Abstract
During a Ru-catalyzed reaction of an olefin with an alkylidene moiety that leads to a metallacycle intermediate, the cis insertion of the olefin can occur from two different directions, namely side and bottom with respect to the phosphine or N-heterocyclic ligand (NHC), depending on the first or second generation Grubbs catalyst. Here, DFT calculations unravel to which extent the bottom coordination of olefins with respect is favored over the side coordination through screening a wide range of catalysts, including first and second generation Grubbs catalysts as well as the subsequent Hoveyda derivatives. The equilibrium between bottom and side coordination is influenced by sterics, electronics, and polarity of the solvent. The side attack is favored for sterically less demanding NHC and/or alkylidene ligands. Moreover the generation of a 14-electron species is also discussed, with either pyridine or phosphine ligands to dissociate.

Graphical Abstract
Introduction
Organic synthesis is based on reactions that drive the formation of carbon–carbon bonds [1]. Olefin metathesis represents a metal-catalyzed redistribution of carbon–carbon double bonds [2-6] and provides a route to unsaturated molecules that are often challenging or impossible to prepare by any other means. Furthermore, the area of ruthenium-catalyzed olefin metathesis reactions is an outstanding field for the synthesis of C–C double bonds [7-9]. After the discovery of well-defined Ru-based (pre)catalysts, such as (PCy3)2Cl2Ru=CHPh [10], first by Grubbs and co-workers the range of these catalysts was broadened because of their tolerancy towards heteroatom ligands and the possibility to work under mild conditions.
The next step was the substitution of one phosphine group by an N-heterocyclic carbene, NHC, which strongly increases the activity [11-15]. And, furthermore, a detailed comprehensive analysis of the chemical mechanics of these Grubbs catalysts was required. Once a better understanding of the performance of such catalysts was achieved, a rational design of new more active catalysts was envisaged [16-19]. Despite experimental [20-24] and theoretical [8,9,25,26] insights during the last two decades, demonstrating the mechanism in Scheme 1, there are still missing parts in the understanding. Anyway, it is also confirmed that the first steps go through a dissociative mechanism instead of an associative, i.e., the entering olefin arrives after the extraction of two pyridine groups. Most of studies are related to phosphine groups instead of pyridine groups, but the function of both chemical groups is the same.
![[1860-5397-11-192-i1]](/bjoc/content/inline/1860-5397-11-192-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Mechanism of the olefin metathesis.
Scheme 1: Mechanism of the olefin metathesis.
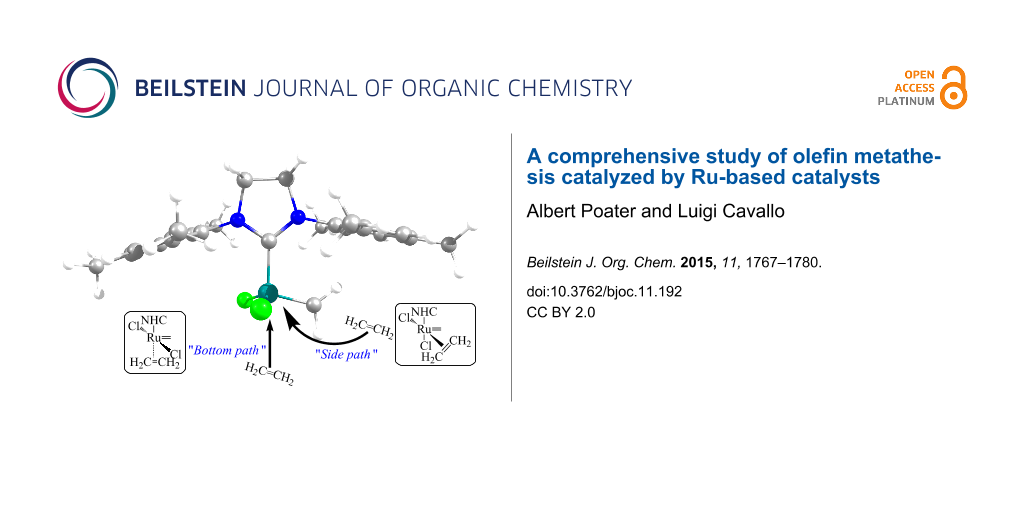
The release of a pyridine or phosphine group generates a 14-electron (14e) species, which binds to an olefin, coordinated cis to the alkylidene. The exchange of the leaving group by an olefin is found to be mainly dissociative [4,27-29], towards the associative or the concerted mechanisms [30]. The next metallacycle intermediate is due to the reaction of the olefin with the alkylidene moiety. Nevertheless in the cis insertion of the olefin, this olefin can enter from two different directions, side and bottom, displayed in Scheme 2. Regarding this cis insertion of the olefin, even though most papers favor the side insertion of the olefin (see Scheme 2) over the bottom insertion [31], both pathways might exist depending on the ligands and type of olefin. Since the year 2000 many papers try to unravel the preference for the bottom attack and how to favor the cis one [8,32-34], with an open debate still for the(NHC)Ru-based catalysts [35-38]. The postulated binding of the substrate can be preferentially trans to the NHC ligand (bottom path in Scheme 2) or cis to this ligand with the simultaneous shift of an halogen group to a trans position (side path in Scheme 2).
![[1860-5397-11-192-i2]](/bjoc/content/inline/1860-5397-11-192-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Possible side or bottom mechanism of the insertion of the olefin.
Scheme 2: Possible side or bottom mechanism of the insertion of the olefin.
Bearing the general acceptance [39-45] that olefin metathesis with Ru-catalysts starts from a bottom-bound olefin complex because of energetics, i.e., reporting higher energies for the possible side-bound olefin complexes, Piers and co-workers demonstrated the bottom-bound geometry for a Ru-cyclobutane model compound by NMR data [46]. However, Grubbs and co-workers supported the side-bound pathway [47]. And this sort of discrepancy is displayed in Scheme 3, where the same catalyst shows two conformations, a and b, bottom and side, respectively. Next, by means of DFT calculations Goddard and co-workers indicated clearly that solvent effects were of paramount relevance to the relatively high stability of b [48], while in the gas phase structure a was much favored [13]. Moreover, Grubbs and co-workers reported the X-ray structure of the model compound c, which clearly indicates that the olefin is side-bound to Ru [49].
![[1860-5397-11-192-i3]](/bjoc/content/inline/1860-5397-11-192-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Ruthenium catalysts, bottom-bound (a) or side-bound (b and c).
Scheme 3: Ruthenium catalysts, bottom-bound (a) or side-bound (b and c).
Next, Correa and Cavallo discussed about the feasibility of the side conformation for the classical olefin metathesis catalysts 7, 16, and 19 displayed in Scheme 4 [8], concluding that the bottom/side equilibrium is based on a delicate balance between electronic, steric, and solvent effects. Particularly sterically demanding substituents of the NHC and bulky olefins clearly favor the side reaction pathway. Moreover this study corroborated the validity of BP86 for these second generation Grubbs catalysts and the conclusion was that any generalization could be done about the side/bottom stability of the coordination intermediate, as well as it is not possible for the first Grubbs catalysts [9]. Overall, the inclusion of a polar solvent and the absence of strong steric effects, i.e., with less bulky ligands (less than SIMes) and/or substrates [50,51], favored the side-bound structures over the bottom-bound ones as suggested by Goddard and Grubbs, respectively [16-18]. This is a possible explanation of why there is experimental evidence for some structures with side confirmation. On the other hand, complex 19 is an example of an asymmetric catalyst, suggesting that the NHC ligand is the source of asymmetry [52].
![[1860-5397-11-192-i4]](/bjoc/content/inline/1860-5397-11-192-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
In this paper we contribute in the understanding of the side- and bottom-bound coordination intermediates and the stability of the corresponding transition states as well as the relative stability of the next metallacycle formed for (NHC)Ru(X)2 catalysts through a DFT approach with the BP86 functional. The comparison with the first generation Grubbs catalysts with a phosphine group instead of an NHC and the game based on the possible electronic and steric possibilities of the NHC will center our interests. All studied systems are displayed in Scheme 4. System 7 has been thoroughly studied because of its simplicity, playing especially with the NHC ligand, replacing one or both mesityl groups by CH3, CF3, t-Bu, H, F, and combinations between them to also observe the effects of an asymmetric NHC ligand. Furthermore this system 7 was also taken to discuss about the evolution of the precatalysts, PRE-II, PRE-I and PRE-0 (see Scheme 1). This can corroborate the dissociative mechanism of the entering olefin, a step which has been taken into account. Then complex 15 reveals a more representative substrate, and is useful to focus on the study of steric effects of the substrate, reinforced the steric hindrance aspect replacing one or both mesityl groups of NHC ligand by methyl groups. Bearing the asymmetric catalysts 17–19, particularly complex 19 can be compared to 16 but introducing asymmetry [17,53]. Then complexes 1 and 14 are representative of the first generation Grubbs catalysts. Finally, several systems are displayed to get insight into the typical properties of free halogen catalysts [54,55].
Computational details
The density functional calculations were performed on all the systems at the GGA level with the Gaussian03 set of programs [56]. Two popular functionals, B3LYP and BP86, were considered. B3LYP calculations utilize Becke's three parameter hybrid exchange functional together with the correlation functional of Lee, Yang and Parr [57-59]. For BP86 calculations, gradient corrections were taken from the work of Becke and Perdew [60-62]. The electronic configuration of the molecular systems was described by the standard SVP basis set, i.e., the split-valence basis set with polarization functions of Ahlrichs and co-worker, for H, C, N, P, O, S, F, Cl and Br [63]. For Ru and I we used the small-core, quasi-relativistic Stuttgart/Dresden effective core potential (standard SDD basis set in Gaussian03) basis set, with an associated (8s7p6d)/[6s5p3d] valence basis set contracted according to a (311111/22111/411) scheme [64-66].
The geometry optimizations were performed without symmetry constraints, and the nature of the extrema was checked by analytical frequency calculations. Furthermore, all extrema were confirmed by calculation of the intrinsic reaction paths. The energies discussed throughout the text contain ZPE corrections. Solvent effects including contributions of non-electrostatic terms have been estimated in single point calculations on the gas phase optimized structures, based on the polarizable continuous solvation model PCM using CH2Cl2 as a solvent [67]. The cavity is created via a series of overlapping spheres.
For the sake of clarity we did not change the functional for the solvent calculations, despite knowing that the dispersion interactions can occur [68-73]. However, here we consider that the qualitative comparison between the set of studied catalysts and even the quantitative trends (side/bottom) should not be affected by this omission.
Results and Discussion
In this section we first discuss the structure and energetics of the key steps of metallacycle formation, starting with dissociation of the leaving L ligand, pyridine in this manuscript, from precatalysts 1–13, and moving to coordination of the C=C double bond of ethene in systems 1–13, or of the C=C bond tethered to the Ru atom in systems 14–19. Then, we will discuss structure and energetics of the four-center transition state for metallacycle formation, and finally structure and energetics of the metallacycle. In all cases, we assumed the naked 14e species as zero of energy.
Structure of the naked 14e species. According to all the calculations reported so far, the naked 14e species is very unstable. The dissociation of PR3 or pyridine ligands is highly endothermic, approximately 15–20 kcal/mol even if solvent effects with implicit methods are considered. An unfavorable entropic term of roughly 8–10 kcal/mol would reduce this internal binding energy to free energies of binding around 5–12 kcal/mol [13].
According to simple Boltzmann statistics, these energetics implies that the fraction of the naked 14e species in solution at 25 °C should be in the range of 10−11 to 10−13 of the total precatalyst. Even considering an overestimation of PR3 or pyridine binding by roughly 5 kcal/mol, an error that would be quite large for this kind of calculations, still the fraction of the active species should be in the range of 10−6 to 10−11. Considering that the precatalyst concentration is usually in the order of 10−1 to 10−3 M, this means that the concentration of the real 14e catalyst should be roughly in the range of 10−7 to 10−13. These numbers suggest that it is very unlikely that the naked 14e species is the “real” active species, and that probably some other species is indeed coordinated to the Ru atom in place of the pyridine or phosphine to first dissociate.
Pyridine and phosphine binding. The binding energy of the first (trans to the PR3 or NHC ligand) and of the second (trans to the ylidene group) pyridine ligands to the naked 14e species of 1–19 are reported in the 2nd and 3rd column of Table 1. These values indicate that the first pyridine is bonded quite strongly to the Ru atom. Indeed, it is competitive with PCy3 binding in the prototype 1st and 2nd generation systems based on 1 and 7 [74-76]. In precatalysts 1–7 the binding energy of the first pyridine is roughly 20 kcal/mol, with a small effect of the ligand bulkiness, which only changes for system 8 bearing more sterically demanding isopropyl groups, displaying a value of 15.0 kcal/mol. The pseudo-halide systems, instead, show a remarkably different behavior. The pyridine is quite weakly bound to 9, E1 = 14.3 kcal/mol, and this binding energy decreases to 7.7 kcal/mol only in 10. Differently, the pyridine is bound very strongly to the Ru atom, by more than 30 kcal/mol, in systems 11 and 12. Of course, the pyridine is coordinated trans to the NHC ligand in 9 and 10, whereas it is cis coordinated in 11 and 12. Considering the pseudo-halide family, our results are in qualitative agreement with the experimental finding of Fogg and co-workers that systems 11 and 12 have to be thermally activated [19], while system 10 is even more active than the prototype 2nd generation system 7. Finally, the C1-symmetric system 13 shows E1 values which are substantially independent from the specific geometry; i.e., whether the methylidene group is on the side of the mesityl or of the methyl group of the NHC ligand, species, 13a and 13b, respectively.
Table 1: Binding energy, in kcal/mol, of the first, E1, and of the second, E2, pyridine/PMe3 molecule to the naked 14e species 1–13.
| system | pyridine | PMe3 | ||||
|---|---|---|---|---|---|---|
| E1 | E2 | E1 + E2 | E1 | E2 | E1 + E2 | |
| 1 | 20.3 | 2.5 | 22.8 | 27.3 | 5.1 | 32.4 |
| 2 | 18.3 | 9.6 | 27.9 | 26.8 | 9.1 | 35.9 |
| 3 | 21.0 | 7.7 | 30.7 | 25.5 | 13.2 | 38.7 |
| 4 | 19.9 | 0.2 | 20.1 | 29.3 | −5.4 | 23.9 |
| 5 | 21.7 | −15.4 | 6.3 | 32.8 | — | — |
| 6 | 20.7 | 7.5 | 28.2 | 28.5 | −4.4 | 26.1 |
| 7 | 19.7 | 6.8 | 26.5 | 27.7 | 3.6 | 24.1 |
| 8 | 15.0 | −6.0 | 21.0 | 23.6 | −4.6 | 19.0 |
| 9 | 14.3 | 0.6 | 14.9 | 17.9 | 3.6 | 21.5 |
| 10 | 7.7 | −5.0 | 2.7 | 11.9 | — | — |
| 11 | 33.2 | −2.9 | 30.3 | 41.1 | — | — |
| 12 | 35.6 | — | — | 45.7 | — | — |
| 13a | 21.1 | 5.8 | 26.9 | 28.9 | 5.0 | 33.9 |
| 13b | 13.5 | 6.1 | 19.6 | 17.9 | 4.9 | 22.8 |
| 14 | 8.7 | 7.3 | 16.0 | 17.9 | −2.8 | 15.1 |
| 15 | 11.1 | 12.6 | 23.7 | 19.5 | 11.9 | 31.4 |
| 16 | 11.5 | 7.8 | 19.3 | 20.8 | — | — |
| 17a | 10.4 | 5.3 | 15.7 | 18.5 | 4.1 | 22.6 |
| 17b | 10.3 | 8.6 | 18.9 | 19.2 | 0.0 | 19.2 |
| 18 | 2.2 | 5.4 | 7.6 | 5.1 | 9.5 | 14.6 |
| 19 | 13.0 | −1.5 | 11.5 | 19.8 | — | — |
Table 1 also includes corresponding values for the phosphine dissociation, PMe3 for this analysis. The trend is the same as the one explained above for the pyridine dissociation. But quantitatively the phosphine dissociation is more expensive.
The double coordination of pyridine or trimethylphosphine is not possible for all systems. Systems 5, 10, 11, 12, 16, and 19 do not accept the second phosphine group because of steric hindrances. We must point out that for system 12 when bonding one Py or PMe3 moiety the octahedral environment around the metal is already obtained. Furthermore there are some other systems that exhibit a negative value for E2, i.e., no stability for the octahedral structure. This instability is related to the elongation of the Ru–P bond distance cis to the NHC ligand. Indeed in all systems the Ru–P bond is much shorter for the phosphine placed trans to the NHC (around 2.45 Å), compared with the Ru–P bond distance cis to the NHC (around 2.60 Å). However for some systems this distance is too long to be more stable than the monophosphine complex. Thus, when this bond distance is over 2.65 Å the bisphosphine precatalyst structure is not stable.
Substrate coordination. The coordination energy of the C=C double bond of the substrate to the Ru atom of the various 14e species are reported in the 3rd column of Table 2, while in the 4th column the coordination energy of the cis isomer relative to the trans isomer is reported. We focus first on C2H4 coordination to the symmetric precatalysts 1–8. For these systems we considered both trans and cis coordination of C2H4 to the PR3 or NHC ligand, denoted as “T” and “C” in Table 2. In agreement with experimental findings on related systems, we found that cis coordination of the substrate is preferred. This preference is influenced by the bulkiness of the NHC ligand, and is smaller for larger N-substituents. Indeed, the calculated energy difference between the cis and trans geometries in the coordination intermediates of 2–5, ΔE of 4th column in Table 2, follows a clear trend with the bulkiness of the N-substituents, F < Me < CF3 < t-Bu, ΔE = −8.0 < −5.2 < −3.4 < −2.1 kcal/mol, respectively.
Table 2: Energies (E) in kcal/mol, of the coordination intermediates, transition states and metallacycles with respect to the 14e species and the uncoordinated C=C double bond. For each system, the energies of both isomers with the C=C trans or cis to the NHC ligand are reported. For each species, ΔE is the energy difference between the cis and the trans isomer, labeled as C and T, respectively.
| system | geometry | I | TS | II | |||
|---|---|---|---|---|---|---|---|
| E | ΔE | E | ΔE | E | ΔE | ||
| 1 P | T | −10.0 | 0 | −4.2 | 0 | −19.6 | 0 |
| C | −11.3 | −1.3 | −6.6 | −2.4 | −19.8 | -0.2 | |
| 2 Me | T | −13.4 | 0 | −9.5 | 0 | −25.7 | 0 |
| C | −18.6 | −5.2 | −10.2 | −0.7 | −23.4 | 2.3 | |
| 3 F | T | −6.6 | 0 | −5.7 | 0 | −17.8 | 0 |
| C | −14.6 | −8.0 | −10.4 | −4.7 | −23.2 | −5.4 | |
| 4 CF3 | T | −11.9 | 0 | −10.9 | 0 | −23.6 | 0 |
| C | −15.3 | −3.4 | −8.7 | 2.2 | −22.5 | 1.1 | |
| 5 t-Bu | T | −17.0 | 0 | −13.1 | 0 | −30.2 | 0 |
| C | −19.1 | −2.1 | −5.6 | 7.5 | −18.0 | 12.2 | |
| 6 IMes | T | −15.5 | 0 | −11.4 | 0 | −25.2 | 0 |
| C | −13.6 | 1.9 | −7.8 | 3.6 | −20.8 | 4.4 | |
| 7 SIMes | T | −11.8 | 0 | −10.1 | 0 | −25.0 | 0 |
| C | −14.7 | −2.9 | −6.2 | 3.9 | −19.0 | 6.0 | |
| 8 SIPr | T | −9.9 | 0 | −7.2 | 0 | −21.9 | 0 |
| C | −14.7 | −4.8 | −6.9 | 0.3 | −19.5 | 2.4 | |
| 9 FgF5 | T | −7.8 | 0 | −6.7 | 0 | −14.2 | 0 |
| C | −16.3 | −8.5 | −9.7 | −3.0 | −20.2 | −6.0 | |
| 10 FgBr | T | 1.1 | 0 | 0.6 | 0 | −9.1 | 0 |
| C1 | −9.7 | −10.8 | −0.9 | −1.5 | −11.4 | −2.3 | |
| C2 | −5.7 | −6.8 | 0.6 | 0.0 | −15.0 | −5.9 | |
| 11 FgO | T | 3.1 | 0 | 3.2 | 0 | −20.6 | 0 |
| C | −33.5 | −36.6 | −24.0 | −27.1 | −39.1 | −18.5 | |
| 12 FgS | C (S1) | −27.5 | 0 | −21.9 | 0 | −35.6 | 0 |
| C(S2) | −34.3 | −6.8 | −26.6 | −4.7 | −37.6 | −2.0 | |
| C(O) | −29.4 | −1.9 | −18.7 | −2.8 | −37.5 | −1.9 | |
| 13a C1 | T | −14.6 | 0 | −13.0 | 0 | −26.3 | 0 |
| C | −19.7 | −5.1 | −9.5 | 3.5 | −23.9 | 2.4 | |
| 13b C1 | T | −8.8 | 0 | −4.9 | 0 | −22.4 | 0 |
| C | −12.7 | −3.9 | −4.8 | 0.1 | −20.1 | 2.3 | |
| 14 PR3 | T | 4.5 | 0 | 19.0 | 0 | 7.7 | 0 |
| C-re | 9.7 | 5.2 | 20.2 | 1.2 | 15.9 | 8.2 | |
| C-si | 14.4 | 9.9 | 20.7 | 1.7 | 17.1 | 9.4 | |
| 15 Me | T | 0.1 | 0 | 1.0 | 0 | −3.3 | 0 |
| C-re | −5.9 | −6.0 | 4.0 | 3.0 | −1.8 | 2.5 | |
| C-si | −8.1 | −8.2 | 4.5 | 3.5 | 2.2 | 5.5 | |
| 16 Mes | T | −1.8 | 0 | 0.6 | 0 | −2.7 | 0 |
| C-re | −1.5 | 0.3 | 12.6 | 12.0 | 8.4 | 11.1 | |
| C-si | 2.4 | 4.2 | 19.9 | 19.3 | 12.1 | 14.8 | |
| 17a C1 | T | 0.0 | 0 | 2.7 | 0 | −0.2 | 0 |
| C-re | −4.6 | −4.6 | 11.2 | 8.5 | 7.4 | 7.6 | |
| C-si | −0.8 | −0.8 | 12.8 | 10.1 | 5.5 | 5.7 | |
| 17b C1 | T | −0.1 | 0 | 0.5 | 0 | −4.7 | 0 |
| C-re | −2.1 | −2.0 | 2.4 | 1.9 | −3.8 | 0.9 | |
| C-si | −2.4 | −2.3 | 6.8 | 6.3 | 7.4 | 12.1 | |
| 18 Me1 | T | 1.3 | 0 | 2.1 | 0 | −2.7 | 0 |
| C-re | −2.7 | −4.0 | 6.9 | 4.8 | 0.9 | 3.6 | |
| C-si | −7.7 | −9.0 | 13.2 | 11.1 | 7.3 | 10.0 | |
| 19 Pr1 | T | −0.7 | 0 | 0.7 | 0 | −4.2 | 0 |
| C-re | −0.9 | −0.2 | 14.7 | 14.0 | 11.8 | 16.0 | |
| C-si | −2.8 | −2.1 | 16.7 | 16.0 | 12.9 | 17.1 | |
Steric effects have little influence on the absolute C2H4 binding energy, E of 3rd column in Table 2, since rather similar values are calculated for 2 and 5 (−18.6 and −19.1 kcal/mol, respectively). Differently, electronic effects influence the ability of the Ru atom to capture C2H4, particularly in the trans isomer. For example, the electron withdrawing F substituents in 3 results in a trans C2H4 coordination energy of only 6.6 kcal/mol, whereas in the sterically similar system 2 the trans C2H4 coordination energy is 13.4 kcal/mol. The highest coordination energies are calculated for 2 and 5, which present electron donating substituents, whereas systems 6 and 7, with aromatic N-substituents, lie in between. This suggests that electron donating N-substituents increase electron density at the carbene C atom of the NHC, which results in a higher electron density on the σ contribution of the HOMO of the NHC. When 1st and 2nd generation catalysts are compared, system 1, with the PCy3 ligand, behaves rather similary to the 2nd generation catalyst 7, with a SIMes NHC ligand.
Substitution of the Cl ligands of 7 with the far bulkier O(C6F5) ligands, such as in 9, increases the preference for cis C2H4 coordination from 2.9 to 8.5 kcal/mol, while the absolute C2H4 binding energy in the cis isomer increases from 14.7 to 16.3 kcal/mol. Interestingly, in the naked 14e species one of the F atoms of the O(C6F5) ligands is engaged in a Ru…F interaction trans to the NHC, which makes the bottom pathway for the entering olefin more difficult, see Figure 1. This kind of Ru…F interactions was first reported by Grubbs and co-workers [77,78]. Finally, replacing just one Cl atom of 7 with a C6Br5 ring, such as in 9, results again in a preferred cis coordination, and the most stable isomer presents the Cl atom cis to the SIMes ligand.
![[1860-5397-11-192-1]](/bjoc/content/figures/1860-5397-11-192-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: a) Naked 14e species for system 9 (distance in Å). b) trans (T); c) cis(S) (C(S)); and d) cis(O) (C(O)) C2H4 coordinated species for 12.
Figure 1: a) Naked 14e species for system 9 (distance in Å). b) trans (T); c) cis(S) (C(S)); and d) cis(O) (C...
Moving to the pseudo-halide systems with a chelating ligand, the most striking difference is in the absolute C2H4 coordination energy, roughly 30 kcal/mol, which is about 15 kcal/mol better than in the non-chelating ligands. The chelating ligand has a minor effect on the cis/trans preference for 12, whereas for 11 trans coordination of C2H4 is not favored. This is easily explained by considering that the 5-membered ring formed through chelation of the cresol group to Ru makes a trans O–Ru–O disposition geometrically difficult, whereas the 6-membered ring formed by chelation of the sulfoxide ligand in 12 allows for a trans O–Ru–O geometry with little steric strain. 12 presents three cis isomers, denoted as C(S1), C(S2), and C(O) in Table 2, which correspond to have the phenolic or the sulfonic O atom trans to the NHC ligand (see Figure 1). Our calculations indicate a preference for the C(S2) isomer, which is 4.9 and 6.8 kcal/mol more stable than the C(O) and the C(S1) isomers, respectively. The preference for the C(S2) isomer can be explained by the weaker donicity of the sulphonic O atom with respect to the phenolic O atom, which results in a softer ligand trans to the SIMes ligand.
Focusing on system 13, bearing a C1-symmetric ligand, we found that the cis coordination still is favored, and the most stable structure corresponds to 13a, which presents the methylidene moiety on the side of the mesityl N-substituent, while isomer 13b, which presents the methylidene moiety on the side of the Me N-substituent is 7.0 kcal/mol higher in energy. The structure of the two cis coordination intermediates clearly indicates that the C2H4 molecule nicely avoids steric repulsion with the NHC ligand in 13a, whereas it experiences repulsive interaction with the methyl on the NHC ligand in 13b (see Figure 2).
![[1860-5397-11-192-2]](/bjoc/content/figures/1860-5397-11-192-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Coordinated species for species a) 13a and b) 13b.
Figure 2: Coordinated species for species a) 13a and b) 13b.
Moving to substrates bulkier than C2H4, systems 14–19, the cis geometry is favored only in the systems that present small methyl N-substituents, i.e., 15, 17a and 18. This indicates that bulkier substrates can be accommodated in the cis position with difficulty. Furthermore, in almost all the cases coordination of the C=C double bond, either in the cis or trans position, is disfavored (positive E values in the 3rd column of Table 2). This striking difference relative to C2H4 coordination, which is always favored (negative E values in the 3rd column of Table 2), is due to the coordination of the O atom of the substrate in 14–19. In other words, with a heteroatom containing substrate the heteroatom can coordinate to the Ru center, as in the Hoveyda-type precatalysts [6], maybe stabilizing the active species, see Figure 3a. In any case, coordination of the C=C double bond requires displacement of the coordinated O atom, and it is likely that the terminal C=C double bond of complex substrates will be dangling, which is in agreement with the NMR experiments of Piers and co-workers on strictly related systems [13]. Focusing on a selection between the two prochiral faces of the C=C bond in 14–19, we always found that coordination takes places in the face that presents the two methyl groups pointing away from the NHC ligand. Of course, this results in reduced steric interactions with the NHC ligand.
![[1860-5397-11-192-3]](/bjoc/content/figures/1860-5397-11-192-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Naked 14e species for system 14 with the O atom of the substrate coordinated to the Ru center (distance in Å), part a; and representative coordination geometries for systems 7, 11 and 14 with a cis coordinated C2H4 molecule, parts b–d.
Figure 3: Naked 14e species for system 14 with the O atom of the substrate coordinated to the Ru center (dist...
In the trans geometries, instead, the C=C double bond of C2H4 is nearly perpendicular to the Ru–methylidene bond, whereas in the bigger substrate the tether forces the coordinated C=C bond to be almost aligned with the Ru–alkylidene bond. However, the molecular dynamics simulations (vide infra) clearly indicate that in the trans geometries the C2H4 molecule can freely rotate around the coordination axis to the Ru center. Finally, the Ru–C bond distances are roughly the same (2.20–2.25 Å) in the case of C2H4 coordination, whereas in the case of the bigger substrate the coordination of the olefin is highly asymmetric, with the internal C roughly 2.50–2.60 Å away from the Ru atom.
Transition state for metallacycle formation. The energy of the various transition states for metallacycle formation with respect to the uncoordinated C=C double bond are reported in the 5th column of Table 2, while in the 6th column it is reported the energy of the cis transition states with respect to the trans transition state. The numbers reported clearly indicate that at the transition state the bulkiness of the N-substituents plays a more remarkable role. Indeed, while cis C2H4 coordination is favored for all the systems we considered, the cis transition state is favored only for some systems. Specifically, for the 1st generation system 1, for symmetric NHC systems with small N-substituents, such as 3, for all the pseudo-halide systems, and for system 13, with a C1-symmetric NHC ligand. Differently, for non pseudo-halide NHC systems with N-substituents bulkier than Me, such as 4, 5, 6, and 7, the trans transition state is clearly favored. The increased role of steric effects can be clearly understood if we consider that at the transition state the C=C double bond must be placed almost parallel to the Ru=CH2 bond, which results in increased steric repulsion with the NHC ligand, whereas in the coordination intermediates the C=C double bond is almost perpendicular to the Ru=CH2 bond, to occupy the free space above the NHC ring, between the N-substituents. The pseudo-halide systems, where the cis coordination is strongly favored, prefer the cis geometry also in the transition state. Finally, for system 9 the Ru…F interaction is preserved, see Figure 4. A similar interaction is also retained in the trans transition state, see again Figure 4.
![[1860-5397-11-192-4]](/bjoc/content/figures/1860-5397-11-192-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: System 9 with a Ru…F interaction in the cis and trans geometries, parts a and b, respectively (distance in Å).
Figure 4: System 9 with a Ru…F interaction in the cis and trans geometries, parts a and b, respectively (dist...
In the case of systems 14–19, with the bulkier substrate, the trans transition state is clearly favored for all the systems. Also for those that present rather stable cis geometry at the level of the coordination intermediate, such as system 15. As already indicated, this is due to the steric pressure of the PCy3 or NHC ligands on the bulkier substrate. Interestingly, in all the systems considered the energy barrier for metallacyle formation, that is the energy difference between the transition states and the coordination intermediates, is quite small, always below 10 kcal except for system 14, and usually below 5 kcal/mol. This indicates that all these systems should be highly active, which unfortunately is not the case. This fact suggests that the origin of the remarkably different catalytic activity shown by these systems lays somewhere else.
Metallacycle. The energy of the various metallacycles with respect to the uncoordinated C=C double bond are reported in the 5th column of Table 2, while in the 6th column the energy of the cis metallacycle with respect to the trans metallacycle is reported. Beside a few cases, the general trend is that ongoing from the coordination intermediates, to the transition states and finally to the metallacycles, there is an energetic shift towards the trans geometry. In fact, besides the pseudo-halide systems, which strongly favor the cis isomer, the cis metallacycle is favored only for system 3, with the small F substituents. The stability of the metallacycles relative to the coordination intermediates is strongly influenced by the nature of the substrate. In fact, with C2H4 as the substrate, systems 1–13, the metallacycle is roughly 10 kcal/mol lower in energy with respect to the coordination intermediates, which suggests that the resting state is the Ru–metallacycle species. This is in agreement with the NMR experiments of Piers and co-workers that found the Ru–metallacycle as the most abundant species in the ethene exchange metathesis promoted by system 7 [32,79]. Differently, with the bigger substrate the metallacycle is of comparable stability or slightly higher in energy with respect to the coordination intermediate, also in agreement with the data of Piers and coworkers [33].
Structurally, the Ru–C(metallacycle) bonds in 1, 2, 3 and 7 are rather similar, around 1.97–1.98 Å, see Figure 5a, while in 5 they are remarkably longer with 2.01 Å, probably due to the steric pressure of the t-Bu N-substituents. In all the cases the C–C bonds of the Ru–cyclobutane are around 1.58–1.59 Å. Differently, in the presence of the bigger substrate the metallacycle is quite asymmetric, see Figure 5b. The C–C length of the former C=C double bond is quite shorter, around 1.57 Å in 14 and 16, than the C–C bond just formed, which is around 1.65–1.66 Å. This finding is in good agreement with the NMR experiments by Piers and co-workers [33].
![[1860-5397-11-192-5]](/bjoc/content/figures/1860-5397-11-192-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Representative geometries of the metallacycles 7 and 15, parts a and b, respectively (distance in Å).
Figure 5: Representative geometries of the metallacycles 7 and 15, parts a and b, respectively (distance in Å...
After this reporting on the energetic and structure of each intermediate separately, we combine this information into the energy profile that connects the naked 14e precatalysts and the uncoordinated substrate with the metallacycles. For reasons of simplicity, we selected the most representative cases, which we believe are the 1st and 2nd generation systems with PCy3 and SIMes as ligands, both for the small substrate C2H4, systems 1 and 7, and the bigger substrate, systems 14 and 16. In the case of C2H4 as substrate, coordination of the olefin is clearly favored both for the 1st and 2nd generation systems 1 and 7, see Figure 6 and Figure 7, although it is clear that the 2nd generation catalysts coordinate to the substrate somewhat better. With the more complex substrate C=C binding has to compete with the O binding, which substantially results in no energy gain for the C=C coordination. Moving to the transition states for metallacycle formation requires climbing the rather low energy barrier of 4.7 and 1.7 kcal/mol for 1 and 7, respectively, and in both cases the transition state is lower in energy than the naked 14e precatalyst, by 6.6 and 10.1 kcal/mol, respectively. This indicates that metallacycle formation is favored with respect to C2H4 dissociation. Finally, both transition states collapse into very stable metallacycles. However, beside an overall similarity there is the sharp difference that in 1 the cis path is favored, whereas in 7 the trans path is favored. Furthermore, the energy gain associated with metallacycle formation is quite higher in the 2nd generation catalyst rather than for the 1st generation catalyst. Moving to the same catalysts with the bigger substrate, systems 14 and 16, metallacycle formation is an overall uphill path. Coordination of the terminal C=C double bond with respect to coordination of the O heteroatom is disfavored by 4.5 kcal/mol in 14 and favored by 1.8 kcal/mol in 16, respectively. The transition states are 20.2 and 19.9 kcal/mol higher in energy with respect to the 14e species, and the metallacycles are 7.7 kcal/mol above and 12.1 kcal/mol above the 14e species.
![[1860-5397-11-192-6]](/bjoc/content/figures/1860-5397-11-192-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Energy profiles for systems 1 and 14.
Figure 6: Energy profiles for systems 1 and 14.
![[1860-5397-11-192-7]](/bjoc/content/figures/1860-5397-11-192-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Energy profiles for systems 7 and 16.
Figure 7: Energy profiles for systems 7 and 16.
To better understand the different stability of the metallacycle relative to the coordination intermediate of the 1st and 2nd generation catalysts we investigated the thermodynamics of the reaction shown in Figure 8. E1 is the energy gain associated to metallacycle formation, already discussed, while E2 is the energy loss due to the hypothetical release of cyclopropane from the Ru–metallacycle species. The larger E1 and E2 values for the 2nd generation system 7 clearly indicate that the Ru–C σ-bonds are stronger in the presence of an NHC ligand. The origin of this difference is in the better ability of the NHC ligand to donate electron density to the Ru center, which formally is in the high formal oxidation state of +4 in the metallacycle.
![[1860-5397-11-192-8]](/bjoc/content/figures/1860-5397-11-192-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Metallocycle and cyclopropane formation energy profile (energies in kcal/mol).
Figure 8: Metallocycle and cyclopropane formation energy profile (energies in kcal/mol).
Conclusion
After discussing a wide range of representative systems of second generation Grubbs catalysts the conclusion is that any compound sterically demanding either through the alkylidene group or the olefin or even the NHC ligand means a stabilization of the bottom-bound isomer with respect to the side-bound. However, with the simplest olefin and alkylidene groups this less favored isomer plays a key role, especially if taking into account the solvent effect. Therefore, calculations indicate that the preferred reaction pathway is an equilibrium described by steric, electronic, and solvent effects. In spite of the expectation that the hydrogen atom is the less sterically demanding it can produce H–H repulsion with the alkylidene group, which the mesityl or even the methyl groups do not. And comparing these last two complexes the possible H–H repulsions are possible when there is a methyl group in the NHC ligand, but never in the case of mesityl groups. Therefore, for the latter substituent, any H–H repulsion is avoided, and there are favorable C–H interactions even though the mesityl group is sterically more demanding. When including t-Bu in the NHC ligand the effect of a high sterically demanding group is present, and for this compound no side-bound isomers occurs.
The differentiation between the power of the three effects, steric, electronic and solvent, has turned out be a hazard to make predictions difficult. However the CF3 groups have become key to explain the electronic effects, as well as the t-Bu for steric effects. Steric effects owing to interaction between bulky NHC ligands [16] and bulky substrates make the bottom reaction pathway more likely surpassing the other effects. Finally the solvent effect helps the stabilization of the side-bound isomer, but at a higher degree for the high sterically demanding complexes. As already stated by Goddard, polar solvents favor the side reaction pathway or at least reduce the electronic preference for the bottom reaction pathway [13]. Therefore, bearing less sterically demanding substrates and/or ligands, the side reaction pathway, as suggested by Grubbs and co-workers [12,14], might be competitive.
Even though over the last three decades thousands of papers have presented and described the olefin metathesis catalysis, neither a general catalyst for any metathesis reaction has been found [80-82], nor are perfect rules available to predict the behavior of a given catalyst [83-92], although great efforts in characterizing the decomposition reactions [93-97], to validate computational protocols [37,45,68,98-102], and in the experimental synthesis and characterization [33,103] have been taken. Thus, this study provides valuable insight and yields at least a general recipe to obtain a side attack of the olefin towards the NHC ligand.
References
-
Trnka, T. M.; Grubbs, R. H. Acc. Chem. Res. 2001, 34, 18–29. doi:10.1021/ar000114f
Return to citation in text: [1] -
Chauvin, Y. Angew. Chem., Int. Ed. 2006, 45, 3740–3747. doi:10.1002/anie.200601234
Return to citation in text: [1] -
Grubbs, R. H. Angew. Chem., Int. Ed. 2006, 45, 3760–3765. doi:10.1002/anie.200600680
Return to citation in text: [1] -
Schrock, R. R. Angew. Chem., Int. Ed. 2006, 45, 3748–3759. doi:10.1002/anie.200600085
Return to citation in text: [1] [2] -
Hérisson, P. J.-L.; Chauvin, Y. Makromol. Chem. 1971, 141, 161–176. doi:10.1002/macp.1971.021410112
Return to citation in text: [1] -
Fustero, S.; Bello, P.; Miró, J.; Sánchez-Roselló, M.; Haufe, G.; del Pozo, C. Beilstein J. Org. Chem. 2013, 9, 2688–2695. doi:10.3762/bjoc.9.305
Return to citation in text: [1] [2] -
Yuan, W.; Wei, Y.; Shi, M. ChemistryOpen 2013, 2, 63–68. doi:10.1002/open.201300002
Return to citation in text: [1] -
Perfetto, A.; Costabile, C.; Longo, P.; Bertolasi, V.; Grisi, F. Chem. – Eur. J. 2013, 19, 10492–10496. doi:10.1002/chem.201301540
Return to citation in text: [1] [2] [3] [4] -
Perfetto, A.; Costabile, C.; Longo, P.; Grisi, F. Organometallics 2014, 33, 2747–2759. doi:10.1021/om5001452
Return to citation in text: [1] [2] [3] -
Nguyen, S. T.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1993, 115, 9858–9859. doi:10.1021/ja00074a086
Return to citation in text: [1] -
Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org. Lett. 1999, 1, 953–956. doi:10.1021/ol990909q
Return to citation in text: [1] -
Huang, J.; Stevens, E. D.; Nolan, S. P.; Peterson, J. L. J. Am. Chem. Soc. 1999, 121, 2674–2678. doi:10.1021/ja9831352
Return to citation in text: [1] [2] -
Weskamp, T.; Kohl, F. J.; Hieringer, W.; Gleich, D.; Herrmann, W. A. Angew. Chem., Int. Ed. 1999, 38, 2416–2419. doi:10.1002/(SICI)1521-3773(19990816)38:16<2416::AID-ANIE2416>3.0.CO;2-#
Return to citation in text: [1] [2] [3] [4] [5] -
Bielawski, C. W.; Grubbs, R. H. Angew. Chem., Int. Ed. 2000, 39, 2903–2906. doi:10.1002/1521-3773(20000818)39:16<2903::AID-ANIE2903>3.0.CO;2-Q
Return to citation in text: [1] [2] -
Samojłowicz, C.; Bieniek, M.; Grela, K. Chem. Rev. 2009, 109, 3708–3742. doi:10.1021/cr800524f
Return to citation in text: [1] -
Hoveyda, A. H.; Schrock, R. R. Chem. – Eur. J. 2001, 7, 945–950. doi:10.1002/1521-3765(20010302)7:5<945::AID-CHEM945>3.0.CO;2-3
Return to citation in text: [1] [2] [3] -
Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 42, 4592–4633. doi:10.1002/anie.200300576
Return to citation in text: [1] [2] [3] -
Fürstner, A. Angew. Chem., Int. Ed. 2000, 39, 3012–3043. doi:10.1002/1521-3773(20000901)39:17<3012::AID-ANIE3012>3.0.CO;2-G
Return to citation in text: [1] [2] -
Grubbs, R. H., Ed. Handbook of Olefin Metathesis; Wiley-VCH: Weinheim, Germany, 2003. doi:10.1002/9783527619481
Return to citation in text: [1] [2] -
Dias, E. L.; Nguyen, S. T.; Grubbs, R. H. J. Am. Chem. Soc. 1997, 119, 3887–3897. doi:10.1021/ja963136z
Return to citation in text: [1] -
Ulman, M.; Grubbs, R. H. Organometallics 1998, 17, 2484–2489. doi:10.1021/om9710172
Return to citation in text: [1] -
Adlhart, C.; Hinderling, C.; Baumann, H.; Chen, P. J. Am. Chem. Soc. 2000, 122, 8204–8214. doi:10.1021/ja9938231
Return to citation in text: [1] -
Adlhart, C.; Volland, M. A. O.; Hofmann, P.; Chen, P. Helv. Chim. Acta 2000, 83, 3306–3311. doi:10.1002/1522-2675(20001220)83:12<3306::AID-HLCA3306>3.0.CO;2-7
Return to citation in text: [1] -
Adlhart, C.; Chen, P. Helv. Chim. Acta 2000, 83, 2192–2196. doi:10.1002/1522-2675(20000906)83:9<2192::AID-HLCA2192>3.0.CO;2-G
Return to citation in text: [1] -
Aagaard, O. M.; Meier, R. J.; Buda, F. J. Am. Chem. Soc. 1998, 120, 7174–7182. doi:10.1021/ja974131k
Return to citation in text: [1] -
Cavallo, L. J. Am. Chem. Soc. 2002, 124, 8965–8973. doi:10.1021/ja016772s
Return to citation in text: [1] -
Tallarico, J. A.; Bonitatebus, P. J., Jr.; Snapper, M. L. J. Am. Chem. Soc. 1997, 119, 7157–7158. doi:10.1021/ja971285r
Return to citation in text: [1] -
Chen, P. Angew. Chem., Int. Ed. 2003, 42, 2832–2847. doi:10.1002/anie.200200560
Return to citation in text: [1] -
Correa, A.; Cavallo, A. J. Am. Chem. Soc. 2006, 128, 13352–13353. doi:10.1021/ja064924j
Return to citation in text: [1] -
Urbina-Blanco, C. A.; Poater, A.; Lebl, T.; Manzini, S.; Slawin, A. M. Z.; Cavallo, L.; Nolan, S. P. J. Am. Chem. Soc. 2013, 135, 7073–7079. doi:10.1021/ja402700p
Return to citation in text: [1] -
Poater, A.; Ragone, F.; Correa, A.; Szadkowska, A.; Barbasiewicz, M.; Grela, K.; Cavallo, L. Chem. – Eur. J. 2010, 16, 14354–14364. doi:10.1002/chem.201001849
Return to citation in text: [1] -
Romero, P. E.; Piers, W. E. J. Am. Chem. Soc. 2007, 129, 1698–1704. doi:10.1021/ja0675245
Return to citation in text: [1] [2] -
Pump, E.; Poater, A.; Zirngast, M.; Torvisco, A.; Fischer, R.; Cavallo, L.; Slugovc, C. Organometallics 2014, 33, 2806–2813. doi:10.1021/om500315t
Return to citation in text: [1] [2] [3] [4] -
Poater, A.; Correa, A.; Pump, E.; Cavallo, L. Chem. Heterocycl. Compd. 2014, 50, 389–395. doi:10.1007/s10593-014-1491-6
Return to citation in text: [1] -
Benitez, D.; Tkatchouk, E.; Goddard, W. A., III. Chem. Commun. 2008, 46, 6194–6196. doi:10.1039/b815665d
Return to citation in text: [1] -
Vidavsky, Y.; Anaby, A.; Lemcoff, N. G. Dalton Trans. 2012, 41, 32–43. doi:10.1039/C1DT11404B
Return to citation in text: [1] -
Urbina-Blanco, C. A.; Bantreil, X.; Wappel, J.; Schmid, T. E.; Slawin, A. M. Z.; Slugovc, C.; Cazin, C. S. J. Organometallics 2013, 32, 6240–6247. doi:10.1021/om4004362
Return to citation in text: [1] [2] -
Schmid, T. E.; Bantreil, X.; Citadelle, C. A.; Slawin, A. M. Z.; Cazin, C. S. J. Chem. Commun. 2011, 47, 7060–7062. doi:10.1039/c1cc10825e
Return to citation in text: [1] -
Costabile, C.; Cavallo, L. J. Am. Chem. Soc. 2004, 126, 9592–9600. doi:10.1021/ja0484303
Return to citation in text: [1] -
Adlhart, C.; Chen, P. J. Am. Chem. Soc. 2004, 126, 3496–3510. doi:10.1021/ja0305757
Return to citation in text: [1] -
Adlhart, C.; Chen, P. Angew. Chem., Int. Ed. 2002, 41, 4484–4487. doi:10.1002/1521-3773(20021202)41:23<4484::AID-ANIE4484>3.0.CO;2-Q
Return to citation in text: [1] -
Vyboishchikov, S. F.; Bühl, M.; Thiel, W. Chem. – Eur. J. 2002, 8, 3962–3975. doi:10.1002/1521-3765(20020902)8:17<3962::AID-CHEM3962>3.0.CO;2-X
Return to citation in text: [1] -
Fomine, S.; Martinez Vargas, S.; Tlenkopatchev, M. A. Organometallics 2003, 22, 93–99. doi:10.1021/om020581w
Return to citation in text: [1] -
Suresh, C. H.; Koga, N. Organometallics 2004, 23, 76–80. doi:10.1021/om034011n
Return to citation in text: [1] -
Occhipinti, G.; Bjørsvik, H.-R.; Jensen, V. R. J. Am. Chem. Soc. 2006, 128, 6952–6954. doi:10.1021/ja060832i
Return to citation in text: [1] [2] -
Romero, P. E.; Piers, W. E. J. Am. Chem. Soc. 2005, 127, 5032–5033. doi:10.1021/ja042259d
Return to citation in text: [1] -
Ung, T.; Hejl, A.; Grubbs, R. H.; Schrodi, Y. Organometallics 2004, 23, 5399–5401. doi:10.1021/om0493210
Return to citation in text: [1] -
Benitez, D.; Goddard, W. A., III. J. Am. Chem. Soc. 2005, 127, 12218–12219. doi:10.1021/ja051796a
Return to citation in text: [1] -
Anderson, D. R.; Hickstein, D. D.; O’Leary, D. J.; Grubbs, R. H. J. Am. Chem. Soc. 2006, 128, 8386–8387. doi:10.1021/ja0618090
Return to citation in text: [1] -
Dorta, R.; Stevens, E. D.; Scott, N. M.; Costabile, C.; Cavallo, L.; Hoff, C. D.; Nolan, S. P. J. Am. Chem. Soc. 2005, 127, 2485–2495. doi:10.1021/ja0438821
Return to citation in text: [1] -
Viciu, M. S.; Navarro, O.; Germaneau, R. F.; Kelly, R. A., III; Sommer, W.; Marion, N.; Stevens, E. D.; Cavallo, L.; Nolan, S. P. Organometallics 2004, 23, 1629–1635. doi:10.1021/om034319e
Return to citation in text: [1] -
Seiders, T. J.; Ward, D. W.; Grubbs, R. H. Org. Lett. 2001, 3, 3225–3228. doi:10.1021/ol0165692
Return to citation in text: [1] -
Funk, T. W.; Berlin, J. M.; Grubbs, R. H. J. Am. Chem. Soc. 2006, 128, 1840–1846. doi:10.1021/ja055994d
Return to citation in text: [1] -
Monfette, S.; Fogg, D. E. Organometallics 2006, 25, 1940–1944. doi:10.1021/om050952j
Return to citation in text: [1] -
Conrad, J. C.; Parnas, H. H.; Snelgrove, J. L.; Fogg, D. E. J. Am. Chem. Soc. 2005, 127, 11882–11883. doi:10.1021/ja042736s
Return to citation in text: [1] -
Gaussian 03, Version B.1; Gaussian, Inc.: Pittsburgh, PA, U.S.A., 2003.
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5651. doi:10.1063/1.464913
Return to citation in text: [1] -
Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. J. Phys. Chem. 1994, 98, 11623–11627. doi:10.1021/j100096a001
Return to citation in text: [1] -
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/PhysRevB.37.785
Return to citation in text: [1] -
Becke, A. D. Phys. Rev. A 1988, 38, 3098–3100. doi:10.1103/PhysRevA.38.3098
Return to citation in text: [1] -
Perdew, J. P. Phys. Rev. B 1986, 33, 8822–8824. doi:10.1103/PhysRevB.33.8822
Return to citation in text: [1] -
Perdew, J. P. Phys. Rev. B 1986, 34, 7406. doi:10.1103/PhysRevB.34.7406
Return to citation in text: [1] -
Schäfer, A.; Horn, H.; Ahlrichs, R. J. Chem. Phys. 1992, 97, 2571–2577. doi:10.1063/1.463096
Return to citation in text: [1] -
Häusermann, U.; Dolg, M.; Stoll, H.; Preuss, H.; Schwerdtfeger, P.; Pitzer, R. M. Mol. Phys. 1993, 78, 1211–1224. doi:10.1080/00268979300100801
Return to citation in text: [1] -
Küchle, W.; Dolg, M.; Stoll, H.; Preuss, H. J. Chem. Phys. 1994, 100, 7535–7542. doi:10.1063/1.466847
Return to citation in text: [1] -
Leininger, T.; Nicklass, A.; Stoll, H.; Dolg, M.; Schwerdtfeger, P. J. Chem. Phys. 1996, 105, 1052–1059. doi:10.1063/1.471950
Return to citation in text: [1] -
Barone, V.; Cossi, M. J. Phys. Chem. A 1998, 102, 1995–2001. doi:10.1021/jp9716997
Return to citation in text: [1] -
Zhao, Y.; Truhlar, D. G. Org. Lett. 2007, 9, 1967–1970. doi:10.1021/ol0705548
Return to citation in text: [1] [2] -
Pandian, S.; Hillier, I. H.; Vincent, M. A.; Burton, N. A.; Ashworth, I. W.; Nelson, D. J.; Percy, J. M.; Rinaudo, G. Chem. Phys. Lett. 2009, 476, 37–40. doi:10.1016/j.cplett.2009.06.021
Return to citation in text: [1] -
Minenkov, Y.; Occhipinti, G.; Heyndrickx, W.; Jensen, V. R. Eur. J. Inorg. Chem. 2012, 2012, 1507–1516. doi:10.1002/ejic.201100932
Return to citation in text: [1] -
Fey, N.; Ridgway, B. M.; Jover, J.; McMullin, C. L.; Harvey, J. N. Dalton Trans. 2011, 40, 11184–11191. doi:10.1039/c1dt10909j
Return to citation in text: [1] -
Pump, E.; Slugovc, C.; Cavallo, L.; Poater, A. Organometallics 2015, 34, 3107–3111. doi:10.1021/om501246q
Return to citation in text: [1] -
Poater, A.; Pump, E.; Vummaleti, S. V. C.; Cavallo, L. J. Chem. Theory Comput. 2014, 10, 4442–4448. doi:10.1021/ct5003863
Return to citation in text: [1] -
Credendino, R.; Poater, A.; Ragone, F.; Cavallo, L. Catal. Sci. Technol. 2011, 1, 1287–1297. doi:10.1039/c1cy00052g
Return to citation in text: [1] -
Poater, A.; Ragone, F.; Correa, A.; Cavallo, L. Dalton Trans. 2011, 40, 11066–11069. doi:10.1039/c1dt10959f
Return to citation in text: [1] -
Manzini, S.; Urbina-Blanco, C. A.; Nelson, D. J.; Poater, A.; Lebl, T.; Meiries, S.; Slawin, A. M. Z.; Falivene, L.; Cavallo, L.; Nolan, S. P. J. Organomet. Chem. 2015, 780, 43–48. doi:10.1016/j.jorganchem.2014.12.040
Return to citation in text: [1] -
Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746–1787. doi:10.1021/cr9002424
Return to citation in text: [1] -
Schwab, P.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1996, 118, 100–110. doi:10.1021/ja952676d
Return to citation in text: [1] -
van der Eide, E. F.; Piers, W. E. Nat. Chem. 2010, 2, 571–576. doi:10.1038/nchem.653
Return to citation in text: [1] -
Falivene, L.; Poater, A.; Cazin, C. S. J.; Slugovc, C.; Cavallo, L. Dalton Trans. 2013, 42, 7312–7317. doi:10.1039/C2DT32277C
Return to citation in text: [1] -
Leitgeb, A.; Abbas, M.; Fischer, R. C.; Poater, A.; Cavallo, L.; Slugovc, C. Catal. Sci. Technol. 2012, 2, 1640–1643. doi:10.1039/c2cy20311a
Return to citation in text: [1] -
Bantreil, X.; Poater, A.; Urbina-Blanco, C. A.; Bidal, Y. D.; Falivene, L.; Randall, R. A. M.; Cavallo, L.; Slawin, A. M. Z.; Cazin, C. S. Organometallics 2012, 31, 7415–7426. doi:10.1021/om300703p
Return to citation in text: [1] -
Szczepaniak, G.; Kosinski, K.; Grela, K. Green Chem. 2014, 16, 4474–4492. doi:10.1039/C4GC00705K
Return to citation in text: [1] -
Minenkov, Y.; Singstad, A.; Occhipinti, G.; Jensen, V. R. Dalton Trans. 2012, 41, 5526–5541. doi:10.1039/c2dt12232d
Return to citation in text: [1] -
Minenkov, Y.; Occhipinti, G.; Heyndrickx, W.; Jensen, V. R. Eur. J. Inorg. Chem. 2012, 2012, 1507–1516. doi:10.1002/ejic.201100932
Return to citation in text: [1] -
Minenkov, Y.; Occhipinti, G.; Jensen, V. R. Organometallics 2013, 32, 2099–2111. doi:10.1021/om301192a
Return to citation in text: [1] -
Paredes-Gil, K.; Solans-Monfort, X.; Rodriguez-Santiago, L.; Sodupe, M.; Jaque, P. Organometallics 2014, 33, 6065–6075. doi:10.1021/om500718a
Return to citation in text: [1] -
Poater, A.; Pump, E.; Vummaleti, S. V. C.; Cavallo, L. Chem. Phys. Lett. 2014, 610–611, 29–32. doi:10.1016/j.cplett.2014.06.063
Return to citation in text: [1] -
Poater, A.; Vummaleti, S. V. C.; Pump, E.; Cavallo, L. Dalton Trans. 2014, 43, 11216–11220. doi:10.1039/c4dt00325j
Return to citation in text: [1] -
Poater, A.; Credendino, R.; Slugovc, C.; Cavallo, L. Dalton Trans. 2013, 42, 7271–7275. doi:10.1039/c3dt32884h
Return to citation in text: [1] -
Poater, A.; Falivene, L.; Urbina-Blanco, C. A.; Manzini, S.; Nolan, S. P.; Cavallo, L. Dalton Trans. 2013, 42, 7433–7439. doi:10.1039/c3dt32980a
Return to citation in text: [1] -
Nuñez-Zarur, F.; Solans-Monfort, X.; Rodríguez-Santiago, L.; Sodupe, M. Organometallics 2012, 31, 4203–4215. doi:10.1021/om300150d
Return to citation in text: [1] -
Poater, A.; Cavallo, L. J. Mol. Catal. A: Chem. 2010, 324, 75–79. doi:10.1016/j.molcata.2010.02.023
Return to citation in text: [1] -
Poater, A.; Bahri-Lalehac, N.; Cavallo, L. Chem. Commun. 2011, 47, 6674–6676. doi:10.1039/c1cc11594d
Return to citation in text: [1] -
Manzini, S.; Poater, A.; Nelson, D. J.; Cavallo, L.; Nolan, S. P. Chem. Sci. 2014, 5, 180–188. doi:10.1039/C3SC52612G
Return to citation in text: [1] -
Manzini, S.; Poater, A.; Nelson, D. J.; Cavallo, L.; Slawin, A. M. Z.; Nolan, S. P. Angew. Chem., Int. Ed. 2014, 53, 8995–8999. doi:10.1002/anie.201403770
Return to citation in text: [1] -
Manzini, S.; Nelson, D. J.; Lebl, T.; Poater, A.; Cavallo, L.; Slawin, A. M. Z.; Nolan, S. P. Chem. Commun. 2014, 50, 2205–2207. doi:10.1039/c3cc49481k
Return to citation in text: [1] -
Kulkarni, A. D.; Truhlar, D. G. J. Chem. Theory Comput. 2011, 7, 2325–2332. doi:10.1021/ct200188n
Return to citation in text: [1] -
Zhao, Y.; Truhlar, D. G. J. Chem. Theory Comput. 2009, 5, 324–333. doi:10.1021/ct800386d
Return to citation in text: [1] -
Zhao, Y.; Truhlar, D. G. Chem. Phys. Lett. 2011, 502, 1–13. doi:10.1016/j.cplett.2010.11.060
Return to citation in text: [1] -
Zhao, Y.; Truhlar, D. G. Acc. Chem. Res. 2008, 41, 157–167. doi:10.1021/ar700111a
Return to citation in text: [1] -
Tsipis, A. C.; Orpen, A. G.; Harvey, J. N. Dalton Trans. 2005, 2849–2858. doi:10.1039/b506929g
Return to citation in text: [1] -
Bantreil, X.; Nolan, S. P. Nat. Protoc. 2011, 6, 69–77. doi:10.1038/nprot.2010.177
Return to citation in text: [1]
| 57. | Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5651. doi:10.1063/1.464913 |
| 58. | Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. J. Phys. Chem. 1994, 98, 11623–11627. doi:10.1021/j100096a001 |
| 59. | Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/PhysRevB.37.785 |
| 60. | Becke, A. D. Phys. Rev. A 1988, 38, 3098–3100. doi:10.1103/PhysRevA.38.3098 |
| 61. | Perdew, J. P. Phys. Rev. B 1986, 33, 8822–8824. doi:10.1103/PhysRevB.33.8822 |
| 62. | Perdew, J. P. Phys. Rev. B 1986, 34, 7406. doi:10.1103/PhysRevB.34.7406 |
| 63. | Schäfer, A.; Horn, H.; Ahlrichs, R. J. Chem. Phys. 1992, 97, 2571–2577. doi:10.1063/1.463096 |
| 77. | Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746–1787. doi:10.1021/cr9002424 |
| 78. | Schwab, P.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1996, 118, 100–110. doi:10.1021/ja952676d |
| 6. | Fustero, S.; Bello, P.; Miró, J.; Sánchez-Roselló, M.; Haufe, G.; del Pozo, C. Beilstein J. Org. Chem. 2013, 9, 2688–2695. doi:10.3762/bjoc.9.305 |
| 74. | Credendino, R.; Poater, A.; Ragone, F.; Cavallo, L. Catal. Sci. Technol. 2011, 1, 1287–1297. doi:10.1039/c1cy00052g |
| 75. | Poater, A.; Ragone, F.; Correa, A.; Cavallo, L. Dalton Trans. 2011, 40, 11066–11069. doi:10.1039/c1dt10959f |
| 76. | Manzini, S.; Urbina-Blanco, C. A.; Nelson, D. J.; Poater, A.; Lebl, T.; Meiries, S.; Slawin, A. M. Z.; Falivene, L.; Cavallo, L.; Nolan, S. P. J. Organomet. Chem. 2015, 780, 43–48. doi:10.1016/j.jorganchem.2014.12.040 |
| 19. | Grubbs, R. H., Ed. Handbook of Olefin Metathesis; Wiley-VCH: Weinheim, Germany, 2003. doi:10.1002/9783527619481 |
| 68. | Zhao, Y.; Truhlar, D. G. Org. Lett. 2007, 9, 1967–1970. doi:10.1021/ol0705548 |
| 69. | Pandian, S.; Hillier, I. H.; Vincent, M. A.; Burton, N. A.; Ashworth, I. W.; Nelson, D. J.; Percy, J. M.; Rinaudo, G. Chem. Phys. Lett. 2009, 476, 37–40. doi:10.1016/j.cplett.2009.06.021 |
| 70. | Minenkov, Y.; Occhipinti, G.; Heyndrickx, W.; Jensen, V. R. Eur. J. Inorg. Chem. 2012, 2012, 1507–1516. doi:10.1002/ejic.201100932 |
| 71. | Fey, N.; Ridgway, B. M.; Jover, J.; McMullin, C. L.; Harvey, J. N. Dalton Trans. 2011, 40, 11184–11191. doi:10.1039/c1dt10909j |
| 72. | Pump, E.; Slugovc, C.; Cavallo, L.; Poater, A. Organometallics 2015, 34, 3107–3111. doi:10.1021/om501246q |
| 73. | Poater, A.; Pump, E.; Vummaleti, S. V. C.; Cavallo, L. J. Chem. Theory Comput. 2014, 10, 4442–4448. doi:10.1021/ct5003863 |
| 13. | Weskamp, T.; Kohl, F. J.; Hieringer, W.; Gleich, D.; Herrmann, W. A. Angew. Chem., Int. Ed. 1999, 38, 2416–2419. doi:10.1002/(SICI)1521-3773(19990816)38:16<2416::AID-ANIE2416>3.0.CO;2-# |
| 64. | Häusermann, U.; Dolg, M.; Stoll, H.; Preuss, H.; Schwerdtfeger, P.; Pitzer, R. M. Mol. Phys. 1993, 78, 1211–1224. doi:10.1080/00268979300100801 |
| 65. | Küchle, W.; Dolg, M.; Stoll, H.; Preuss, H. J. Chem. Phys. 1994, 100, 7535–7542. doi:10.1063/1.466847 |
| 66. | Leininger, T.; Nicklass, A.; Stoll, H.; Dolg, M.; Schwerdtfeger, P. J. Chem. Phys. 1996, 105, 1052–1059. doi:10.1063/1.471950 |
| 67. | Barone, V.; Cossi, M. J. Phys. Chem. A 1998, 102, 1995–2001. doi:10.1021/jp9716997 |
| 13. | Weskamp, T.; Kohl, F. J.; Hieringer, W.; Gleich, D.; Herrmann, W. A. Angew. Chem., Int. Ed. 1999, 38, 2416–2419. doi:10.1002/(SICI)1521-3773(19990816)38:16<2416::AID-ANIE2416>3.0.CO;2-# |
| 32. | Romero, P. E.; Piers, W. E. J. Am. Chem. Soc. 2007, 129, 1698–1704. doi:10.1021/ja0675245 |
| 79. | van der Eide, E. F.; Piers, W. E. Nat. Chem. 2010, 2, 571–576. doi:10.1038/nchem.653 |
| 33. | Pump, E.; Poater, A.; Zirngast, M.; Torvisco, A.; Fischer, R.; Cavallo, L.; Slugovc, C. Organometallics 2014, 33, 2806–2813. doi:10.1021/om500315t |
| 93. | Poater, A.; Cavallo, L. J. Mol. Catal. A: Chem. 2010, 324, 75–79. doi:10.1016/j.molcata.2010.02.023 |
| 94. | Poater, A.; Bahri-Lalehac, N.; Cavallo, L. Chem. Commun. 2011, 47, 6674–6676. doi:10.1039/c1cc11594d |
| 95. | Manzini, S.; Poater, A.; Nelson, D. J.; Cavallo, L.; Nolan, S. P. Chem. Sci. 2014, 5, 180–188. doi:10.1039/C3SC52612G |
| 96. | Manzini, S.; Poater, A.; Nelson, D. J.; Cavallo, L.; Slawin, A. M. Z.; Nolan, S. P. Angew. Chem., Int. Ed. 2014, 53, 8995–8999. doi:10.1002/anie.201403770 |
| 97. | Manzini, S.; Nelson, D. J.; Lebl, T.; Poater, A.; Cavallo, L.; Slawin, A. M. Z.; Nolan, S. P. Chem. Commun. 2014, 50, 2205–2207. doi:10.1039/c3cc49481k |
| 37. | Urbina-Blanco, C. A.; Bantreil, X.; Wappel, J.; Schmid, T. E.; Slawin, A. M. Z.; Slugovc, C.; Cazin, C. S. J. Organometallics 2013, 32, 6240–6247. doi:10.1021/om4004362 |
| 45. | Occhipinti, G.; Bjørsvik, H.-R.; Jensen, V. R. J. Am. Chem. Soc. 2006, 128, 6952–6954. doi:10.1021/ja060832i |
| 68. | Zhao, Y.; Truhlar, D. G. Org. Lett. 2007, 9, 1967–1970. doi:10.1021/ol0705548 |
| 98. | Kulkarni, A. D.; Truhlar, D. G. J. Chem. Theory Comput. 2011, 7, 2325–2332. doi:10.1021/ct200188n |
| 99. | Zhao, Y.; Truhlar, D. G. J. Chem. Theory Comput. 2009, 5, 324–333. doi:10.1021/ct800386d |
| 100. | Zhao, Y.; Truhlar, D. G. Chem. Phys. Lett. 2011, 502, 1–13. doi:10.1016/j.cplett.2010.11.060 |
| 101. | Zhao, Y.; Truhlar, D. G. Acc. Chem. Res. 2008, 41, 157–167. doi:10.1021/ar700111a |
| 102. | Tsipis, A. C.; Orpen, A. G.; Harvey, J. N. Dalton Trans. 2005, 2849–2858. doi:10.1039/b506929g |
| 80. | Falivene, L.; Poater, A.; Cazin, C. S. J.; Slugovc, C.; Cavallo, L. Dalton Trans. 2013, 42, 7312–7317. doi:10.1039/C2DT32277C |
| 81. | Leitgeb, A.; Abbas, M.; Fischer, R. C.; Poater, A.; Cavallo, L.; Slugovc, C. Catal. Sci. Technol. 2012, 2, 1640–1643. doi:10.1039/c2cy20311a |
| 82. | Bantreil, X.; Poater, A.; Urbina-Blanco, C. A.; Bidal, Y. D.; Falivene, L.; Randall, R. A. M.; Cavallo, L.; Slawin, A. M. Z.; Cazin, C. S. Organometallics 2012, 31, 7415–7426. doi:10.1021/om300703p |
| 83. | Szczepaniak, G.; Kosinski, K.; Grela, K. Green Chem. 2014, 16, 4474–4492. doi:10.1039/C4GC00705K |
| 84. | Minenkov, Y.; Singstad, A.; Occhipinti, G.; Jensen, V. R. Dalton Trans. 2012, 41, 5526–5541. doi:10.1039/c2dt12232d |
| 85. | Minenkov, Y.; Occhipinti, G.; Heyndrickx, W.; Jensen, V. R. Eur. J. Inorg. Chem. 2012, 2012, 1507–1516. doi:10.1002/ejic.201100932 |
| 86. | Minenkov, Y.; Occhipinti, G.; Jensen, V. R. Organometallics 2013, 32, 2099–2111. doi:10.1021/om301192a |
| 87. | Paredes-Gil, K.; Solans-Monfort, X.; Rodriguez-Santiago, L.; Sodupe, M.; Jaque, P. Organometallics 2014, 33, 6065–6075. doi:10.1021/om500718a |
| 88. | Poater, A.; Pump, E.; Vummaleti, S. V. C.; Cavallo, L. Chem. Phys. Lett. 2014, 610–611, 29–32. doi:10.1016/j.cplett.2014.06.063 |
| 89. | Poater, A.; Vummaleti, S. V. C.; Pump, E.; Cavallo, L. Dalton Trans. 2014, 43, 11216–11220. doi:10.1039/c4dt00325j |
| 90. | Poater, A.; Credendino, R.; Slugovc, C.; Cavallo, L. Dalton Trans. 2013, 42, 7271–7275. doi:10.1039/c3dt32884h |
| 91. | Poater, A.; Falivene, L.; Urbina-Blanco, C. A.; Manzini, S.; Nolan, S. P.; Cavallo, L. Dalton Trans. 2013, 42, 7433–7439. doi:10.1039/c3dt32980a |
| 92. | Nuñez-Zarur, F.; Solans-Monfort, X.; Rodríguez-Santiago, L.; Sodupe, M. Organometallics 2012, 31, 4203–4215. doi:10.1021/om300150d |
| 13. | Weskamp, T.; Kohl, F. J.; Hieringer, W.; Gleich, D.; Herrmann, W. A. Angew. Chem., Int. Ed. 1999, 38, 2416–2419. doi:10.1002/(SICI)1521-3773(19990816)38:16<2416::AID-ANIE2416>3.0.CO;2-# |
| 12. | Huang, J.; Stevens, E. D.; Nolan, S. P.; Peterson, J. L. J. Am. Chem. Soc. 1999, 121, 2674–2678. doi:10.1021/ja9831352 |
| 14. | Bielawski, C. W.; Grubbs, R. H. Angew. Chem., Int. Ed. 2000, 39, 2903–2906. doi:10.1002/1521-3773(20000818)39:16<2903::AID-ANIE2903>3.0.CO;2-Q |
| 33. | Pump, E.; Poater, A.; Zirngast, M.; Torvisco, A.; Fischer, R.; Cavallo, L.; Slugovc, C. Organometallics 2014, 33, 2806–2813. doi:10.1021/om500315t |
| 16. | Hoveyda, A. H.; Schrock, R. R. Chem. – Eur. J. 2001, 7, 945–950. doi:10.1002/1521-3765(20010302)7:5<945::AID-CHEM945>3.0.CO;2-3 |
| 33. | Pump, E.; Poater, A.; Zirngast, M.; Torvisco, A.; Fischer, R.; Cavallo, L.; Slugovc, C. Organometallics 2014, 33, 2806–2813. doi:10.1021/om500315t |
| 103. | Bantreil, X.; Nolan, S. P. Nat. Protoc. 2011, 6, 69–77. doi:10.1038/nprot.2010.177 |
| 1. | Trnka, T. M.; Grubbs, R. H. Acc. Chem. Res. 2001, 34, 18–29. doi:10.1021/ar000114f |
| 11. | Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org. Lett. 1999, 1, 953–956. doi:10.1021/ol990909q |
| 12. | Huang, J.; Stevens, E. D.; Nolan, S. P.; Peterson, J. L. J. Am. Chem. Soc. 1999, 121, 2674–2678. doi:10.1021/ja9831352 |
| 13. | Weskamp, T.; Kohl, F. J.; Hieringer, W.; Gleich, D.; Herrmann, W. A. Angew. Chem., Int. Ed. 1999, 38, 2416–2419. doi:10.1002/(SICI)1521-3773(19990816)38:16<2416::AID-ANIE2416>3.0.CO;2-# |
| 14. | Bielawski, C. W.; Grubbs, R. H. Angew. Chem., Int. Ed. 2000, 39, 2903–2906. doi:10.1002/1521-3773(20000818)39:16<2903::AID-ANIE2903>3.0.CO;2-Q |
| 15. | Samojłowicz, C.; Bieniek, M.; Grela, K. Chem. Rev. 2009, 109, 3708–3742. doi:10.1021/cr800524f |
| 46. | Romero, P. E.; Piers, W. E. J. Am. Chem. Soc. 2005, 127, 5032–5033. doi:10.1021/ja042259d |
| 10. | Nguyen, S. T.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1993, 115, 9858–9859. doi:10.1021/ja00074a086 |
| 47. | Ung, T.; Hejl, A.; Grubbs, R. H.; Schrodi, Y. Organometallics 2004, 23, 5399–5401. doi:10.1021/om0493210 |
| 7. | Yuan, W.; Wei, Y.; Shi, M. ChemistryOpen 2013, 2, 63–68. doi:10.1002/open.201300002 |
| 8. | Perfetto, A.; Costabile, C.; Longo, P.; Bertolasi, V.; Grisi, F. Chem. – Eur. J. 2013, 19, 10492–10496. doi:10.1002/chem.201301540 |
| 9. | Perfetto, A.; Costabile, C.; Longo, P.; Grisi, F. Organometallics 2014, 33, 2747–2759. doi:10.1021/om5001452 |
| 35. | Benitez, D.; Tkatchouk, E.; Goddard, W. A., III. Chem. Commun. 2008, 46, 6194–6196. doi:10.1039/b815665d |
| 36. | Vidavsky, Y.; Anaby, A.; Lemcoff, N. G. Dalton Trans. 2012, 41, 32–43. doi:10.1039/C1DT11404B |
| 37. | Urbina-Blanco, C. A.; Bantreil, X.; Wappel, J.; Schmid, T. E.; Slawin, A. M. Z.; Slugovc, C.; Cazin, C. S. J. Organometallics 2013, 32, 6240–6247. doi:10.1021/om4004362 |
| 38. | Schmid, T. E.; Bantreil, X.; Citadelle, C. A.; Slawin, A. M. Z.; Cazin, C. S. J. Chem. Commun. 2011, 47, 7060–7062. doi:10.1039/c1cc10825e |
| 2. | Chauvin, Y. Angew. Chem., Int. Ed. 2006, 45, 3740–3747. doi:10.1002/anie.200601234 |
| 3. | Grubbs, R. H. Angew. Chem., Int. Ed. 2006, 45, 3760–3765. doi:10.1002/anie.200600680 |
| 4. | Schrock, R. R. Angew. Chem., Int. Ed. 2006, 45, 3748–3759. doi:10.1002/anie.200600085 |
| 5. | Hérisson, P. J.-L.; Chauvin, Y. Makromol. Chem. 1971, 141, 161–176. doi:10.1002/macp.1971.021410112 |
| 6. | Fustero, S.; Bello, P.; Miró, J.; Sánchez-Roselló, M.; Haufe, G.; del Pozo, C. Beilstein J. Org. Chem. 2013, 9, 2688–2695. doi:10.3762/bjoc.9.305 |
| 39. | Costabile, C.; Cavallo, L. J. Am. Chem. Soc. 2004, 126, 9592–9600. doi:10.1021/ja0484303 |
| 40. | Adlhart, C.; Chen, P. J. Am. Chem. Soc. 2004, 126, 3496–3510. doi:10.1021/ja0305757 |
| 41. | Adlhart, C.; Chen, P. Angew. Chem., Int. Ed. 2002, 41, 4484–4487. doi:10.1002/1521-3773(20021202)41:23<4484::AID-ANIE4484>3.0.CO;2-Q |
| 42. | Vyboishchikov, S. F.; Bühl, M.; Thiel, W. Chem. – Eur. J. 2002, 8, 3962–3975. doi:10.1002/1521-3765(20020902)8:17<3962::AID-CHEM3962>3.0.CO;2-X |
| 43. | Fomine, S.; Martinez Vargas, S.; Tlenkopatchev, M. A. Organometallics 2003, 22, 93–99. doi:10.1021/om020581w |
| 44. | Suresh, C. H.; Koga, N. Organometallics 2004, 23, 76–80. doi:10.1021/om034011n |
| 45. | Occhipinti, G.; Bjørsvik, H.-R.; Jensen, V. R. J. Am. Chem. Soc. 2006, 128, 6952–6954. doi:10.1021/ja060832i |
| 4. | Schrock, R. R. Angew. Chem., Int. Ed. 2006, 45, 3748–3759. doi:10.1002/anie.200600085 |
| 27. | Tallarico, J. A.; Bonitatebus, P. J., Jr.; Snapper, M. L. J. Am. Chem. Soc. 1997, 119, 7157–7158. doi:10.1021/ja971285r |
| 28. | Chen, P. Angew. Chem., Int. Ed. 2003, 42, 2832–2847. doi:10.1002/anie.200200560 |
| 29. | Correa, A.; Cavallo, A. J. Am. Chem. Soc. 2006, 128, 13352–13353. doi:10.1021/ja064924j |
| 31. | Poater, A.; Ragone, F.; Correa, A.; Szadkowska, A.; Barbasiewicz, M.; Grela, K.; Cavallo, L. Chem. – Eur. J. 2010, 16, 14354–14364. doi:10.1002/chem.201001849 |
| 8. | Perfetto, A.; Costabile, C.; Longo, P.; Bertolasi, V.; Grisi, F. Chem. – Eur. J. 2013, 19, 10492–10496. doi:10.1002/chem.201301540 |
| 9. | Perfetto, A.; Costabile, C.; Longo, P.; Grisi, F. Organometallics 2014, 33, 2747–2759. doi:10.1021/om5001452 |
| 25. | Aagaard, O. M.; Meier, R. J.; Buda, F. J. Am. Chem. Soc. 1998, 120, 7174–7182. doi:10.1021/ja974131k |
| 26. | Cavallo, L. J. Am. Chem. Soc. 2002, 124, 8965–8973. doi:10.1021/ja016772s |
| 8. | Perfetto, A.; Costabile, C.; Longo, P.; Bertolasi, V.; Grisi, F. Chem. – Eur. J. 2013, 19, 10492–10496. doi:10.1002/chem.201301540 |
| 32. | Romero, P. E.; Piers, W. E. J. Am. Chem. Soc. 2007, 129, 1698–1704. doi:10.1021/ja0675245 |
| 33. | Pump, E.; Poater, A.; Zirngast, M.; Torvisco, A.; Fischer, R.; Cavallo, L.; Slugovc, C. Organometallics 2014, 33, 2806–2813. doi:10.1021/om500315t |
| 34. | Poater, A.; Correa, A.; Pump, E.; Cavallo, L. Chem. Heterocycl. Compd. 2014, 50, 389–395. doi:10.1007/s10593-014-1491-6 |
| 20. | Dias, E. L.; Nguyen, S. T.; Grubbs, R. H. J. Am. Chem. Soc. 1997, 119, 3887–3897. doi:10.1021/ja963136z |
| 21. | Ulman, M.; Grubbs, R. H. Organometallics 1998, 17, 2484–2489. doi:10.1021/om9710172 |
| 22. | Adlhart, C.; Hinderling, C.; Baumann, H.; Chen, P. J. Am. Chem. Soc. 2000, 122, 8204–8214. doi:10.1021/ja9938231 |
| 23. | Adlhart, C.; Volland, M. A. O.; Hofmann, P.; Chen, P. Helv. Chim. Acta 2000, 83, 3306–3311. doi:10.1002/1522-2675(20001220)83:12<3306::AID-HLCA3306>3.0.CO;2-7 |
| 24. | Adlhart, C.; Chen, P. Helv. Chim. Acta 2000, 83, 2192–2196. doi:10.1002/1522-2675(20000906)83:9<2192::AID-HLCA2192>3.0.CO;2-G |
| 16. | Hoveyda, A. H.; Schrock, R. R. Chem. – Eur. J. 2001, 7, 945–950. doi:10.1002/1521-3765(20010302)7:5<945::AID-CHEM945>3.0.CO;2-3 |
| 17. | Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 42, 4592–4633. doi:10.1002/anie.200300576 |
| 18. | Fürstner, A. Angew. Chem., Int. Ed. 2000, 39, 3012–3043. doi:10.1002/1521-3773(20000901)39:17<3012::AID-ANIE3012>3.0.CO;2-G |
| 19. | Grubbs, R. H., Ed. Handbook of Olefin Metathesis; Wiley-VCH: Weinheim, Germany, 2003. doi:10.1002/9783527619481 |
| 30. | Urbina-Blanco, C. A.; Poater, A.; Lebl, T.; Manzini, S.; Slawin, A. M. Z.; Cavallo, L.; Nolan, S. P. J. Am. Chem. Soc. 2013, 135, 7073–7079. doi:10.1021/ja402700p |
| 49. | Anderson, D. R.; Hickstein, D. D.; O’Leary, D. J.; Grubbs, R. H. J. Am. Chem. Soc. 2006, 128, 8386–8387. doi:10.1021/ja0618090 |
| 48. | Benitez, D.; Goddard, W. A., III. J. Am. Chem. Soc. 2005, 127, 12218–12219. doi:10.1021/ja051796a |
| 13. | Weskamp, T.; Kohl, F. J.; Hieringer, W.; Gleich, D.; Herrmann, W. A. Angew. Chem., Int. Ed. 1999, 38, 2416–2419. doi:10.1002/(SICI)1521-3773(19990816)38:16<2416::AID-ANIE2416>3.0.CO;2-# |
| 54. | Monfette, S.; Fogg, D. E. Organometallics 2006, 25, 1940–1944. doi:10.1021/om050952j |
| 55. | Conrad, J. C.; Parnas, H. H.; Snelgrove, J. L.; Fogg, D. E. J. Am. Chem. Soc. 2005, 127, 11882–11883. doi:10.1021/ja042736s |
| 52. | Seiders, T. J.; Ward, D. W.; Grubbs, R. H. Org. Lett. 2001, 3, 3225–3228. doi:10.1021/ol0165692 |
| 17. | Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 42, 4592–4633. doi:10.1002/anie.200300576 |
| 53. | Funk, T. W.; Berlin, J. M.; Grubbs, R. H. J. Am. Chem. Soc. 2006, 128, 1840–1846. doi:10.1021/ja055994d |
| 50. | Dorta, R.; Stevens, E. D.; Scott, N. M.; Costabile, C.; Cavallo, L.; Hoff, C. D.; Nolan, S. P. J. Am. Chem. Soc. 2005, 127, 2485–2495. doi:10.1021/ja0438821 |
| 51. | Viciu, M. S.; Navarro, O.; Germaneau, R. F.; Kelly, R. A., III; Sommer, W.; Marion, N.; Stevens, E. D.; Cavallo, L.; Nolan, S. P. Organometallics 2004, 23, 1629–1635. doi:10.1021/om034319e |
| 16. | Hoveyda, A. H.; Schrock, R. R. Chem. – Eur. J. 2001, 7, 945–950. doi:10.1002/1521-3765(20010302)7:5<945::AID-CHEM945>3.0.CO;2-3 |
| 17. | Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 42, 4592–4633. doi:10.1002/anie.200300576 |
| 18. | Fürstner, A. Angew. Chem., Int. Ed. 2000, 39, 3012–3043. doi:10.1002/1521-3773(20000901)39:17<3012::AID-ANIE3012>3.0.CO;2-G |
| 8. | Perfetto, A.; Costabile, C.; Longo, P.; Bertolasi, V.; Grisi, F. Chem. – Eur. J. 2013, 19, 10492–10496. doi:10.1002/chem.201301540 |
| 9. | Perfetto, A.; Costabile, C.; Longo, P.; Grisi, F. Organometallics 2014, 33, 2747–2759. doi:10.1021/om5001452 |
© 2015 Poater and Cavallo; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)