Abstract
The novel dicationic metathesis catalyst [(RuCl2(H2ITapMe2)(=CH–2-(2-PrO)-C6H4))2+ (OTf−)2] (Ru-2, H2ITapMe2 = 1,3-bis(2’,6’-dimethyl-4’-trimethylammoniumphenyl)-4,5-dihydroimidazol-2-ylidene, OTf− = CF3SO3−) based on a dicationic N-heterocyclic carbene (NHC) ligand was prepared. The reactivity was tested in ring opening metathesis polymerization (ROMP) under biphasic conditions using a nonpolar organic solvent (toluene) and the ionic liquid (IL) 1-butyl-2,3-dimethylimidazolium tetrafluoroborate [BDMIM+][BF4−]. The structure of Ru-2 was confirmed by single crystal X-ray analysis.
Graphical Abstract
Introduction
Ionic metathesis catalysts offer access to metathesis reactions in either aqueous solution [1-10] or under biphasic conditions [11-14]. Particularly the latter aspect is of utmost relevance in case of ionic liquids (ILs) can be used as the phase in which the catalyst is dissolved. The ionic character of both the IL and the ionic catalyst effectively block any crossover of catalyst into the second (organic) phase. This offers access to metathesis reactions in which the products have a low ruthenium contamination [11]. Equally important, reactions can be run under biphasic, continuous conditions applying supported ionic liquid phase (SILP) technology [11]. We recently reported on different Ru-based ionic metathesis catalysts that can be used for these purposes. In these systems, the charge is either located directly at the ruthenium [11,12] or at the 1-methylpyridinium-4-carboxylate ligands that are introduced via anion metathesis [13,14]. These novel catalytic systems have successfully been used under SILP conditions [11,15]. Furthermore, they allow running ring-opening metathesis polymerization (ROMP) reactions under biphasic conditions, an approach that offers access to both ROMP-derived polymers with unprecedented low Ru contamination (typically 25–80 ppm) and to a regeneration of the initiator [14]. With all that systems at hand it also became apparent that reactivity of a certain catalyst strongly depends on the location of the charge. In principle, ionic Ru-based metathesis catalysts can also be prepared with the aid of N-heterocyclic carbenes (NHCs) that bear pendant ionic groups (Figure 1) [10,16-19]. We addressed that issue by preparing a novel ionic Ru-NHC-alkylidene using NHCs with ionic groups. Here we report our results.
![[1860-5397-11-178-1]](/bjoc/content/figures/1860-5397-11-178-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Catalysts synthesized by post-assembly tagging (Mes = mesitylene).
Figure 1: Catalysts synthesized by post-assembly tagging (Mes = mesitylene).
Results and Discussion
Catalyst synthesis
We were attracted by NHC ligands containing a diamino function at the aromatic ring as realized in 1 [20-22] since such ligands can be permanently quaternized to the corresponding dicationic species via double alkylation. Additionally, they remain structurally closely related to mesitylene-based NHC ligands. Attempts to synthesize ionic Ru-based olefin metathesis catalysts using imidazolinium salts bearing two quaternary ammonium groups turned out to be unsuccessful, probably due to their insolubility in common organic solvents. However, quaternization of RuCl2(H2ITap)(=CH–(2-(2-PrO-C6H4))) (Ru-1, Scheme 1) [21] turned out to be successful.
![[1860-5397-11-178-i1]](/bjoc/content/inline/1860-5397-11-178-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Improved synthesis of Ru-1 and quarternization with methyl trifluoromethanesulfonate to Ru-2.
Scheme 1: Improved synthesis of Ru-1 and quarternization with methyl trifluoromethanesulfonate to Ru-2.
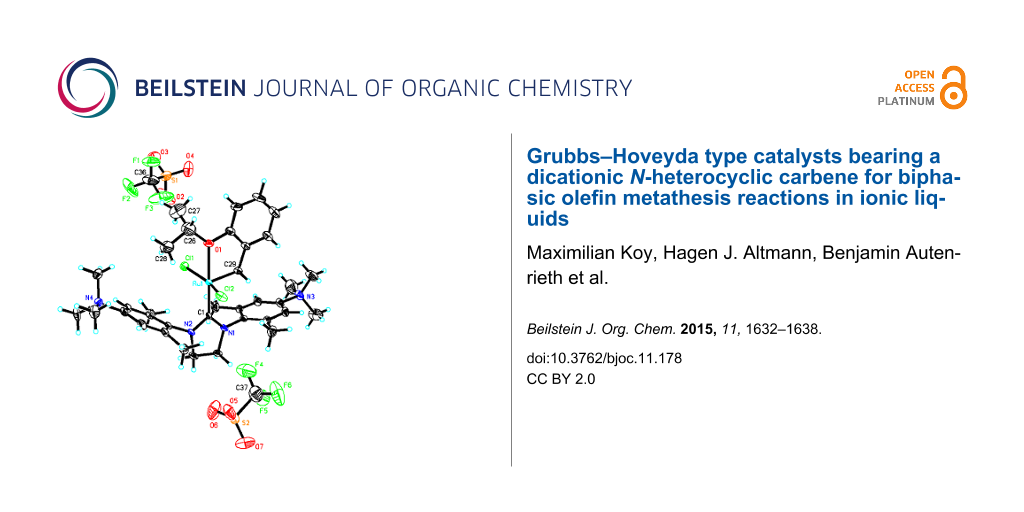
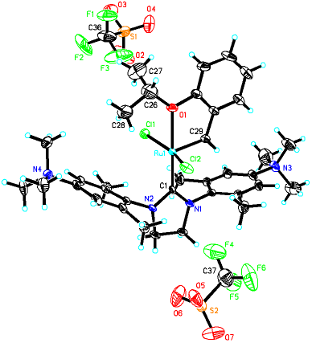
To ensure the solubility in common organic solvents until the last step of the synthesis, the neutral precursor Ru-1 [21] was prepared in an improved one-step synthesis in 82% yield. Quarternization using 2.05 equiv of methyl trifluoromethanesulfonate gave the dicationic ruthenium alkylidene Ru-2 in 90% isolated yield (Scheme 1). This is to the best of our knowledge the first example of a ruthenium alkylidene bearing an NHC ligand with permanent dicationic charge. Crystals of Ru-2 suitable for single-crystal X-ray analysis were obtained from DMF/diethyl ether. Catalyst Ru-2 crystallizes in the triclinic space group ![[Graphic 1]](/bjoc/content/inline/1860-5397-11-178-i3.svg?max-width=637&scale=1.18182) , a = 1398.08(6) pm, b = 1399.41(7) pm, c = 1750.26(13) pm, α = 106.079(3)°, β = 112.209(3)°, γ = 99.300(2)°, Z = 2 (Figure 2, Supporting Information File 1).
, a = 1398.08(6) pm, b = 1399.41(7) pm, c = 1750.26(13) pm, α = 106.079(3)°, β = 112.209(3)°, γ = 99.300(2)°, Z = 2 (Figure 2, Supporting Information File 1).
![[1860-5397-11-178-2]](/bjoc/content/figures/1860-5397-11-178-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Single crystal X-ray structure of Ru-2. Co-solvent and disordered triflates have been omitted for clarity.
Figure 2: Single crystal X-ray structure of Ru-2. Co-solvent and disordered triflates have been omitted for c...
Selected bond lengths are summarized in Table 1. For purposes of comparison, the corresponding distances of the parent system Ru-1 are provided, too. As can be seen, the dicationic charge does influence the binding situation in Ru-2, though not dramatically. Interestingly only a slight increase in the Ru–NHC bond length is observed, accompanied by a very minor decrease in the Ru–O bond. The Ru–Cl bonds remain unaffected. The most dramatic effect is observed in the Ru–alkylidene bond, which is about 9 pm longer in Ru-2 than in Ru-1. This increase in the alkylidene’s length points towards a substantially reduced polarization of the Ru=C bond and accounts for a reduced activity of Ru-2 compared to standard Grubbs- and Grubbs–Hoveyda catalysts. Thus, Ru-2 delivers only turn-over numbers well below 100 in the biphasic ring-closing metathesis (RCM) of 1,7-octadiene, diethyl diallylmalonate and N,N-diallyl p-toluoenesulfonamide using [BDMIM+][BF4−] as IL and toluene as the organic phase (see Supporting Information File 1). It is thus also in line with the fact that Ru–alkylidenes based on electron-rich NHCs, e.g., based on tetrahydropyrimidin-2-ylidenes [23], strongly promote olefin metathesis.
Table 1: Selected bond lengths (pm) for Ru-2 and Ru-1 [21].
| Ru-2 | Ru-1 | |
|---|---|---|
| Ru1–C1 | 198.2(4) | 196.6(7) |
| Ru1–C29 | 182.8(4) | 173.5(9) |
| Ru1–O1 | 225.2(3) | 226.0(5) |
| Ru1–Cl1 | 233.89(12) | 233.0(2) |
| Ru1–Cl2 | 233.50(12) | 233.9(2) |
Biphasic ring-opening metathesis polymerization (ROMP) reactions
To test the reactivity of Ru-2, various ROMP reactions were run under biphasic conditions using [BDMIM+][BF4−] [24] as IL and toluene as the organic phase. The structure of monomers M1–M6 that were used are shown in Figure 3 [14]. Results are summarized in Table 2. At this point it is worth stressing that the purity of the IL used is of utmost importance, the more since imidazolium-based ILs can contain substantial amounts of free base [25], which in turn can negatively affect catalyst performance.
![[1860-5397-11-178-3]](/bjoc/content/figures/1860-5397-11-178-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Monomers used for biphasic ROMP reactions.
Figure 3: Monomers used for biphasic ROMP reactions.
Table 2: ROMP reactions under biphasic conditions.a,b
| Monomer | T [°C] | Time [h] | Yield [%]c |
Mtheo
[g/mol] |
Mn
[g/mol]d |
PDId |
|---|---|---|---|---|---|---|
| M1 | 50 | 2 | 93 | 6,600 | 258,000 | 3.8 |
| M2 | 50 | 2 | 89 | 14,700 | 94,500 | 2.3 |
| M3 | 50 | 2 | 80 | 16,800 | 15,800 | 1.2 |
| M4 | 50 | 2 | 86 | 20,600 | 907,000 | 2.8 |
| M5 | 50 | 1 | 100 | 6,500 | –e | –e |
| M6 | 70 | 3 | 36 | 7,700 | 186,000 | 1.50 |
aRu-2, toluene, [BDMIM+][BF4−], 50−70 °C, 1−3 h. bRu content (measured by ICP–OES) was lower than the limit of detection, which allows for calculating a Ru content <2.5 ppm. cDetermined after precipitation in methanol. dMeasured by GPC in THF. eInsoluble because of crosslinking.
As can be seen, M1–M6 can be polymerized via ROMP under biphasic conditions in good yields, except for M6. This low yield is attributed to the comparable low ring strain in M6. Polydispersity indices (PDIs) were in the range of 1.2 to 3.8. Together with the high molecular weights, this is indicative for substantial chain transfer and potentially incomplete initiation of the Ru–alkylidene, particularly with unsubstituted norbornene (M1) and cis-cyclooctene (M6) but also with M4. However, in turn it allows for the synthesis of high molecular weight polymers. The most striking feature, however, is related to the Ru content of the resulting polymers, which were all obtained as white powders. Unlike in many other Ru–alkylidene-triggered metathesis-based polymerizations, Ru contamination was very low (<2.5 ppm) and even outrivals earlier reported systems bearing two pyridinium carboxylates by at least a factor of 10 [14]. Clearly, both the initiator and any Ru-containing decomposition products selectively stay in the IL phase while after termination, the polymer stays selectively in the organic (toluene) phase. Notably, polymers were simply precipitated from methanol and not subjected to any further purification steps. We believe that particularly for biomedical applications such virtually Ru-free polymers will be of utmost interest.
Recycling experiments carried out with M1 revealed that with the aid of 2-(2-PrO)-styrene, Ru-2 could be used in three consecutive cycles (Scheme 2). Over these three cycles, the number-average molecular weight, Mn, significantly decreased while PDIs increased from 2.1 to 3.0. Again, Ru leaching into the product was below the limit of detection, i.e., <2.5 ppm. The results are summarized in Table 3.
![[1860-5397-11-178-i2]](/bjoc/content/inline/1860-5397-11-178-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Recycling of Ru-2 for continuous ROMP reactions.
Scheme 2: Recycling of Ru-2 for continuous ROMP reactions.
Table 3: ROMP of M1 under biphasic conditions with recycling.a
| Cycle | Yield [%]b | Mn [g/mol]c | PDIc |
|---|---|---|---|
| 1 | 89 | 1,340,000 | 2.1 |
| 2 | 83 | 230,000 | 3.0 |
| 3 | 75 | 120,000 | 3.0 |
a1. Ru-2, M1, toluene, [BDMIM+][BF4−], 50 °C, 1.5 h; 2. 2-(2-PrO)-styrene, toluene, 50 °C, 1 h; 3. M1, toluene, 50 °C, 1.5 h; following cycles: repeat 2 + 3; last cycle: quenched with ethyl vinyl ether. bDetermined after precipitation in methanol. cMeasured by GPC in THF, Mn, theor: 6,600 g/mol.
Conclusion
The first dicationic Ru–alkylidene catalyst based on an N-heterocyclic carbene bearing two quaternary ammonium groups, [(RuCl2(H2ITapMe2)(=CH–2-(2-PrO)-C6H4))2+ (OTf−)2] (Ru-2), was prepared from the neutral precursor RuCl2(H2ITap)(=CH–2-(2-PrO)-C6H4)) (Ru-1) and methyl trifluoromethanesulfonate. Also, an improved, high-yield synthesis of Ru-1 has been presented. Ru-2 was tested for its reactivity in ROMP under biphasic conditions using [BDMIM+][BF4−] as the ionic liquid and toluene as the organic solvent. While Ru-2 showed low RCM activity, it turned out to be active in ROMP reactions of strained cyclic olefins like norbornenes, 7-oxanorbornenes, norbornadiene and cis-cyclooctene allowing for the synthesis of the corresponding polymers with unprecedented low metal contamination (<2.5 ppm) without any additional purification steps.
Experimental
General: Unless noted otherwise, all manipulations were performed in a Labmaster 130 glovebox (MBraun; Garching, Germany) or by standard Schlenk techniques under N2 atmosphere. CH2Cl2 and toluene were purchased from J. T. Baker (Devender, Netherlands) and were dried by using an MBraun SPS-800 solvent purification system. Hexane was purchased from VWR and distilled from sodium/benzophenone under N2. Starting materials were purchased from ABCR, Aldrich, Alfa Aesar, Fluka and TCI Europe and used without further purification. KOCMe2Et was purchased from Alfa Aesar as a 25 wt % solution in toluene. Toluene was co-evaporated with pentane in vacuo prior to use.
NMR spectra were recorded on a Bruker Avance III 400 spectrometer in the indicated solvent at 25 °C and are listed in parts per million downfield from tetramethylsilane as an internal standard. IR spectra were measured on a Bruker ATR/FT-IR IFS 128. GPC measurements were carried out on a system, consisting of a Waters 515 HPLC pump, a Waters 2707 autosampler, Polypore columns (300 × 7.5 mm, Agilent technologies, Böblingen, Germany), a Waters 2489 UV–vis and a Waters 2414 refractive index detector. For calibration, polystyrene standards with 800 < Mn < 2,000,000 g/mol were used. ICP–OES measurements were carried out using a Spectro Acros device (Ametek GmbH; Meerbusch, Germany). Calibration was done with Ru standards containing 0.1, 0.5, 1.0 and 5.0 ppm. Mass spectra were recorded on a Bruker Daltonics Microtof Q mass spectrometer at the Institute of Organic Chemistry at the University of Stuttgart. 1,3-Bis(2,6-dimethyl-4-dimethylaminophenyl)-4,5-dihydroimidazol-2-ylidene (1) [21], 2-(2-PrO)-styrene [23,24], M3 [26] and M4 [27] were prepared according to the literature.
RuCl2(H2ITap)(=CH-2-(2-PrO)-C6H4-O) (Ru-1) [27,28]: Inside a glovebox, 1 (281 mg, 0.70 mmol), KOCMe2Et (88 mg, 0.70 mmol) and hexane (9 mL) were added to a 50 mL Schlenk flask equipped with a magnetic stir bar. The reaction mixture was allowed to stir for 1 h at room temperature, during which time a brownish orange suspension formed. The 1st-generation Grubbs–Hoveyda catalyst (400 mg, 0.67 mmol) dissolved in hexane (6 mL) was added to the reaction mixture. The reaction mixture was removed from the glovebox and was heated to 60 °C for 4 h. The formation of a green solid was observed. After cooling to room temperature, all solids were filtered off and washed with pentane (3 × 10 mL) and diethyl ether (3 × 10 mL); then the product was redissolved in CH2Cl2. Purification was accomplished by chromatography using silica G60 and CH2Cl2/hexane. Drying in vacuo gave the product as a dark green solid (375 mg, 0.55 mmol, 82%). Analytical data were in accordance with the literature [20].
[(RuCl2(H2ITapMe2)(=CH–2-(2-PrO)-C6H4))2+ (OTf-)2] (Ru-2): At −30 °C, methyl trifluormethanesulfonate (99 mg, 601 µmol) dissolved in CH2Cl2 (3 mL) was added to Ru-1 (200.5 mg, 293 µmol) dissolved in CH2Cl2 (5 mL). The mixture was stirred for 18 h at room temperature and then, CH2Cl2 was removed under reduced pressure. The residue was washed with CH2Cl2 (3 × 3 mL) and ethyl acetate (3 × 3 mL), allowing for the isolation of the target compound as a light-green solid (267 mg, 264 µmol, 90%). 1H NMR (DMF-d7) δ 16.47 (s, 1H, Ru=CH), 8.17 (s, 4H, NHC-Ar), 7.70–7.66 (m, 1H, C6H4), 7.20 (d, J = 8.4 Hz, 1H, C6H4), 7.06–7.04 (m, 1H, C6H4), 6.96 (t, J = 7.4 Hz, 1H, C6H4), 5.10 (hept, J = 7.4 Hz, 1H, O-CH-(CH3)2), 4.42 (s, 4H, N-CH2), 4.00 (s, 18H, N-(CH3)3), 2.66 (s, 12H, NHC-Ar-CH3), 1.27 (d, J = 6.1 Hz, 6H, O-CH-CH3); 13C NMR (DMF-d7) δ 292.9 (Ru=CH), 211.6 (N-C=N), 153.5, 148.7, 145.9, 143.1, 141.2, 131.3, 123.6, 123.5, 121.8, 114.5 (C6H2, C6H4), 127.3, 124.1, 120.9, 117.7 (CF3-SO3−, q, JC-F = 322.5 Hz), 76.4 (CH(CH3)2), 57.9 (N(CH3)3), 52.6, 21.9, 20.3 (CH(CH3)2, Ar-CH3); 19F NMR (DMF-d7) δ −78.5; FTIR (ATR, cm−1) ![[Graphic 2]](/bjoc/content/inline/1860-5397-11-178-i4.svg?max-width=637&scale=1.18182) : 1589 (s), 1491 (m), 1251 (m), 1152 (m), 1114 (m), 1028 (s), 923 (m), 840 (s), 801 (s), 754 (s), 637 (s), 572 (s), 517 (s) cm-1; MS (ESI) m/z: calcd. for C35H50Cl2N4ORu (dication, z = 2): 357.1199, found: 357.1214; m/z calcd. for C36H50F3Cl2N4O4RuS: 863.1923, found: 863.1905. Crystals suitable for X-ray diffraction were obtained by layering diethyl ether over a solution of Ru-2 in anhydrous DMF.
: 1589 (s), 1491 (m), 1251 (m), 1152 (m), 1114 (m), 1028 (s), 923 (m), 840 (s), 801 (s), 754 (s), 637 (s), 572 (s), 517 (s) cm-1; MS (ESI) m/z: calcd. for C35H50Cl2N4ORu (dication, z = 2): 357.1199, found: 357.1214; m/z calcd. for C36H50F3Cl2N4O4RuS: 863.1923, found: 863.1905. Crystals suitable for X-ray diffraction were obtained by layering diethyl ether over a solution of Ru-2 in anhydrous DMF.
General ROMP-procedure: Ru-2 (5.6 mg, 5 µmol or 11.13 mg, 10 µmol) and [BDMIM+][BF4−] (400 mg) were placed inside a flame-dried Schlenk tube (25 mL) equipped with a magnetic stir bar. The reaction mixture was heated to the indicated temperature. The monomer (350 µmol or 700 µmol) and toluene (2 mL) were added to a separate flame-dried Schlenk tube. The monomer solution was added via syringe in one portion and the reaction mixture was allowed to stir at the indicated temperature for the indicated time. After cooling to room temperature, ethyl vinyl ether (1 mL) was added and the reaction mixture was allowed to stir for another 30 min. Finally, the reaction mixture was poured into methanol. The polymer was obtained as a white or off-white solid.
General ROMP-procedure with recycling: Ru-2 (11.1 mg, 10 µmol) and [BDMIM+][BF4−] (400 mg) were placed inside a flame-dried Schlenk tube (25 mL) equipped with a magnetic stir bar. The reaction mixture was heated to the indicated temperature. M1 (65.9 mg, 700 µmol) and toluene (5 mL) were added to a separate flame-dried Schlenk tube. The monomer solution was added via syringe in one portion and the reaction mixture was allowed to stir at 50 °C for 1.5 h. Then a solution of 2-(2-PrO)-styrene in anhydrous toluene (1 mL, 1 M) was added. The reaction mixture was stirred at 50 °C for 1 h. The two phases were allowed to separate. The organic phase was poured into methanol. The IL phase was extracted with toluene (4 × 2 mL). The extracted organic phases were also poured into methanol. Poly-M1 was obtained as a white solid. New M1 was added to the IL phase and the procedure was repeated. After the last cycle, the reaction was quenched with ethyl vinyl ether (1 mL).
ICP–OES measurements: The corresponding polymer (20 mg) was added to high-pressure Teflon tubes. Digestion was performed under microwave conditions using aqua regia (10 mL). The mixture was cooled to room temperature, diluted with deionized water (approx. 40 mL), filtered and subjected to ICP–OES for Ru with λ = 240.272 nm ion line and background lines at λ1 = 240.254 nm and λ2 = 240.295 nm.
Poly-M1: 1H NMR (CDCl3) δ 5.35 (s, 1H), 5.21 (s, 1H), 2.79 (bs, 1H), 2.44 (bs, 1H), 1.88–1.79 (2bs, 3H), 1.35 (bs, 2H), 1.09–1.02 (m, 2H); 13C NMR (CDCl3) δ 134.1, 134.0, 133.9, 133.3, 133.2, 133.0, 43.6, 43.3, 42.9, 42.2, 41.5, 38.8, 38.6, 33.3, 33.1, 32.5, 32.4; FTIR (ATR, cm−1) : 2941 (s), 2863 (m), 1446 (w), 1260 (s), 1189 (w), 1081 (s), 1020 (s), 965 (s), 862 (w), 798 (s), 737 (m), 688 (w).
Poly-M2: 1H NMR (CDCl3) δ 5.52 (bs, 2H), 3.64–3.60 (2bs, 6H), 3.13–2.85 (4bs, 4H), 1.89 (2bs, 2H); 13C NMR (CDCl3) δ 172.9, 172.5, 131.9, 131.1, 130.5, 51.7, 51.6, 51.5, 51.4, 51.1, 44.5, 39.2, 38.8, 38.1; FTIR (ATR, cm−1) : 3020 (w), 2951 (w), 1726 (s), 1434 (m), 1386 (w), 1347 (w), 1194 (m), 1169 (m), 1153 (m), 1095 (w), 1041 (w), 978 (w), 957 (w), 807 (w), 747 (m), 666 (w), 603 (w).
Poly-M3: 1H NMR (CDCl3) δ 5.74 (bs, 1H), 5.56 (bs, 1H), 4.59–4.49 (2bs, 1H), 4.17–4.11 (2bs, 5H), 2.39 (bs, 2H), 2.04–2.02 (2s, 6H); 13C NMR (CDCl3) δ 170.8, 170.7, 133.3, 132.9, 132.4, 131.8, 81.5, 81.2, 62.0, 61.9, 61.8, 46.5, 46.2, 46.1, 45.9, 45.6, 21.0, 20.9; FTIR (ATR, cm−1) : 3017 (w), 2958 (w), 2902 (w), 1733 (s), 1468 (w), 1434 (w), 1389 (w), 1366 (m), 1221 (s), 1119 (w), 1030 (s), 968 (m), 834 (w), 750 (m), 667 (w), 604 (m), 506 (w), 471 (w).
Poly-M4: 1H NMR (CDCl3) δ 5.27–5.17 (2bs, 2H), 3.40–3.35 (2bs, 8H), 2.69 (bs, 1H), 2.32 (bs, 1H), 1.96 (bs, 3H), 1.55 (bs, 4H), 1.32–1.13 (2bs, 10H), 0.90 (bs, 6H); 13C NMR (CDCl3) δ 134.0, 133.8, 71.3, 71.2, 70.8, 70.6, 48.1, 47.8, 47.6, 47.0, 45.6, 45.3, 41.2, 40.1, 29.7, 28.7, 22.7, 14.2; FTIR (ATR, cm−1) : 3005 (w), 2919 (s), 2850 (s), 1465 (m), 1438 (w), 1261 (w), 1091 (w), 1071 (w), 1026 (w), 965 (s), 805 (w), 720 (w).
Poly-M6: 1H NMR (CDCl3) δ 5.43–5.31 (m, 2H), 2.02–1.97 (m, 4H), 1.33–1.29 (bs, 8H); 13C NMR (CDCl3) δ 130.5, 130.0, 32.8, 29.9, 29.8, 29.4, 29.3, 29.2, 27.4; FTIR (ATR, cm−1) : 2954 (m), 2929 (m), 2853 (m), 1795 (w), 1482 (w), 1465 (w), 1367 (w), 1104 (s), 1066 (m), 1010 (w), 966 (w), 741 (m).
Supporting Information
| Supporting Information File 1: Analytical data for Ru-2, the polymers prepared, details on the single crystal X-ray structural analysis of Ru-2, results for biphasic RCM. | ||
| Format: PDF | Size: 1.1 MB | Download |
References
-
Gallivan, J. P.; Jordan, J. P.; Grubbs, R. H. Tetrahedron Lett. 2005, 46, 2577–2580. doi:10.1016/j.tetlet.2005.02.096
Return to citation in text: [1] -
Hong, S. H.; Grubbs, R. H. J. Am. Chem. Soc. 2006, 128, 3508–3509. doi:10.1021/ja058451c
Return to citation in text: [1] -
Jordan, J. P.; Grubbs, R. H. Angew. Chem., Int. Ed. 2007, 119, 5244–5247. doi:10.1002/ange.200701258
Return to citation in text: [1] -
Kirkland, T. A.; Lynn, D. M.; Grubbs, R. H. J. Org. Chem. 1998, 63, 9904–9909. doi:10.1021/jo981678o
Return to citation in text: [1] -
Lynn, D. M.; Mohr, B.; Grubbs, R. H. J. Am. Chem. Soc. 1998, 120, 1627–1628. doi:10.1021/ja9736323
Return to citation in text: [1] -
Lynn, D. M.; Mohr, B.; Grubbs, R. H. Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.) 1998, 39, 278–279.
Return to citation in text: [1] -
Lynn, D. M.; Mohr, B.; Grubbs, R. H.; Henling, L. M.; Day, M. W. J. Am. Chem. Soc. 2000, 122, 6601–6609. doi:10.1021/ja0003167
Return to citation in text: [1] -
Mohr, B.; Lynn, D. M.; Grubbs, R. H. Organometallics 1996, 15, 4317–4325. doi:10.1021/om9603373
Return to citation in text: [1] -
Wagaman, M. W.; Grubbs, R. H. Macromolecules 1997, 30, 3978–3985. doi:10.1021/ma9701595
Return to citation in text: [1] -
Klučiar, M.; Grela, K.; Mauduit, M. Dalton Trans. 2013, 42, 7354–7358. doi:10.1039/c2dt32856a
Return to citation in text: [1] [2] -
Autenrieth, B.; Frey, W.; Buchmeiser, M. R. Chem. – Eur. J. 2012, 18, 14069–14078. doi:10.1002/chem.201201199
Return to citation in text: [1] [2] [3] [4] [5] -
Autenrieth, B.; Anderson, E. B.; Wang, D.; Buchmeiser, M. R. Macromol. Chem. Phys. 2013, 214, 33–40. doi:10.1002/macp.201200544
Return to citation in text: [1] [2] -
Autenrieth, B.; Willig, F.; Pursley, D.; Naumann, S.; Buchmeiser, M. R. ChemCatChem 2013, 5, 3033–3040. doi:10.1002/cctc.201300199
Return to citation in text: [1] [2] -
Ferraz, C. P.; Autenrieth, B.; Frey, W.; Buchmeiser, M. R. ChemCatChem 2014, 6, 191–198. doi:10.1002/cctc.201300751
Return to citation in text: [1] [2] [3] [4] [5] -
Zhao, J.; Wang, D.; Autenrieth, B.; Buchmeiser, M. R. Macromol. Rapid Commun. 2015, 36, 190–194. doi:10.1002/marc.201400413
Return to citation in text: [1] -
Skowerski, K.; Szczepaniak, G.; Wierzbicka, C.; Gułajski, Ł.; Bieniek, M.; Grela, K. Catal. Sci. Technol. 2012, 2, 2424–2427. doi:10.1039/c2cy20320k
Return to citation in text: [1] -
Skowerski, K.; Wierzbicka, C.; Szczepaniak, G.; Gułajski, L.; Bienieka, M.; Grela, K. Green Chem. 2012, 14, 3264–3268. doi:10.1039/c2gc36015b
Return to citation in text: [1] -
Szczepaniak, G.; Kosiński, K.; Grela, K. Green Chem. 2014, 16, 4474–4492. doi:10.1039/C4GC00705K
Return to citation in text: [1] -
Kośnik, W.; Grela, K. Dalton Trans. 2013, 42, 7463–7467. doi:10.1039/C3DT33010A
Return to citation in text: [1] -
Leuthäußer, S.; Schwarz, D.; Plenio, H. Chem. – Eur. J. 2007, 13, 7195–7209. doi:10.1002/chem.200700228
Return to citation in text: [1] [2] -
Balof, S. L.; P'Pool, S. J.; Berger, N. J.; Valente, E. J.; Shiller, A. M.; Schanz, H.-J. Dalton Trans. 2008, 5791–5799. doi:10.1039/B809793C
Return to citation in text: [1] [2] [3] [4] [5] -
Balof, S. L.; Yu, B.; Lowe, A. B.; Ling, Y.; Zhang, Y.; Schanz, H.-J. Eur. J. Inorg. Chem. 2009, 1717–1722. doi:10.1002/ejic.200801145
Return to citation in text: [1] -
Krause, J. O.; Wurst, K.; Nuyken, O.; Buchmeiser, M. R. Chem. – Eur. J. 2004, 10, 777–784. doi:10.1002/chem.200305031
Return to citation in text: [1] [2] -
Vygodskii, Y. S.; Shaplov, A. S.; Lozinskaya, E. I.; Filippov, O. A.; Shubina, E. A.; Bandari, R.; Buchmeiser, M. R. Macromolecules 2006, 39, 7821–7830. doi:10.1021/ma061456p
Return to citation in text: [1] [2] -
Aitken, B. S.; Lee, M.; Hunley, M. T.; Gibson, H. W.; Wagener, K. B. Macromolecules 2010, 43, 1699–1701. doi:10.1021/ma9024174
Return to citation in text: [1] -
Takao, K.-i.; Yasui, H.; Yamamoto, S.; Sasaki, D.; Kawasaki, S.; Watanabe, G.; Tadano, K.-i. J. Org. Chem. 2004, 69, 8789–8795. doi:10.1021/jo048566j
Return to citation in text: [1] -
Endo, K.; Grubbs, R. H. J. Am. Chem. Soc. 2011, 133, 8525–8527. doi:10.1021/ja202818v
Return to citation in text: [1] [2] -
Jafarpour, L.; Hillier, A. C.; Nolan, S. P. Organometallics 2002, 21, 442–444. doi:10.1021/om0109511
Return to citation in text: [1]
| 21. | Balof, S. L.; P'Pool, S. J.; Berger, N. J.; Valente, E. J.; Shiller, A. M.; Schanz, H.-J. Dalton Trans. 2008, 5791–5799. doi:10.1039/B809793C |
| 25. | Aitken, B. S.; Lee, M.; Hunley, M. T.; Gibson, H. W.; Wagener, K. B. Macromolecules 2010, 43, 1699–1701. doi:10.1021/ma9024174 |
| 14. | Ferraz, C. P.; Autenrieth, B.; Frey, W.; Buchmeiser, M. R. ChemCatChem 2014, 6, 191–198. doi:10.1002/cctc.201300751 |
| 1. | Gallivan, J. P.; Jordan, J. P.; Grubbs, R. H. Tetrahedron Lett. 2005, 46, 2577–2580. doi:10.1016/j.tetlet.2005.02.096 |
| 2. | Hong, S. H.; Grubbs, R. H. J. Am. Chem. Soc. 2006, 128, 3508–3509. doi:10.1021/ja058451c |
| 3. | Jordan, J. P.; Grubbs, R. H. Angew. Chem., Int. Ed. 2007, 119, 5244–5247. doi:10.1002/ange.200701258 |
| 4. | Kirkland, T. A.; Lynn, D. M.; Grubbs, R. H. J. Org. Chem. 1998, 63, 9904–9909. doi:10.1021/jo981678o |
| 5. | Lynn, D. M.; Mohr, B.; Grubbs, R. H. J. Am. Chem. Soc. 1998, 120, 1627–1628. doi:10.1021/ja9736323 |
| 6. | Lynn, D. M.; Mohr, B.; Grubbs, R. H. Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.) 1998, 39, 278–279. |
| 7. | Lynn, D. M.; Mohr, B.; Grubbs, R. H.; Henling, L. M.; Day, M. W. J. Am. Chem. Soc. 2000, 122, 6601–6609. doi:10.1021/ja0003167 |
| 8. | Mohr, B.; Lynn, D. M.; Grubbs, R. H. Organometallics 1996, 15, 4317–4325. doi:10.1021/om9603373 |
| 9. | Wagaman, M. W.; Grubbs, R. H. Macromolecules 1997, 30, 3978–3985. doi:10.1021/ma9701595 |
| 10. | Klučiar, M.; Grela, K.; Mauduit, M. Dalton Trans. 2013, 42, 7354–7358. doi:10.1039/c2dt32856a |
| 11. | Autenrieth, B.; Frey, W.; Buchmeiser, M. R. Chem. – Eur. J. 2012, 18, 14069–14078. doi:10.1002/chem.201201199 |
| 12. | Autenrieth, B.; Anderson, E. B.; Wang, D.; Buchmeiser, M. R. Macromol. Chem. Phys. 2013, 214, 33–40. doi:10.1002/macp.201200544 |
| 24. | Vygodskii, Y. S.; Shaplov, A. S.; Lozinskaya, E. I.; Filippov, O. A.; Shubina, E. A.; Bandari, R.; Buchmeiser, M. R. Macromolecules 2006, 39, 7821–7830. doi:10.1021/ma061456p |
| 11. | Autenrieth, B.; Frey, W.; Buchmeiser, M. R. Chem. – Eur. J. 2012, 18, 14069–14078. doi:10.1002/chem.201201199 |
| 14. | Ferraz, C. P.; Autenrieth, B.; Frey, W.; Buchmeiser, M. R. ChemCatChem 2014, 6, 191–198. doi:10.1002/cctc.201300751 |
| 11. | Autenrieth, B.; Frey, W.; Buchmeiser, M. R. Chem. – Eur. J. 2012, 18, 14069–14078. doi:10.1002/chem.201201199 |
| 23. | Krause, J. O.; Wurst, K.; Nuyken, O.; Buchmeiser, M. R. Chem. – Eur. J. 2004, 10, 777–784. doi:10.1002/chem.200305031 |
| 20. | Leuthäußer, S.; Schwarz, D.; Plenio, H. Chem. – Eur. J. 2007, 13, 7195–7209. doi:10.1002/chem.200700228 |
| 11. | Autenrieth, B.; Frey, W.; Buchmeiser, M. R. Chem. – Eur. J. 2012, 18, 14069–14078. doi:10.1002/chem.201201199 |
| 12. | Autenrieth, B.; Anderson, E. B.; Wang, D.; Buchmeiser, M. R. Macromol. Chem. Phys. 2013, 214, 33–40. doi:10.1002/macp.201200544 |
| 13. | Autenrieth, B.; Willig, F.; Pursley, D.; Naumann, S.; Buchmeiser, M. R. ChemCatChem 2013, 5, 3033–3040. doi:10.1002/cctc.201300199 |
| 14. | Ferraz, C. P.; Autenrieth, B.; Frey, W.; Buchmeiser, M. R. ChemCatChem 2014, 6, 191–198. doi:10.1002/cctc.201300751 |
| 21. | Balof, S. L.; P'Pool, S. J.; Berger, N. J.; Valente, E. J.; Shiller, A. M.; Schanz, H.-J. Dalton Trans. 2008, 5791–5799. doi:10.1039/B809793C |
| 10. | Klučiar, M.; Grela, K.; Mauduit, M. Dalton Trans. 2013, 42, 7354–7358. doi:10.1039/c2dt32856a |
| 16. | Skowerski, K.; Szczepaniak, G.; Wierzbicka, C.; Gułajski, Ł.; Bieniek, M.; Grela, K. Catal. Sci. Technol. 2012, 2, 2424–2427. doi:10.1039/c2cy20320k |
| 17. | Skowerski, K.; Wierzbicka, C.; Szczepaniak, G.; Gułajski, L.; Bienieka, M.; Grela, K. Green Chem. 2012, 14, 3264–3268. doi:10.1039/c2gc36015b |
| 18. | Szczepaniak, G.; Kosiński, K.; Grela, K. Green Chem. 2014, 16, 4474–4492. doi:10.1039/C4GC00705K |
| 19. | Kośnik, W.; Grela, K. Dalton Trans. 2013, 42, 7463–7467. doi:10.1039/C3DT33010A |
| 21. | Balof, S. L.; P'Pool, S. J.; Berger, N. J.; Valente, E. J.; Shiller, A. M.; Schanz, H.-J. Dalton Trans. 2008, 5791–5799. doi:10.1039/B809793C |
| 27. | Endo, K.; Grubbs, R. H. J. Am. Chem. Soc. 2011, 133, 8525–8527. doi:10.1021/ja202818v |
| 14. | Ferraz, C. P.; Autenrieth, B.; Frey, W.; Buchmeiser, M. R. ChemCatChem 2014, 6, 191–198. doi:10.1002/cctc.201300751 |
| 21. | Balof, S. L.; P'Pool, S. J.; Berger, N. J.; Valente, E. J.; Shiller, A. M.; Schanz, H.-J. Dalton Trans. 2008, 5791–5799. doi:10.1039/B809793C |
| 27. | Endo, K.; Grubbs, R. H. J. Am. Chem. Soc. 2011, 133, 8525–8527. doi:10.1021/ja202818v |
| 28. | Jafarpour, L.; Hillier, A. C.; Nolan, S. P. Organometallics 2002, 21, 442–444. doi:10.1021/om0109511 |
| 11. | Autenrieth, B.; Frey, W.; Buchmeiser, M. R. Chem. – Eur. J. 2012, 18, 14069–14078. doi:10.1002/chem.201201199 |
| 15. | Zhao, J.; Wang, D.; Autenrieth, B.; Buchmeiser, M. R. Macromol. Rapid Commun. 2015, 36, 190–194. doi:10.1002/marc.201400413 |
| 23. | Krause, J. O.; Wurst, K.; Nuyken, O.; Buchmeiser, M. R. Chem. – Eur. J. 2004, 10, 777–784. doi:10.1002/chem.200305031 |
| 24. | Vygodskii, Y. S.; Shaplov, A. S.; Lozinskaya, E. I.; Filippov, O. A.; Shubina, E. A.; Bandari, R.; Buchmeiser, M. R. Macromolecules 2006, 39, 7821–7830. doi:10.1021/ma061456p |
| 13. | Autenrieth, B.; Willig, F.; Pursley, D.; Naumann, S.; Buchmeiser, M. R. ChemCatChem 2013, 5, 3033–3040. doi:10.1002/cctc.201300199 |
| 14. | Ferraz, C. P.; Autenrieth, B.; Frey, W.; Buchmeiser, M. R. ChemCatChem 2014, 6, 191–198. doi:10.1002/cctc.201300751 |
| 20. | Leuthäußer, S.; Schwarz, D.; Plenio, H. Chem. – Eur. J. 2007, 13, 7195–7209. doi:10.1002/chem.200700228 |
| 21. | Balof, S. L.; P'Pool, S. J.; Berger, N. J.; Valente, E. J.; Shiller, A. M.; Schanz, H.-J. Dalton Trans. 2008, 5791–5799. doi:10.1039/B809793C |
| 22. | Balof, S. L.; Yu, B.; Lowe, A. B.; Ling, Y.; Zhang, Y.; Schanz, H.-J. Eur. J. Inorg. Chem. 2009, 1717–1722. doi:10.1002/ejic.200801145 |
| 26. | Takao, K.-i.; Yasui, H.; Yamamoto, S.; Sasaki, D.; Kawasaki, S.; Watanabe, G.; Tadano, K.-i. J. Org. Chem. 2004, 69, 8789–8795. doi:10.1021/jo048566j |
© 2015 Koy et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)