, 190.0, 210.6, 288.2 ppm; IR (KBr) ν: 3003, 2952, 2912, 2854, 2734, 2232, 1950, 1734, 1694, 1605, 1584, 1551, 1480, 1419, 1401, 1379, 1319, 1294, 1261, 1222, 1174, 1154, 1138, 1092, 1036, 987, 929, 910, 887, 846, 813, 794, 775, 732, 699, 680, 644, 591, 578, 535, 419 cm−1; HRMS (ESI) (m/z): [M − 2Cl + H]+ calcd, 578.1745; found, 578.1732.
Supporting Information
| Supporting Information File 1: Full experimental section along with all the synthetic procedures and analytical data of the obtained compounds. | ||
| Format: PDF | Size: 3.2 MB | Download |
Acknowledgements
KŻ thanks for the „Diamond Grant” research project financed from the governmental funds for science for 2012–2015. Dr. M. Kędziorek is acknowledged for providing the precursor of 5a. EP gratefully acknowledges to “Chemical Monthly” for financial support of this work. Authors acknowledge for the WTZ PL10/2014 cooperation.
References
-
Grela, K., Ed. Olefin Metathesis: Theory and Practice; John Wiley & Sons, Inc.: Hoboken, 2014.
Return to citation in text: [1] -
Bieniek, M.; Michrowska, A.; Usanov, D. L.; Grela, K. Chem. – Eur. J. 2008, 14, 806–818. doi:10.1002/chem.200701340
Return to citation in text: [1] -
Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 42, 4592–4633. doi:10.1002/anie.200300576
Return to citation in text: [1] -
Leitgeb, A.; Wappel, J.; Slugovc, C. Polymer 2010, 51, 2927–2946. doi:10.1016/j.polymer.2010.05.002
Return to citation in text: [1] -
Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746–1787. doi:10.1021/cr9002424
Return to citation in text: [1] -
Samojłowicz, C.; Bieniek, M.; Grela, K. Chem. Rev. 2009, 109, 3708–3742. doi:10.1021/cr800524f
Return to citation in text: [1] -
Garber, B.; Kingsbury, J. S.; Gray, B. L.; Hoveyda, A. H. J. Am. Chem. Soc. 2000, 122, 8168–8179. doi:10.1021/ja001179g
Return to citation in text: [1] -
Vidavsky, Y.; Anaby, A.; Lemcoff, N. G. Dalton Trans. 2012, 41, 32–43. doi:10.1039/C1DT11404B
Return to citation in text: [1] -
Ben-Asuly, A.; Tzur, E.; Diesendruck, C. E.; Sigalov, M.; Goldberg, I.; Lemcoff, N. G. Organometallics 2008, 27, 811–813. doi:10.1021/om701180z
Return to citation in text: [1] -
Szadkowska, A.; Makal, A.; Woźniak, K.; Kadyrov, R.; Grela, K. Organometallics 2009, 28, 2693–2700. doi:10.1021/om801183g
Return to citation in text: [1] -
Szadkowska, A.; Żukowska, K.; Pazio, A. E.; Woźniak, K.; Kadyrov, R.; Grela, K. Organometallics 2011, 30, 1130–1138. doi:10.1021/om101129b
Return to citation in text: [1] -
van der Schaaf, P. A.; Kolly, R.; Kirner, H.-J.; Rime, F.; Mühlebach, A.; Hafner, A. J. Organomet. Chem. 2000, 606, 65–74. doi:10.1016/S0022-328X(00)00289-8
Return to citation in text: [1] -
Szadkowska, A.; Gstrein, X.; Burtscher, D.; Jarzembska, K.; Woźniak, K.; Slugovc, C.; Grela, K. Organometallics 2010, 29, 117–124. doi:10.1021/om900857w
Return to citation in text: [1] -
Barbasiewicz, M.; Szadkowska, A.; Bujok, R.; Grela, K. Organometallics 2006, 25, 3599–3604. doi:10.1021/om060091u
Return to citation in text: [1] [2] [3] [4] [5] -
Peeck, L. H.; Savka, R. D.; Plenio, H. Chem. – Eur. J. 2012, 18, 12845–12853. doi:10.1002/chem.201201010
Return to citation in text: [1] -
Diesendruck, C. E.; Tzur, E.; Ben-Asuly, A.; Goldberg, I.; Straub, B. F.; Lemcoff, N. G. Inorg. Chem. 2009, 48, 10819–10825. doi:10.1021/ic901444c
Return to citation in text: [1] [2] -
Żukowska, K.; Szadkowska, A.; Pazio, A. E.; Woźniak, K.; Grela, K. Organometallics 2012, 31, 462–469. doi:10.1021/om2011062
Return to citation in text: [1] -
Hejl, A.; Day, M. W.; Grubbs, R. H. Organometallics 2006, 25, 6149–6154. doi:10.1021/om060620u
Return to citation in text: [1] -
Slugovc, C.; Burtscher, D.; Stelzer, F.; Mereiter, K. Organometallics 2005, 24, 2255–2258. doi:10.1021/om050141f
Return to citation in text: [1] -
De Clercq, B.; Verpoort, F. Adv. Synth. Catal. 2002, 344, 639–648.
Return to citation in text: [1] -
Jordaan, M.; Vosloo, H. C. M. Adv. Synth. Catal. 2007, 349, 184–192. doi:10.1002/adsc.200600474
Return to citation in text: [1] -
Slugovc, C.; Perner, B.; Stelzer, F.; Mereiter, K. Organometallics 2004, 23, 3622–3626. doi:10.1021/om049877n
Return to citation in text: [1] -
Poater, A.; Ragone, F.; Correa, A.; Szadkowska, A.; Barbasiewicz, M.; Grela, K.; Cavallo, L. Chem. – Eur. J. 2010, 16, 14354–14364. doi:10.1002/chem.201001849
Return to citation in text: [1] [2] [3] -
Pump, E.; Cavallo, L.; Slugovc, C. Monatsh. Chem. 2015, 146, 1131–1141. doi:10.1007/s00706-015-1433-8
Return to citation in text: [1] [2] [3] -
Ung, T.; Hejl, A.; Grubbs, R. H.; Schrodi, Y. Organometallics 2004, 23, 5399–5401. doi:10.1021/om0493210
Return to citation in text: [1] -
Guidone, S.; Songis, O.; Nahra, F.; Cazin, C. S. J. ACS Catal. 2015, 5, 2697–2701. doi:10.1021/acscatal.5b00197
Return to citation in text: [1] -
Aharoni, A.; Vidavsky, Y.; Diesendruck, C. E.; Ben-Asuly, A.; Goldberg, I.; Lemcoff, N. G. Organometallics 2011, 30, 1607–1615. doi:10.1021/om1011402
Return to citation in text: [1] -
Zaja, M.; Connon, S. J.; Dunne, A. M.; Rivard, M.; Buschmann, N.; Jiricek, J.; Blechert, S. Tetrahedron 2003, 59, 6545–6558. doi:10.1016/S0040-4020(03)01029-9
Return to citation in text: [1] -
Michrowska, A.; Bujok, R.; Harutyunyan, S.; Sashuk, V.; Dolgonos, G.; Grela, K. J. Am. Chem. Soc. 2004, 126, 9318–9325. doi:10.1021/ja048794v
Return to citation in text: [1] [2] -
Pump, E.; Poater, A.; Zirngast, M.; Torvisco, A.; Fischer, R.; Cavallo, L.; Slugovc, C. Organometallics 2014, 33, 2806–2813. doi:10.1021/om500315t
Return to citation in text: [1] -
Grela, K.; Barbasiewicz, M.; Szadkowska, A. Complexes of Ruthenium and osmium, method of production thereof and use thereof as (pre)catalysts of the metathesis reaction. WO Patent WO140954, Dec 13, 2007.
Return to citation in text: [1] -
Puentener, K.; Scalone, M. New ruthenium complexes as catalysts for metathesis reactions. WO Patent WO2008000644, Jan 3, 2008.
Return to citation in text: [1] [2] -
Nelson, D. J.; Manzini, S.; Urbina-Blanco, C. A.; Nolan, S. P. Chem. Commun. 2014, 50, 10355–10375. doi:10.1039/C4CC02515F
Return to citation in text: [1] -
Lummiss, J. A. M.; Beach, N. J.; Smith, J. C.; Fogg, D. E. Catal. Sci. Technol. 2012, 2, 1630–1632. doi:10.1039/C2CY20213A
Return to citation in text: [1] -
Sanford, M. S.; Love, J. A.; Grubbs, R. H. J. Am. Chem. Soc. 2001, 123, 6543–6554. doi:10.1021/ja010624k
Return to citation in text: [1] -
Hong, S. H.; Wenzel, A. G.; Salguero, T. T.; Day, M. W.; Grubbs, R. H. J. Am. Chem. Soc. 2007, 129, 7961–7968. doi:10.1021/ja0713577
Return to citation in text: [1] -
Nelson, D. J.; Percy, J. M. Dalton Trans. 2014, 43, 4674–4679. doi:10.1039/C4DT00007B
Return to citation in text: [1] -
Lloyd-Jones, G. C. Org. Biomol. Chem. 2003, 1, 215–236. doi:10.1039/B209175P
Return to citation in text: [1] [2] -
van Rensburg, W. J.; Steynberg, P. J.; Meyer, W. H.; Kirk, M. M.; Forman, G. S. J. Am. Chem. Soc. 2004, 126, 14332–14333. doi:10.1021/ja0453174
Return to citation in text: [1] -
van Rensburg, W. J.; Steynberg, P. J.; Kirk, M. M.; Meyer, W. H.; Forman, G. S. J. Organomet. Chem. 2006, 691, 5312–5325. doi:10.1016/j.jorganchem.2006.08.075
Return to citation in text: [1] -
Bourgeois, D.; Pancrazi, A.; Nolan, S. P.; Prunet, J. J. Organomet. Chem. 2002, 643–644, 247–252. doi:10.1016/S0022-328X(01)01269-4
Return to citation in text: [1] -
Lehman, S. E., Jr.; Schwendeman, J. E.; O’Donnel, P. M.; Wagener, K. B. Inorg. Chim. Acta 2003, 345, 190–198. doi:10.1016/S0020-1693(02)01307-5
Return to citation in text: [1] -
Feng, C.; Wang, X.; Wang, B.-Q.; Zhao, K.-Q.; Hu, P.; Shi, Z.-J. Chem. Commun. 2012, 48, 356–358. doi:10.1039/C1CC15835J
Return to citation in text: [1] -
Lysenko, Z.; Maughon, B. R.; Mokhtar-Zadeh, T.; Tulchinsky, M. L. J. Organomet. Chem. 2006, 691, 5197–5203. doi:10.1016/j.jorganchem.2006.08.031
Return to citation in text: [1] -
Monfette, S.; Eyholzer, M.; Roberge, D. M.; Fogg, D. E. Chem. – Eur. J. 2010, 16, 11720–11725. doi:10.1002/chem.201001210
Return to citation in text: [1] -
Skowerski, K.; Czarnocki, S. J.; Knapkiewicz, P. ChemSusChem 2014, 7, 536–542. doi:10.1002/cssc.201300829
Return to citation in text: [1] -
Stewart, I. C.; Keitz, B. K.; Kuhn, K. M.; Thomas, R. M.; Grubbs, R. H. J. Am. Chem. Soc. 2010, 132, 8534–8535. doi:10.1021/ja1029045
Return to citation in text: [1] -
Slugovc, C.; Demel, S.; Riegler, S.; Hobisch, J.; Stelzer, F. Macromol. Rapid Commun. 2004, 25, 475–480. doi:10.1002/marc.200300196
Return to citation in text: [1] -
Kozłowska, A.; Dranka, M.; Zachara, J.; Pump, E.; Slugovc, C.; Skowerski, K.; Grela, K. Chem. – Eur. J. 2014, 20, 14120–14125. doi:10.1002/chem.201403580
Return to citation in text: [1] -
Urbina-Blanco, C. A.; Bantreil, X.; Wappel, J.; Schmid, T. E.; Slawin, A. M. Z.; Slugovc, C.; Cazin, C. S. J. Organometallics 2013, 32, 6240–6247. doi:10.1021/om4004362
Return to citation in text: [1] -
Pump, E.; Fischer, R. C.; Slugovc, C. Organometallics 2012, 31, 6972–6979. doi:10.1021/om300785t
Return to citation in text: [1] -
Urbina-Blanco, C. A.; Leitgeb, A.; Slugovc, C.; Bantreil, X.; Clavier, H.; Slawin, A. M. Z.; Nolan, S. P. Chem. – Eur. J. 2011, 17, 5045–5053. doi:10.1002/chem.201003082
Return to citation in text: [1] -
Broggi, J.; Urbina-Blanco, C. A.; Clavier, H.; Leitgeb, A.; Slugovc, C.; Slawin, A. M. Z.; Nolan, S. P. Chem. – Eur. J. 2010, 16, 9215–9225. doi:10.1002/chem.201000659
Return to citation in text: [1] -
Strasser, S.; Pump, E.; Fischer, R. C.; Slugovc, C. Monatsh. Chem. 2015, 146, 1143–1151. doi:10.1007/s00706-015-1484-x
Return to citation in text: [1] -
Diesendruck, C. E.; Vidavsky, V.; Ben-Asuly, A.; Lemcoff, N. G. J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 4209–4213. doi:10.1002/pola.23476
Return to citation in text: [1] -
Żukowska, K.; Szadkowska, A.; Trzaskowski, B.; Pazio, A.; Pączek, Ł.; Woźniak, K.; Grela, K. Organometallics 2013, 32, 2192–2198. doi:10.1021/om400064c
Return to citation in text: [1] -
Trzaskowski, B.; Grela, K. Organometallics 2013, 32, 3625–3630. doi:10.1021/om400233s
Return to citation in text: [1] -
Leitgeb, A.; Wappel, J.; Urbina-Blanco, C. A.; Strasser, S.; Wappl, C.; Cazin, C. S. J.; Slugovc, C. Monatsh. Chem. 2014, 145, 1513–1517. doi:10.1007/s00706-014-1249-y
Return to citation in text: [1]
| 44. | Lysenko, Z.; Maughon, B. R.; Mokhtar-Zadeh, T.; Tulchinsky, M. L. J. Organomet. Chem. 2006, 691, 5197–5203. doi:10.1016/j.jorganchem.2006.08.031 |
| 45. | Monfette, S.; Eyholzer, M.; Roberge, D. M.; Fogg, D. E. Chem. – Eur. J. 2010, 16, 11720–11725. doi:10.1002/chem.201001210 |
| 46. | Skowerski, K.; Czarnocki, S. J.; Knapkiewicz, P. ChemSusChem 2014, 7, 536–542. doi:10.1002/cssc.201300829 |
| 47. | Stewart, I. C.; Keitz, B. K.; Kuhn, K. M.; Thomas, R. M.; Grubbs, R. H. J. Am. Chem. Soc. 2010, 132, 8534–8535. doi:10.1021/ja1029045 |
| 1. | Grela, K., Ed. Olefin Metathesis: Theory and Practice; John Wiley & Sons, Inc.: Hoboken, 2014. |
| 8. | Vidavsky, Y.; Anaby, A.; Lemcoff, N. G. Dalton Trans. 2012, 41, 32–43. doi:10.1039/C1DT11404B |
| 23. | Poater, A.; Ragone, F.; Correa, A.; Szadkowska, A.; Barbasiewicz, M.; Grela, K.; Cavallo, L. Chem. – Eur. J. 2010, 16, 14354–14364. doi:10.1002/chem.201001849 |
| 24. | Pump, E.; Cavallo, L.; Slugovc, C. Monatsh. Chem. 2015, 146, 1131–1141. doi:10.1007/s00706-015-1433-8 |
| 7. | Garber, B.; Kingsbury, J. S.; Gray, B. L.; Hoveyda, A. H. J. Am. Chem. Soc. 2000, 122, 8168–8179. doi:10.1021/ja001179g |
| 27. | Aharoni, A.; Vidavsky, Y.; Diesendruck, C. E.; Ben-Asuly, A.; Goldberg, I.; Lemcoff, N. G. Organometallics 2011, 30, 1607–1615. doi:10.1021/om1011402 |
| 5. | Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746–1787. doi:10.1021/cr9002424 |
| 6. | Samojłowicz, C.; Bieniek, M.; Grela, K. Chem. Rev. 2009, 109, 3708–3742. doi:10.1021/cr800524f |
| 14. | Barbasiewicz, M.; Szadkowska, A.; Bujok, R.; Grela, K. Organometallics 2006, 25, 3599–3604. doi:10.1021/om060091u |
| 58. | Leitgeb, A.; Wappel, J.; Urbina-Blanco, C. A.; Strasser, S.; Wappl, C.; Cazin, C. S. J.; Slugovc, C. Monatsh. Chem. 2014, 145, 1513–1517. doi:10.1007/s00706-014-1249-y |
| 2. | Bieniek, M.; Michrowska, A.; Usanov, D. L.; Grela, K. Chem. – Eur. J. 2008, 14, 806–818. doi:10.1002/chem.200701340 |
| 3. | Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 42, 4592–4633. doi:10.1002/anie.200300576 |
| 4. | Leitgeb, A.; Wappel, J.; Slugovc, C. Polymer 2010, 51, 2927–2946. doi:10.1016/j.polymer.2010.05.002 |
| 14. | Barbasiewicz, M.; Szadkowska, A.; Bujok, R.; Grela, K. Organometallics 2006, 25, 3599–3604. doi:10.1021/om060091u |
| 18. | Hejl, A.; Day, M. W.; Grubbs, R. H. Organometallics 2006, 25, 6149–6154. doi:10.1021/om060620u |
| 19. | Slugovc, C.; Burtscher, D.; Stelzer, F.; Mereiter, K. Organometallics 2005, 24, 2255–2258. doi:10.1021/om050141f |
| 20. | De Clercq, B.; Verpoort, F. Adv. Synth. Catal. 2002, 344, 639–648. |
| 21. | Jordaan, M.; Vosloo, H. C. M. Adv. Synth. Catal. 2007, 349, 184–192. doi:10.1002/adsc.200600474 |
| 22. | Slugovc, C.; Perner, B.; Stelzer, F.; Mereiter, K. Organometallics 2004, 23, 3622–3626. doi:10.1021/om049877n |
| 23. | Poater, A.; Ragone, F.; Correa, A.; Szadkowska, A.; Barbasiewicz, M.; Grela, K.; Cavallo, L. Chem. – Eur. J. 2010, 16, 14354–14364. doi:10.1002/chem.201001849 |
| 24. | Pump, E.; Cavallo, L.; Slugovc, C. Monatsh. Chem. 2015, 146, 1131–1141. doi:10.1007/s00706-015-1433-8 |
| 29. | Michrowska, A.; Bujok, R.; Harutyunyan, S.; Sashuk, V.; Dolgonos, G.; Grela, K. J. Am. Chem. Soc. 2004, 126, 9318–9325. doi:10.1021/ja048794v |
| 55. | Diesendruck, C. E.; Vidavsky, V.; Ben-Asuly, A.; Lemcoff, N. G. J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 4209–4213. doi:10.1002/pola.23476 |
| 16. | Diesendruck, C. E.; Tzur, E.; Ben-Asuly, A.; Goldberg, I.; Straub, B. F.; Lemcoff, N. G. Inorg. Chem. 2009, 48, 10819–10825. doi:10.1021/ic901444c |
| 17. | Żukowska, K.; Szadkowska, A.; Pazio, A. E.; Woźniak, K.; Grela, K. Organometallics 2012, 31, 462–469. doi:10.1021/om2011062 |
| 25. | Ung, T.; Hejl, A.; Grubbs, R. H.; Schrodi, Y. Organometallics 2004, 23, 5399–5401. doi:10.1021/om0493210 |
| 26. | Guidone, S.; Songis, O.; Nahra, F.; Cazin, C. S. J. ACS Catal. 2015, 5, 2697–2701. doi:10.1021/acscatal.5b00197 |
| 14. | Barbasiewicz, M.; Szadkowska, A.; Bujok, R.; Grela, K. Organometallics 2006, 25, 3599–3604. doi:10.1021/om060091u |
| 56. | Żukowska, K.; Szadkowska, A.; Trzaskowski, B.; Pazio, A.; Pączek, Ł.; Woźniak, K.; Grela, K. Organometallics 2013, 32, 2192–2198. doi:10.1021/om400064c |
| 57. | Trzaskowski, B.; Grela, K. Organometallics 2013, 32, 3625–3630. doi:10.1021/om400233s |
| 12. | van der Schaaf, P. A.; Kolly, R.; Kirner, H.-J.; Rime, F.; Mühlebach, A.; Hafner, A. J. Organomet. Chem. 2000, 606, 65–74. doi:10.1016/S0022-328X(00)00289-8 |
| 13. | Szadkowska, A.; Gstrein, X.; Burtscher, D.; Jarzembska, K.; Woźniak, K.; Slugovc, C.; Grela, K. Organometallics 2010, 29, 117–124. doi:10.1021/om900857w |
| 14. | Barbasiewicz, M.; Szadkowska, A.; Bujok, R.; Grela, K. Organometallics 2006, 25, 3599–3604. doi:10.1021/om060091u |
| 15. | Peeck, L. H.; Savka, R. D.; Plenio, H. Chem. – Eur. J. 2012, 18, 12845–12853. doi:10.1002/chem.201201010 |
| 48. | Slugovc, C.; Demel, S.; Riegler, S.; Hobisch, J.; Stelzer, F. Macromol. Rapid Commun. 2004, 25, 475–480. doi:10.1002/marc.200300196 |
| 9. | Ben-Asuly, A.; Tzur, E.; Diesendruck, C. E.; Sigalov, M.; Goldberg, I.; Lemcoff, N. G. Organometallics 2008, 27, 811–813. doi:10.1021/om701180z |
| 10. | Szadkowska, A.; Makal, A.; Woźniak, K.; Kadyrov, R.; Grela, K. Organometallics 2009, 28, 2693–2700. doi:10.1021/om801183g |
| 11. | Szadkowska, A.; Żukowska, K.; Pazio, A. E.; Woźniak, K.; Kadyrov, R.; Grela, K. Organometallics 2011, 30, 1130–1138. doi:10.1021/om101129b |
| 16. | Diesendruck, C. E.; Tzur, E.; Ben-Asuly, A.; Goldberg, I.; Straub, B. F.; Lemcoff, N. G. Inorg. Chem. 2009, 48, 10819–10825. doi:10.1021/ic901444c |
| 49. | Kozłowska, A.; Dranka, M.; Zachara, J.; Pump, E.; Slugovc, C.; Skowerski, K.; Grela, K. Chem. – Eur. J. 2014, 20, 14120–14125. doi:10.1002/chem.201403580 |
| 50. | Urbina-Blanco, C. A.; Bantreil, X.; Wappel, J.; Schmid, T. E.; Slawin, A. M. Z.; Slugovc, C.; Cazin, C. S. J. Organometallics 2013, 32, 6240–6247. doi:10.1021/om4004362 |
| 51. | Pump, E.; Fischer, R. C.; Slugovc, C. Organometallics 2012, 31, 6972–6979. doi:10.1021/om300785t |
| 52. | Urbina-Blanco, C. A.; Leitgeb, A.; Slugovc, C.; Bantreil, X.; Clavier, H.; Slawin, A. M. Z.; Nolan, S. P. Chem. – Eur. J. 2011, 17, 5045–5053. doi:10.1002/chem.201003082 |
| 53. | Broggi, J.; Urbina-Blanco, C. A.; Clavier, H.; Leitgeb, A.; Slugovc, C.; Slawin, A. M. Z.; Nolan, S. P. Chem. – Eur. J. 2010, 16, 9215–9225. doi:10.1002/chem.201000659 |
| 54. | Strasser, S.; Pump, E.; Fischer, R. C.; Slugovc, C. Monatsh. Chem. 2015, 146, 1143–1151. doi:10.1007/s00706-015-1484-x |
| 30. | Pump, E.; Poater, A.; Zirngast, M.; Torvisco, A.; Fischer, R.; Cavallo, L.; Slugovc, C. Organometallics 2014, 33, 2806–2813. doi:10.1021/om500315t |
| 23. | Poater, A.; Ragone, F.; Correa, A.; Szadkowska, A.; Barbasiewicz, M.; Grela, K.; Cavallo, L. Chem. – Eur. J. 2010, 16, 14354–14364. doi:10.1002/chem.201001849 |
| 24. | Pump, E.; Cavallo, L.; Slugovc, C. Monatsh. Chem. 2015, 146, 1131–1141. doi:10.1007/s00706-015-1433-8 |
| 28. | Zaja, M.; Connon, S. J.; Dunne, A. M.; Rivard, M.; Buschmann, N.; Jiricek, J.; Blechert, S. Tetrahedron 2003, 59, 6545–6558. doi:10.1016/S0040-4020(03)01029-9 |
| 29. | Michrowska, A.; Bujok, R.; Harutyunyan, S.; Sashuk, V.; Dolgonos, G.; Grela, K. J. Am. Chem. Soc. 2004, 126, 9318–9325. doi:10.1021/ja048794v |
| 39. | van Rensburg, W. J.; Steynberg, P. J.; Meyer, W. H.; Kirk, M. M.; Forman, G. S. J. Am. Chem. Soc. 2004, 126, 14332–14333. doi:10.1021/ja0453174 |
| 40. | van Rensburg, W. J.; Steynberg, P. J.; Kirk, M. M.; Meyer, W. H.; Forman, G. S. J. Organomet. Chem. 2006, 691, 5312–5325. doi:10.1016/j.jorganchem.2006.08.075 |
| 41. | Bourgeois, D.; Pancrazi, A.; Nolan, S. P.; Prunet, J. J. Organomet. Chem. 2002, 643–644, 247–252. doi:10.1016/S0022-328X(01)01269-4 |
| 42. | Lehman, S. E., Jr.; Schwendeman, J. E.; O’Donnel, P. M.; Wagener, K. B. Inorg. Chim. Acta 2003, 345, 190–198. doi:10.1016/S0020-1693(02)01307-5 |
| 43. | Feng, C.; Wang, X.; Wang, B.-Q.; Zhao, K.-Q.; Hu, P.; Shi, Z.-J. Chem. Commun. 2012, 48, 356–358. doi:10.1039/C1CC15835J |
| 37. | Nelson, D. J.; Percy, J. M. Dalton Trans. 2014, 43, 4674–4679. doi:10.1039/C4DT00007B |
| 33. | Nelson, D. J.; Manzini, S.; Urbina-Blanco, C. A.; Nolan, S. P. Chem. Commun. 2014, 50, 10355–10375. doi:10.1039/C4CC02515F |
| 32. | Puentener, K.; Scalone, M. New ruthenium complexes as catalysts for metathesis reactions. WO Patent WO2008000644, Jan 3, 2008. |
| 34. | Lummiss, J. A. M.; Beach, N. J.; Smith, J. C.; Fogg, D. E. Catal. Sci. Technol. 2012, 2, 1630–1632. doi:10.1039/C2CY20213A |
| 35. | Sanford, M. S.; Love, J. A.; Grubbs, R. H. J. Am. Chem. Soc. 2001, 123, 6543–6554. doi:10.1021/ja010624k |
| 36. | Hong, S. H.; Wenzel, A. G.; Salguero, T. T.; Day, M. W.; Grubbs, R. H. J. Am. Chem. Soc. 2007, 129, 7961–7968. doi:10.1021/ja0713577 |
| 14. | Barbasiewicz, M.; Szadkowska, A.; Bujok, R.; Grela, K. Organometallics 2006, 25, 3599–3604. doi:10.1021/om060091u |
| 31. | Grela, K.; Barbasiewicz, M.; Szadkowska, A. Complexes of Ruthenium and osmium, method of production thereof and use thereof as (pre)catalysts of the metathesis reaction. WO Patent WO140954, Dec 13, 2007. |
| 32. | Puentener, K.; Scalone, M. New ruthenium complexes as catalysts for metathesis reactions. WO Patent WO2008000644, Jan 3, 2008. |
© 2015 Żukowska et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)