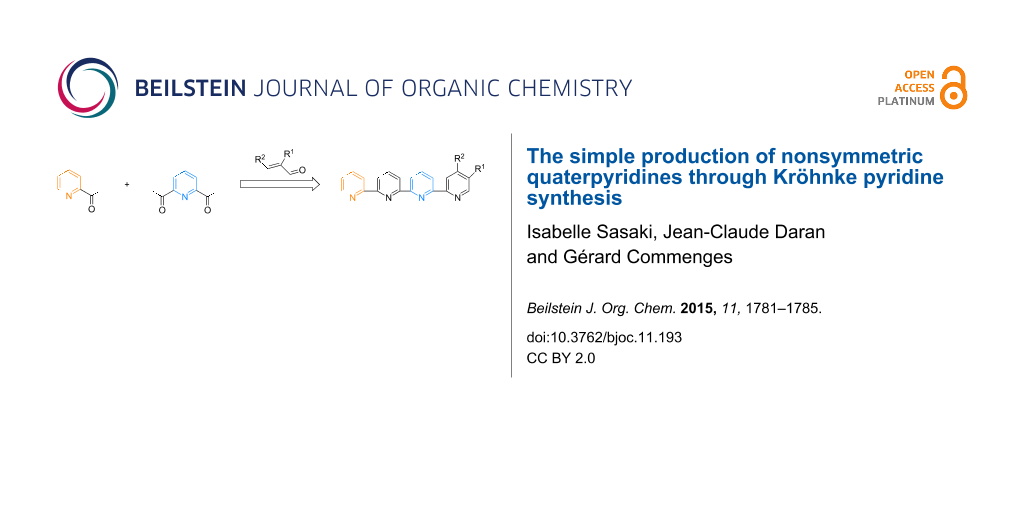
Abstract
Quaterpyridines have been demonstrated to be useful building blocks in metallo-supramolecular chemistry; however, their synthesis requires the preparation of sensitive building blocks. We present here three examples of nonsymmetric quaterpyridines that were easily obtained in yields of 70–85% by condensation of commercially available enones with 6-acetyl-2,2’:6’,2’’-terpyridine through a Kröhnke pyridine synthesis. Easy access to 6-acetyl-2,2’:6’,2’’-terpyridine starting from 2,6-diacetylpyridine and 2-acetylpyridine is described. The X-ray analysis of a chiral quaterpyridine and its Pt(II) complex is presented.

Graphical Abstract
Findings
Polypyridines have been demonstrated to be useful building blocks in metallo-supramolecular chemistry [1-3]. In particular, polypyridines joined at the 2,6-position have the ability to accommodate different coordination numbers preferred by a particular metal. These systems are of particular interest since they offer the possibility to control the assembly of helical systems to grid-like superstructures [1,4-8]. Depending on the metal ions, complexes with 2,2’:6’,2”:6”,2’’’-quaterpyridines as ligands can either adopt square planar, octahedral [3] or tetrahedral dinuclear double-helical geometries [9,10].
Different synthetic methods have been proposed for the preparation of these ligands but the first symmetric quaterpyridines were obtained by Kröhnke [11]. Constable et al. modified this methodology to prepare quaterpyridines bearing phenyl rings on the central pyridine units [7,12,13], then this method was further used by Grätzel et al. [14]. Potts et al. reported a high yield synthesis [15] that requires the cleavage of alkylthio-substituents before the final ligands can be obtained and precludes the preparation of asymmetrical analogues. Various coupling methodologies have also been developed: (i) the Ullmann reaction with copper powder at high temperature [9]; (ii) coupling of 6-halo-2,2’-bipyridines in the presence of nickel reagents [16,17], some of which are chiral [18,19]; and (iii) a Stille-based synthetic pathway [17,20], or a Suzuki–Miyaura cross-coupling reaction [21]. The use of triazine derivatives (which under particular conditions undergo an inverse Diels–Alder reaction) can also produce oligopyridines [22]. On the other hand, there are a limited number of reports dealing with the preparation of nonsymmetric quaterpyridines; Constable et al. proposed a multistep synthesis of 4’-(alkylthio)quaterpyridines [23] to avoid the Stille palladium-catalyzed coupling, whereas Fallahpour obtained the 4’-nitroquaterpyridine by employing the Stille coupling method [24]. Sauer et al. extended the use of triazine derivatives to the synthesis of 4-bromooligopyridines [25].
Constable et al. investigated the coordination behaviour of chiral 2,2’:6’,2”:6”,2”’-quaterpyridines bearing fused chiral groups in the 5,6- and 5”,6”-positions [26]. Recently, a synthesis strategy of asymmetrically substituted 2,2’:6’,2”:6”,2”’-quaterpyridines was described that required a multistep synthesis producing elaborate intermediates [24,27].
This short survey of the different ways to produce quaterpyridines (and in particular, nonsymmetric quaterpyridines) shows that a new simple synthesis can be quite relevant.
The synthetic approach adopted is based on the Kröhnke pyridine method [11]. The asymmetrical quaterpyridines were obtained in a four-step synthesis (Scheme 1).
![[1860-5397-11-193-i1]](/bjoc/content/inline/1860-5397-11-193-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of the quaterpyridines 6–8 and of the platinum complex 9.
Scheme 1: Synthesis of the quaterpyridines 6–8 and of the platinum complex 9.
Starting from 2,6-diacetylpyridine (1), it was possible to carry out the reaction with Eschenmoser’s salt on only one acetyl group leading to the nonsymmetric Mannich salt 3 as the masked enone. Pure compound 3 was only obtained in the presence of an excess of Eschenmoser’s salt, by addition of 2 equiv of (N,N-dimethyl)methyliminium chloride to 1, in acetonitrile at room temperature. Then, compound 3 was condensed with N-[2-oxo-2-(2-pyridinyl)ethyl]pyridinium iodide (2) in the presence of ammonium acetate and gave 6-acetyl-2,2’:6’,2’’-terpyridine (4). This intermediate was cited by Potts [28] but to our knowledge, was not described. During the preparation of nonsymmetric quaterpyridines, 6-acetyl-2,2’:6’,2’’-terpyridine (4) is the key intermediate.
Recently Potvin et al. proposed a simple synthesis of 6,6”-diacetyl-4’-aryl-2,2’:6’,2”- terpyridines [29]. The 6,6”-diacetyl-4’-aryl-2,2’:6’,2”-terpyridine was obtained in a one-step synthesis from commercially available reactants in a 70% yield. Later, Solan et al. proposed a general strategy to prepare oligopyridylimine ligands using Stille-type coupling methodologies. They described different formyl or acetyl-oligopyridyl derivatives obtained by a Stille-type cross-coupling route requiring the preparation of stannylated derivatives [30], and namely, they described compound 4.
After a further Kröhnke synthesis step leading to the corresponding acylpyridinium iodide 5, the asymmetric ligands 6–8 were obtained by condensation with various α,β-unsaturated enones with good to excellent yield (see Supporting Information File 1 for the full experimental data). Von Zelewsky et al. showed that pinene-based chirality is easily introduced on the 2,2’-bipyridine or 2,2’:6’,2”:6”-terpyridine moieties [31,32]. In fact, the use of a chiral enone (i.e., (−)-myrtenal) led to the chiral 5,6-substituted quaterpyridine 8. The corresponding platinum complex 9 was obtained by a classical synthesis, which entails the reaction of the platinum salt in the presence of the ligand in a mixture of acetonitrile and water under reflux for some hours (see Supporting Information File 1).
The quaterpyridines and the platinum complex 9 were fully characterized by NMR, EIMS, and elemental analysis. In particular, for 5, 8 and 9, the chemical shifts of all the signals were identified from two dimensional NMR COSY, HMBC and HSQC spectra. Whereas all the chemical shifts of the protons in the free ligand 8 were different, in the corresponding complex 9, the chemical shifts of some protons and of some carbons were not discernible, and in particular, some in the aromatic region. The 1H spectrum of the complex showed a general deshielding varying from 0.8 to 0.2 ppm, where the most important values were observed on the aromatic protons (H5’’’ and H6) and the much lower values on the aliphatic protons. We observed a deshielding of the aromatic carbons from 2 to 29 ppm, whereas only weak shielding was observed on the aliphatic carbons. We noticed that the most affected carbons are those on the meta- or para-positions to the nitrogen atoms that are coordinated to the Pt atom. The most important deshielding instances are observed on C5 and C4, belonging to the aromatic ring substituted by the pinene moieties. These may impose some strain in the Pt complex, resulting in a slightly different electronic environment as compared to the other nonsubstituted aromatic rings.
Crystals of compounds 5, 8 and 9 were obtained and their X-ray structures are described (for compound 5, see Supporting Information File 1 for a description of the structure). The X-ray structures for the chiral quaterpyridine 8 and its Pt complex 9 are presented in Figure 1 and Figure 2 (for details, see Supporting Information File 1).
Compound 8 is comprised of 3 pyridine rings and a fused pyridine pinene fragment (Figure 1). The first 3 pyridine rings are roughly planar with the largest deviation from the mean plane being −0.052(2) at C33. The fourth pyridine ring and the first five atoms of the pinene part are also roughly planar with the largest deviation being −0.052(2) at C134. These two planes make a dihedral angle of 20.48(5)°. The carbon atoms C133 and C135 of the pinene fragment are roughly symmetrically distributed above and below the N1-C11-C12-C13-C14-C15-C131-C132-C134 planes by −1.118(3) and 1.008(3). Thus, both the C6 rings adopt a half-chair conformation. The packing is stabilised by van der Waals contacts and weak π–π interactions between symmetry related N2-C21-C22-C23-C24-C25 and N4-C41-C42-C43-C44-C45 pyridine rings (1+x, y, z) with a centroid-to-centroid distance of 3.761(2) Å, and an average interatomic distance between planes of 3.425(1) Å, resulting in a slippage of 1.54 Å. The absolute configuration is established by the occurrence of the known chirality of the starting material as (−)-(1R,5S)-myrtenal was used.
![[1860-5397-11-193-1]](/bjoc/content/figures/1860-5397-11-193-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Molecular view of compound 8 with the atom labelling scheme. Ellipsoids are drawn at the 50% probability level. H atoms are represented as small circles of arbitrary radii.
Figure 1: Molecular view of compound 8 with the atom labelling scheme. Ellipsoids are drawn at the 50% probab...
Crystals of complex 9 were obtained by slow evaporation of a solution of the compound with trifluoromethanesulfonate as the counter ion. The complex 9 results from the coordination of the chiral quaterpyridine on the Pt atom (Figure 2). The asymmetric unit contains two triflate anions to equilibrate the charges. The four nitrogen atoms of the quaterpyridine are coordinated to the platinum resulting in a distorted, square planar environment. The PtN4 framework is nearly planar with the largest deviations being −0.008(4) Å at the Pt atom. However, to accommodate the tetracoordination, the N-Pt-N angles range from 80.3(3)° to 116.8(3)°. Because of the presence of the Pt atom, the absolute configuration can be reliably determined by X-ray analysis [33], confirming the (R,S)-configuration on the pinene part.
![[1860-5397-11-193-2]](/bjoc/content/figures/1860-5397-11-193-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular view of compound 9 using the atom labelling scheme. Selected values of bonds (Å) and angles (°) are given: Pt(1)-N(1): 2.046(6); Pt(1)-N(2): 1.936(7); Pt(1)-N(3): 1.934(6); Pt(1)-N(4):2.052(6); N(3)-Pt(1)-N(2): 82.2(3); N(3)-Pt(1)-N(4):80.3(3); N(3)-Pt(1)-N(1): 162.9(3); N(2)-Pt(1)-N(4):162.4(3); N(2)-Pt(1)-N(1): 80.8(3); N(1)-Pt(1)-N(4): 116.8(3). Ellipsoids are drawn at the 50% probability level. H atoms are represented as small circle of arbitrary radii.
Figure 2: Molecular view of compound 9 using the atom labelling scheme. Selected values of bonds (Å) and angl...
Conclusion
The primary result of this work was the development of a novel, simple, synthetic route for nonsymmetric quaterpyridines and circumventing the production of complex intermediates. This was accomplished through the simple synthesis of 6-acetyl-2,2’:6’,2”-terpyridine. After the preparation of the corresponding pyridinium salt, the subsequent condensation with enones led to the desired quaterpyridines. The methyl groups on the pyridine ring are potential grafting points, which can be used without modifying the chelating properties of the polydentate ligands.
Supporting Information
| Supporting Information File 1:
Experimental procedures, characterisation data for all new compounds and X-ray analysis of compound 5.
Crystallographic data (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1401819-1401821. Copies of the data can be obtained free of charge on application to the Director at CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (FAX: (+44) 1223-336-033; email: deposit@ccdc.cam.ac.uk). |
||
| Format: PDF | Size: 1.4 MB | Download |
References
-
Constable, E. C. Prog. Inorg. Chem. 1994, 42, 67–138. doi:10.1002/9780470166437.ch2
Return to citation in text: [1] [2] -
Lehn, J.-M. Supramolecular Chemistry; VCH: Weinheim, 1995. doi:10.1002/3527607439
Return to citation in text: [1] -
Von Zelewsky, A. Stereochemistry of Coordination Compounds; John Wiley and Sons Ltd: Chichester, 1996.
Return to citation in text: [1] [2] -
Constable, E. C. Tetrahedron 1992, 48, 10013–10059. doi:10.1016/S0040-4020(01)89035-9
Return to citation in text: [1] -
Piguet, C.; Bernardinelli, G.; Hopfgartner, G. Chem. Rev. 1997, 97, 2005–2062. doi:10.1021/cr960053s
Return to citation in text: [1] -
Ward, M. D. Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem. 1999, 95, 261–312. doi:10.1039/A804897E
Return to citation in text: [1] -
Constable, E. C.; Hannon, M. J.; Harverson, P.; Neuburger, M.; Smith, D. R.; Wanner, V. F.; Whall, L. A.; Zehnder, M. Polyhedron 2000, 19, 23–34. doi:10.1016/S0277-5387(99)00318-6
Return to citation in text: [1] [2] -
Machado, V. G.; Baxter, P. N. W.; Lehn, J.-M. J. Braz. Chem. Soc. 2001, 12, 431–462. doi:10.1590/S0103-50532001000400002
Return to citation in text: [1] -
Lehn, J. M.; Sauvage, J. P.; Simon, J.; Ziessel, R.; Piccinni-Leopardi, C.; Germain, G.; Declercq, J. P.; Van Meerssche, M. Nouv. J. Chim. 1983, 7, 413–420.
Return to citation in text: [1] [2] -
Constable, E. C.; Hannon, M. J.; Martin, P. A.; Raithby, P. R.; Tocher, D. A. Polyhedron 1992, 11, 2967–2971. doi:10.1016/S0277-5387(00)83604-9
Return to citation in text: [1] -
Kröhnke, F. Synthesis 1976, 1–24. doi:10.1055/s-1976-23941
Return to citation in text: [1] [2] -
Constable, E. C.; Harverson, P.; Smith, D. R.; Whall, L. A. Tetrahedron 1994, 50, 7799–7806. doi:10.1016/S0040-4020(01)85263-7
Return to citation in text: [1] -
Constable, E. C.; Harverson, P. Polyhedron 1999, 18, 3093–3106. doi:10.1016/S0277-5387(99)00224-7
Return to citation in text: [1] -
Renouard, T.; Grätzel, M. Tetrahedron 2001, 57, 8145–8150. doi:10.1016/S0040-4020(01)00801-8
Return to citation in text: [1] -
Potts, K. T.; Gheysen Raiford, K. A.; Keshavarz-K, M. J. Am. Chem. Soc. 1993, 115, 2793–2807. doi:10.1021/ja00060a029
Return to citation in text: [1] -
Constable, E. C.; Elder, S. M.; Healy, J.; Tocher, D. A. J. Chem. Soc., Dalton Trans. 1990, 1669–1674. doi:10.1039/dt9900001669
Return to citation in text: [1] -
Renouard, T.; Fallahpour, R.-A.; Nazeeruddin, M. K.; Humphry-Baker, R.; Gorelsky, S. I.; Lever, A. B. P.; Grätzel, M. Inorg. Chem. 2002, 41, 367–378. doi:10.1021/ic010512u
Return to citation in text: [1] [2] -
Kwong, H.-L.; Yeung, H.-L.; Lee, W.-S.; Wong, W.-T. Chem. Commun. 2006, 4841–4843. doi:10.1039/b608481h
Return to citation in text: [1] -
Yeung, H.-L.; Wong, W.-Y.; Wong, C.-Y.; Kwong, H.-L. Inorg. Chem. 2009, 48, 4108–4117. doi:10.1021/ic802249f
Return to citation in text: [1] -
Barolo, C.; Nazeeruddin, M. K.; Fantacci, S.; Di Censo, D.; Comte, P.; Liska, P.; Viscardi, G.; Quagliotto, P.; De Angelis, F.; Ito, S.; Grätzel, M. Inorg. Chem. 2006, 45, 4642–4653. doi:10.1021/ic051970w
Return to citation in text: [1] -
Coluccini, C.; Manfredi, N.; Salamone, M. M.; Ruffo, R.; Lobello, M. G.; De Angelis, F.; Abbotto, A. J. Org. Chem. 2012, 77, 7945–7956. doi:10.1021/jo301226z
Return to citation in text: [1] -
Pabst, G. R.; Schmid, K.; Sauer, J. Tetrahedron Lett. 1998, 39, 6691–6694. doi:10.1016/S0040-4039(98)01438-5
Return to citation in text: [1] -
Constable, E. C.; Heirtzler, F.; Neuburger, M.; Zehnder, M. J. Am. Chem. Soc. 1997, 119, 5606–5617. doi:10.1021/ja9623626
Return to citation in text: [1] -
Fallahpour, R. Helv. Chim. Acta 2000, 83, 384–393. doi:10.1002/(SICI)1522-2675(20000216)83:2<384::AID-HLCA384>3.0.CO;2-F
Return to citation in text: [1] [2] -
Pabst, G. R.; Sauer, J. Tetrahedron 1999, 55, 5067–5088. doi:10.1016/S0040-4020(99)00179-9
Return to citation in text: [1] -
Constable, E. C.; Kulke, T.; Baum, G.; Fenske, D. Inorg. Chem. Commun. 1998, 1, 80–82. doi:10.1016/S1387-7003(98)00020-3
Return to citation in text: [1] -
Hougen, I. A. Breaking the symmetry with helicating oligopyridines. Ph.D. Thesis, University of Basel, Switzerland, 2004.
Return to citation in text: [1] -
Potts, K. T. Bull. Soc. Chim. Belg. 1990, 99, 741–768. doi:10.1002/bscb.19900990911
Return to citation in text: [1] -
Masciello, L.; Potvin, P. G. Can. J. Chem. 2003, 81, 209–218. doi:10.1139/v03-020
Return to citation in text: [1] -
Champouret, Y. D. M.; Chaggar, R. K.; Dadhiwala, I.; Fawcett, J.; Solan, G. A. Tetrahedron 2006, 62, 79–89. doi:10.1016/j.tet.2005.09.137
Return to citation in text: [1] -
Hayoz, P.; von Zelewsky, A. Tetrahedron Lett. 1992, 33, 5165–5168. doi:10.1016/S0040-4039(00)79123-4
Return to citation in text: [1] -
Hayoz, P.; von Zelewsky, A.; Stoeckli-Evans, H. J. Am. Chem. Soc. 1993, 113, 5111–5114. doi:10.1021/ja00065a023
Return to citation in text: [1] -
Flack, H. D. Acta Crystallogr., Sect. A: Found. Crystallogr. 1983, 39, 876–881. doi:10.1107/S0108767383001762
Return to citation in text: [1]
| 1. | Constable, E. C. Prog. Inorg. Chem. 1994, 42, 67–138. doi:10.1002/9780470166437.ch2 |
| 2. | Lehn, J.-M. Supramolecular Chemistry; VCH: Weinheim, 1995. doi:10.1002/3527607439 |
| 3. | Von Zelewsky, A. Stereochemistry of Coordination Compounds; John Wiley and Sons Ltd: Chichester, 1996. |
| 23. | Constable, E. C.; Heirtzler, F.; Neuburger, M.; Zehnder, M. J. Am. Chem. Soc. 1997, 119, 5606–5617. doi:10.1021/ja9623626 |
| 9. | Lehn, J. M.; Sauvage, J. P.; Simon, J.; Ziessel, R.; Piccinni-Leopardi, C.; Germain, G.; Declercq, J. P.; Van Meerssche, M. Nouv. J. Chim. 1983, 7, 413–420. |
| 10. | Constable, E. C.; Hannon, M. J.; Martin, P. A.; Raithby, P. R.; Tocher, D. A. Polyhedron 1992, 11, 2967–2971. doi:10.1016/S0277-5387(00)83604-9 |
| 24. | Fallahpour, R. Helv. Chim. Acta 2000, 83, 384–393. doi:10.1002/(SICI)1522-2675(20000216)83:2<384::AID-HLCA384>3.0.CO;2-F |
| 3. | Von Zelewsky, A. Stereochemistry of Coordination Compounds; John Wiley and Sons Ltd: Chichester, 1996. |
| 21. | Coluccini, C.; Manfredi, N.; Salamone, M. M.; Ruffo, R.; Lobello, M. G.; De Angelis, F.; Abbotto, A. J. Org. Chem. 2012, 77, 7945–7956. doi:10.1021/jo301226z |
| 1. | Constable, E. C. Prog. Inorg. Chem. 1994, 42, 67–138. doi:10.1002/9780470166437.ch2 |
| 4. | Constable, E. C. Tetrahedron 1992, 48, 10013–10059. doi:10.1016/S0040-4020(01)89035-9 |
| 5. | Piguet, C.; Bernardinelli, G.; Hopfgartner, G. Chem. Rev. 1997, 97, 2005–2062. doi:10.1021/cr960053s |
| 6. | Ward, M. D. Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem. 1999, 95, 261–312. doi:10.1039/A804897E |
| 7. | Constable, E. C.; Hannon, M. J.; Harverson, P.; Neuburger, M.; Smith, D. R.; Wanner, V. F.; Whall, L. A.; Zehnder, M. Polyhedron 2000, 19, 23–34. doi:10.1016/S0277-5387(99)00318-6 |
| 8. | Machado, V. G.; Baxter, P. N. W.; Lehn, J.-M. J. Braz. Chem. Soc. 2001, 12, 431–462. doi:10.1590/S0103-50532001000400002 |
| 22. | Pabst, G. R.; Schmid, K.; Sauer, J. Tetrahedron Lett. 1998, 39, 6691–6694. doi:10.1016/S0040-4039(98)01438-5 |
| 9. | Lehn, J. M.; Sauvage, J. P.; Simon, J.; Ziessel, R.; Piccinni-Leopardi, C.; Germain, G.; Declercq, J. P.; Van Meerssche, M. Nouv. J. Chim. 1983, 7, 413–420. |
| 18. | Kwong, H.-L.; Yeung, H.-L.; Lee, W.-S.; Wong, W.-T. Chem. Commun. 2006, 4841–4843. doi:10.1039/b608481h |
| 19. | Yeung, H.-L.; Wong, W.-Y.; Wong, C.-Y.; Kwong, H.-L. Inorg. Chem. 2009, 48, 4108–4117. doi:10.1021/ic802249f |
| 15. | Potts, K. T.; Gheysen Raiford, K. A.; Keshavarz-K, M. J. Am. Chem. Soc. 1993, 115, 2793–2807. doi:10.1021/ja00060a029 |
| 17. | Renouard, T.; Fallahpour, R.-A.; Nazeeruddin, M. K.; Humphry-Baker, R.; Gorelsky, S. I.; Lever, A. B. P.; Grätzel, M. Inorg. Chem. 2002, 41, 367–378. doi:10.1021/ic010512u |
| 20. | Barolo, C.; Nazeeruddin, M. K.; Fantacci, S.; Di Censo, D.; Comte, P.; Liska, P.; Viscardi, G.; Quagliotto, P.; De Angelis, F.; Ito, S.; Grätzel, M. Inorg. Chem. 2006, 45, 4642–4653. doi:10.1021/ic051970w |
| 14. | Renouard, T.; Grätzel, M. Tetrahedron 2001, 57, 8145–8150. doi:10.1016/S0040-4020(01)00801-8 |
| 7. | Constable, E. C.; Hannon, M. J.; Harverson, P.; Neuburger, M.; Smith, D. R.; Wanner, V. F.; Whall, L. A.; Zehnder, M. Polyhedron 2000, 19, 23–34. doi:10.1016/S0277-5387(99)00318-6 |
| 12. | Constable, E. C.; Harverson, P.; Smith, D. R.; Whall, L. A. Tetrahedron 1994, 50, 7799–7806. doi:10.1016/S0040-4020(01)85263-7 |
| 13. | Constable, E. C.; Harverson, P. Polyhedron 1999, 18, 3093–3106. doi:10.1016/S0277-5387(99)00224-7 |
| 16. | Constable, E. C.; Elder, S. M.; Healy, J.; Tocher, D. A. J. Chem. Soc., Dalton Trans. 1990, 1669–1674. doi:10.1039/dt9900001669 |
| 17. | Renouard, T.; Fallahpour, R.-A.; Nazeeruddin, M. K.; Humphry-Baker, R.; Gorelsky, S. I.; Lever, A. B. P.; Grätzel, M. Inorg. Chem. 2002, 41, 367–378. doi:10.1021/ic010512u |
| 24. | Fallahpour, R. Helv. Chim. Acta 2000, 83, 384–393. doi:10.1002/(SICI)1522-2675(20000216)83:2<384::AID-HLCA384>3.0.CO;2-F |
| 27. | Hougen, I. A. Breaking the symmetry with helicating oligopyridines. Ph.D. Thesis, University of Basel, Switzerland, 2004. |
| 25. | Pabst, G. R.; Sauer, J. Tetrahedron 1999, 55, 5067–5088. doi:10.1016/S0040-4020(99)00179-9 |
| 26. | Constable, E. C.; Kulke, T.; Baum, G.; Fenske, D. Inorg. Chem. Commun. 1998, 1, 80–82. doi:10.1016/S1387-7003(98)00020-3 |
| 31. | Hayoz, P.; von Zelewsky, A. Tetrahedron Lett. 1992, 33, 5165–5168. doi:10.1016/S0040-4039(00)79123-4 |
| 32. | Hayoz, P.; von Zelewsky, A.; Stoeckli-Evans, H. J. Am. Chem. Soc. 1993, 113, 5111–5114. doi:10.1021/ja00065a023 |
| 33. | Flack, H. D. Acta Crystallogr., Sect. A: Found. Crystallogr. 1983, 39, 876–881. doi:10.1107/S0108767383001762 |
| 29. | Masciello, L.; Potvin, P. G. Can. J. Chem. 2003, 81, 209–218. doi:10.1139/v03-020 |
| 30. | Champouret, Y. D. M.; Chaggar, R. K.; Dadhiwala, I.; Fawcett, J.; Solan, G. A. Tetrahedron 2006, 62, 79–89. doi:10.1016/j.tet.2005.09.137 |
| 28. | Potts, K. T. Bull. Soc. Chim. Belg. 1990, 99, 741–768. doi:10.1002/bscb.19900990911 |
© 2015 Sasaki et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)