
Abstract
A facile and moderately functional-group-tolerant synthetic method for the preparation of 7,8-diaza[5]helicenes has been developed. It comprises of an oxidative ring-closing process of 1,1’-binaphthalene-2,2’-diamine (BINAM) derivatives with a chlorine-containing oxidant (t-BuOCl) in the presence of a base (2,6-lutidine). In addition the basic physicochemical properties of newly synthesized compounds have been investigated.
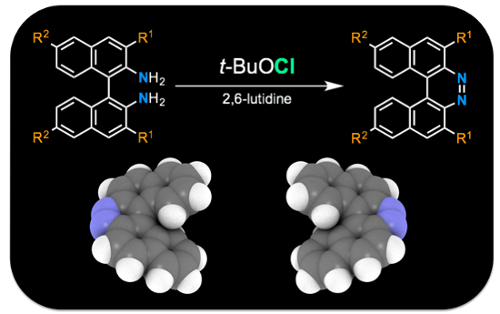
Graphical Abstract
Introduction
Helicenes, ortho-fused polycyclic aromatic compounds, have been fascinating organic chemists over the last century since the first synthesis of azahelicenes was reported by Meisenheimer in 1903 [1], not only because of their aesthetically attractive structures, but also because of their unique properties arising from the helical chirality [2-11]. In fact, helicenes have been finding more and more potential applications such as optoelectronic materials, asymmetric catalysts, and chemosensors [9]. Therefore, the development of synthetic methods for the preparation of helicenes which are difficult to access by conventional methodologies, would offer us many opportunities not only for creating helicene-based novel functional molecules but also uncovering missing pieces of unique aspects of helicenes. A bibliographic survey about helicenes led us focus on diazahelicenes having a diazene (–N=N–) moiety, which is a member of the azahelicene family [5]: These compounds would be a suitable model for probing the effects of the replacement of carbon atoms at the K-region of the carbohelicenes with nitrogen atoms on their structures and physicochemical properties [12-14] and should serve as ligands to metal complexes through the coordination at the diazene unit [15-17]. Taking 7,8-diaza[5]helicene as an example, conventional methodologies of preparing such compounds (Scheme 2) involve i) reductive coupling of 2-nitronaphthalenes using strong reductants like Zn dust [1,18] and PH3 [19], ii) reductive ring-closure of 2,2’-dinitro-1,1’-binaphthalenes using various reductants such as Na2S and LiAlH4 [20,21], and iii) the Scholl-type cyclization of 2,2’-azonaphthalenes using a eutectic melt of AlCl3/NaCl [22]. However, these methods require harsh reaction conditions such as the use of strong reductants or oxidants, leading to low functional group compatibility. More importantly, the reductive methods i) and ii) hardly avoid the production of N-oxide of diazahelicenes as well as azoxynaphthalenes (Scheme 1), the N-oxides are quantitatively reduced by a strong reductant to the corresponding diazahelicenes though. Another simple synthetic route to 7,8-diaza[5]helicenes would involve an oxidative ring-closure of 1,1’-binaphthalene-2,2’-diamines (BINAMs) (Scheme 1, iv). However, interestingly enough, this route remains to be explored. Corbett and Holt reported that the oxidation of BINAM with NaBO3 only gave a trace amount of the corresponding diazahelicene [23]. In this connection, Caronna and co-workers have recently reported a simple protocol for synthesizing 7,8-diaza[5]helicene through oxidation of BINAM with mCPBA and following reduction of its oxides [24]. Although the method is high yielding in 2 steps, it is not straightforward due to the formation of major amounts of its N-oxide and N,N’-dioxide (Scheme 1). Furthermore, functional group compatibility of the method still remains an open question, because of the involvement of the reduction process of the oxides employing LiAlH4. Therefore, in conjunction with recent advances in preparative methods of functionalized BINAMs [25,26], the development of oxidative ring-closing methods that allow for one-step and functional-group-tolerant synthesis of 7,8-diaza[5]helicenes from BINAMs would be desirable.
![[1860-5397-11-2-i1]](/bjoc/content/inline/1860-5397-11-2-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic routes to 7,8-diaza[5]helicene.
Scheme 1: Synthetic routes to 7,8-diaza[5]helicene.
As a part of our project to develop oxidative transformations of aromatic amines for the construction of diverse aza-containing π-conjugated functional molecules [27-30], we have recently discovered an iodine-containing oxidant-induced unusual oxidative rearrangement of BINAMs leading exclusively to U-shaped azaacenes (Scheme 2, the upper reaction) [31]. During a thorough screening of oxidants for the rearrangement reaction, we found out that the use of tert-butyl hypochlorite (t-BuOCl) as an oxidant exclusively gave 7,8-diaza[5]helicene instead of the corresponding rearranged product in good yields. This preliminary finding prompted us to further explore the potential of this oxidative system as a facile and functional-group-tolerant synthetic method of this class of diazahelicenes, since no N-oxide byproducts are formed under this system and thereby any reduction processes using strong reductants are not needed. Herein we present a facile, straightforward, and moderately functional-group-tolerant synthesis of 7,8-diaza[5]helicenes (benzo[f]naphtho[2,1-c]cinnolines) bearing functional substituents on the helical π-conjugated backbone through an oxidative ring-closure of BINAM derivatives (Scheme 2, the lower reaction).
![[1860-5397-11-2-i2]](/bjoc/content/inline/1860-5397-11-2-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Oxidant-controlled transformations of BINAMs.
Scheme 2: Oxidant-controlled transformations of BINAMs.
Results and Discussion
Initially, we began to identify the optimum reaction conditions (oxidants, bases, and solvents) for the oxidative ring-closure of BINAMs using 1a as the model substrate (Table 1, for the full details of the screening of reaction conditions, see Supporting Information File 1). As aforementioned in the Introduction section, we have recognized that the treatment of 1a with 4 equivalents of chlorine-containing oxidant (t-BuOCl) exclusively gave diazahelicene 2a in a high yield (Table 1, entry 1) [31]. As related works to this project, we have reported that the unique iodine-containing reagent t-BuOI, which is readily generated in situ from t-BuOCl and NaI, serves as a powerful oxidant for homo- and cross-dimerization of aromatic amines leading to aromatic azo compounds [27,28,30]. However, the employment of t-BuOI as an oxidant for this intramolecular oxidative N=N forming reaction resulted in much lower yield of 2a (Table 1, entry 2) while the skeletal rearrangement product was obtained as a main product (51% yield) [31], showing the obvious superiority of t-BuOCl as an oxidant. Encouraged by these results, a variety of chlorine-containing oxidants were tested to probe the influence of oxidants on the chemical yields of 2a (Table 1, entries 3–6). While the N-monochlorinated reagents N-chlorosuccinimide (NCS, entry 3) and N-chlorophthalimide (NCPh, entry 5) did not produce 2a at all, di- and tri-chlorinated oxidants 1,3-dichloro-5,5-dimethylhydantoin (DCH, entry 4) and trichloroisocyanuric acid (TCCA, entry 6) gave low yields (ca. 20%) of 2a. Solvents also significantly affected the yields of 2a (Table 1, entries 7–11), and t-BuOH gave the best yield. In terms of the amounts of oxidant, the use of 2 equivalents of t-BuOCl (0.4 mmol) gave a lower yield of 2a (54%, entry 12) with a moderate conversion of 1a, in comparison with the case of using 4 equivalents of t-BuOCl (89%, entry 1). In light of the reaction stoichiometry of the oxidative process, 2 equivalents of HCl are supposed to be concomitantly generated, which could have protonated diamine 1a leading to inhibition of conversion of 1a. To trap the resulting HCl, the efficacy of base addition was investigated applying an ideal stoichiometry (2 equivalents) of t-BuOCl (Table 1, entries 13–17). The addition of an inorganic base (K2CO3) slightly improved the yield (Table 1, entry 13), while strong organic bases like DABCO (pKa 8.93 in DMSO [32]) and DBU (pKa 23.9 in MeCN [33]) gave a lower yield (32%) and no product, respectively (Table 1, entries 14 and 15). As results, the use of the moderately weak organic base 2,6-lutidine (pKa 6.72 in water [34]) successfully afforded 2a in high yield (Table 1, entry 16), and a small excess amount (2.2 equiv) of t-BuOCl and 2,6-lutidine gave diazahelicene 2a quantitatively (Table 1, entry 17). When optically pure (R)-BINAM 1a (99% ee) was applied to the optimized reaction conditions, racemic 2a ([α]D22 = 0.0°, c 1.0, CHCl3) was obtained, losing the chiral information of 1a through rapid racemization of optically active 2a or of the reaction intermediates.
Table 1: The effect of reaction parameters on the oxidative ring-closure of 1a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-2-i6.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Oxidant (X) | Base (Y) | Solvent | Yield (%)a | Recovery of 1a (%)a |
|---|---|---|---|---|---|
| 1 | t-BuOCl (0.8) | — | t-BuOH | 89b | 0 |
| 2 | t-BuOI (0.8) | — | t-BuOH | 6c | 0 |
| 3 | NCS (0.8) | — | t-BuOH | 0 | 63 |
| 4 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-2-i7.svg?max-width=637&scale=1.0)
DCH (0.4) |
— | t-BuOH | 24b | 0 |
| 5 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-2-i8.svg?max-width=637&scale=1.0)
NCPh (0.8) |
— | t-BuOH | 0 | 95 |
| 6 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-2-i9.svg?max-width=637&scale=1.0)
TCCA (0.27) |
— | t-BuOH | 20 | 0 |
| 7 | t-BuOCl (0.8) | — | EtOH | 61 | 0 |
| 8 | t-BuOCl (0.8) | — | MeOH | 58 | 0 |
| 9 | t-BuOCl (0.8) | — | THF | 30 | 0 |
| 10 | t-BuOCl (0.8) | — | MeCN | 22 | 0 |
| 11 | t-BuOCl (0.8) | — | toluene | 68 | 0 |
| 12 | t-BuOCl (0.4) | — | t-BuOH | 54 | 34 |
| 13 | t-BuOCl (0.4) | K2CO3 (0.4) | t-BuOH | 69 | 20 |
| 14 | t-BuOCl (0.4) | DABCO (0.4) | t-BuOH | 32 | 45 |
| 15 | t-BuOCl (0.4) | DBU (0.4) | t-BuOH | 0 | 34 |
| 16 | t-BuOCl (0.4) | 2,6-lutidine (0.4) | t-BuOH | 90 | 10 |
| 17 | t-BuOCl (0.44) | 2,6-lutidine (0.44) | t-BuOH | 97b | 0 |
aDetermined by 1H NMR. bIsolated yields. cDibenzo[a,j]phenazine was obtained in 51% yield.
Having identified the optimized conditions for the oxidative ring-closure of 1a, we then turned our attention to applying this method to the preparation of functionalized 7,8-diaza[5]helicenes (Scheme 3). When diamine 1b bearing 3,3’-dimethyl substituents was subjected to the optimized conditions [t-BuOCl/2,6-lutidine (2.2 equiv) in t-BuOH at room temperature for 3 h], the desired helicene 2b was obtained in a low yield (32%), even though electron-rich benzylic carbons labile to oxidative conditions are present. Modification studies (for the details, see the Supporting Information File 1) revealed that the use of toluene as a solvent slightly improved the yield of 2b (Scheme 3). Notably, 3,3’-dibromo-substituted diamine 1c gave a high yield of dibrominated diazahelicene 2c even in the absence of 2,6-lutidine, albeit in a longer reaction time (Scheme 3). Such brominated helicene would be difficult to prepare by reductive methods using strong reductants like Zn dust, due to competitive over-reduction of the bromo functionality. It should be noted that the reaction efficiency of the oxidative process was not affected by the steric congestion around the aromatic amino moieties (Scheme 3, 2d). Diazahelicene 2e bearing two electron-withdrawing carboxylic ester groups was also successfully obtained in a high yield (84%) without impairing the ester functionality (Scheme 3). Again, it is worth emphasizing that such a type of diazahelicene would be difficult to prepare by conventional reductive methods. Furthermore, a BINAM derivative installed with two alkyl substituents at the 6,6’-position, 1f, was efficiently transformed to the corresponding diazahelicene 2f in a good yield. Bromo functionalities at the 6,6’-position of BINAM also survived the oxidative conditions to give the corresponding diazahelicene 2g (Scheme 3). Regarding the limitation of this oxidative method, diamine substrates having strongly electron-donating substituents (both the 3,3’-MeO and 7,7’-MeO) were not successful, probably due to background side-reactions such as chlorination on the aromatic rings and oxidative oligomerization of BINAMs.
![[1860-5397-11-2-i3]](/bjoc/content/inline/1860-5397-11-2-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Substrate scope of the oxidative ring-closure of BINAMs. Reaction conditions: 1 (0.1 mmol), t-BuOCl (0.22 mmol), 2,6-lutidine (0.22 mmol), and solvent (10 mL) were stirred at the indicated temperature over the time shown below the respective product. Yields shown in parentheses indicate isolated yields. aRun with 4 equivalents of t-BuOCl in the absence of 2,6-lutidine.
Scheme 3: Substrate scope of the oxidative ring-closure of BINAMs. Reaction conditions: 1 (0.1 mmol), t-BuOCl...
Furthermore, this new oxidative method was found to be applicable in the ring-closing reaction of 1,1’-biphenyl-2,2’-diamine (3) leading to benzo[c]cinnoline (4) in a good yield, although the modification of the reaction temperature was required (Scheme 4, for the detailed modification study of reaction conditions, see Supporting Information File 1).
![[1860-5397-11-2-i4]](/bjoc/content/inline/1860-5397-11-2-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
A possible reaction pathway of the oxidative ring-closure of 1a leading to 2a is illustrated in Scheme 5. Chlorination of an amino group of 1a with t-BuOCl would generate N-monochlorinated BINAM (Int-A) and release t-BuOH. The N-chlorination would induce umpolung reactivity of the amino moiety because of the attachment of electronegative halogen species on the N atom [27-30], and thereby an intramolecular nucleophilic unit (–NH2) would attack the electrophilic N-center to form N–N single bond (Int-B). The organic base (2,6-lutidine) would trap HCl generated through this process. Another repetition of a similar N-chlorination/HCl elimination cycle should give 2a via Int-C.
![[1860-5397-11-2-i5]](/bjoc/content/inline/1860-5397-11-2-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: A possible reaction pathway of the oxidative ring-closure of 1a.
Scheme 5: A possible reaction pathway of the oxidative ring-closure of 1a.
Since some of the synthesized diazahelicenes 2 are new members of 7,8-diaza[5]helicenes, basic physicochemical properties of 2a–2g were also investigated (for the detailed data, see the Supporting Information File 1). Diluted dichloromethane solutions of 2 (ca. 10−5 M) exhibited UV–vis absorption spectra featuring weak absorptions ascribed to n–π* transitions in the lower energy region (400–450 nm), an absorption (shoulder at 330–350 nm) and a strong absorption (300–330 nm) ascribed to π–π* transitions, which are typical to aromatic ring-fused cinnoline derivatives [13] (representative spectra are shown in Figure 1; for the full spectra, see Supporting Information File 1). The introduction of two methyl (2b) and phenyl groups (2d) at the 3,3’-position of BINAM skeleton resulted in a red-shift of the absorption spectra over the whole region (Figure 1). In contrast, the introduction of 6,6’-n-Bu substituents (2f) caused a blue-shift of n–π* transitions (380–430 nm), while π–π* transitions (300–340 nm) were red-shifted. On the other hand, diluted dichloromethane solutions (10−5 M) of diazahelicenes 2 did not show photoluminescence (Φf < 0.02, for the detailed values of quantum yields, see the Supporting Information File 1).
![[1860-5397-11-2-1]](/bjoc/content/figures/1860-5397-11-2-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: UV–vis absorption spectra of 2a, 2b, 2d, and 2f.
Figure 1: UV–vis absorption spectra of 2a, 2b, 2d, and 2f.
Due to the presence of the electronegative diazene (–N=N–) moiety, diazahelicenes 2 are expected to possess stabilized LUMO energies. The LUMO energies, which were estimated from cyclic voltammetry (CV) experiments [ELUMO = –(4.8 + redE1/2) eV] using diluted CH2Cl2 solutions (10−4 M) of 2, range from –2.92 to –3.13 eV (for the full cyclic voltammograms and the LUMO energies, see Supporting Information File 1). These LUMO energy levels fall in a similar range of the U-shaped azaacenes that our group previously reported [31] and well-known electron-transporting materials Alq3 [35], suggesting promising electron-accepting abilities of diazahelicenes.
Conclusion
We have developed a facile and moderately functional-group-tolerant method for the synthesis of 7,8-diaza[5]helicenes through an efficient oxidative ring-closure of BINAMs with the combination of t-BuOCl as an oxidant and 2,6-lutidine as a base. This method has been applied to functionalized BINAMs except for highly electron-rich substrates to produce functionalized 7,8-diaza[5]helicenes in a single-step from BINAMs. Furthermore, basic properties of the azahelicenes have also been investigated.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, copies of NMR charts, UV–vis spectra, cyclic voltammograms, and TGA profiles. | ||
| Format: PDF | Size: 2.1 MB | Download |
Acknowledgements
This research was partly supported by the research Grant from The Murata Science Foundation, the Sasakawa Scientific Research Grant from The Japan Science Society, and the Research Encouragement Grants from The Asahi Glass Foundation (to Y.T.). Also, one of the authors (Y.T.) would like to acknowledge all the warm support from the Frontier Research Base for Global Young Researchers, Osaka University, on the Program of MEXT.
References
-
Meisenheimer, J.; Witte, K. Ber. Dtsch. Chem. Ges. 1903, 36, 4153–4164. doi:10.1002/cber.19030360481
Return to citation in text: [1] [2] -
Martin, R. H. Angew. Chem. 1974, 86, 727–738. doi:10.1002/ange.19740862003
Angew. Chem., Int. Ed. Engl. 1974, 13, 649–660. doi:10.1002/anie.197406491
Return to citation in text: [1] -
Urbano, A. Angew. Chem. 2003, 115, 4116–4119. doi:10.1002/ange.200301667
Angew. Chem., Int. Ed. 2003, 42, 3986–3989. doi:10.1002/anie.200301667
Return to citation in text: [1] -
Collins, S. K.; Vachon, M. P. Org. Biomol. Chem. 2006, 4, 2518–2524. doi:10.1039/b603305a
Return to citation in text: [1] -
Dumitrascu, F.; Dumitrescu, D. G.; Aron, I. ARKIVOC 2010, No. i, 1–32.
Return to citation in text: [1] [2] -
Shen, Y.; Chen, C.-F. Chem. Rev. 2012, 112, 1463–1535. doi:10.1021/cr200087r
Return to citation in text: [1] -
Gingras, M. Chem. Soc. Rev. 2012, 42, 968–1006. doi:10.1039/c2cs35154d
Return to citation in text: [1] -
Gingras, M.; Félix, G.; Peresutti, R. Chem. Soc. Rev. 2013, 42, 1007–1050. doi:10.1039/c2cs35111k
Return to citation in text: [1] -
Gingras, M. Chem. Soc. Rev. 2012, 42, 1051–1095. doi:10.1039/c2cs35134j
Return to citation in text: [1] [2] -
Aillard, P.; Voituriez, A.; Marinetti, A. Dalton Trans. 2014, 43, 15263–15278. doi:10.1039/C4DT01935K
Return to citation in text: [1] -
Saleh, N.; Shen, C.; Crassous, J. Chem. Sci. 2014, 5, 3680–3694. doi:10.1039/C4SC01404A
Return to citation in text: [1] -
Corbett, J. F.; Holt, P. F.; Hughes, A. N. J. Chem. Soc. 1961, 1363–1369. doi:10.1039/jr9610001363
Return to citation in text: [1] -
Li, W.; Cai, X.; Hu, Y.; Ye, Y.; Luo, M.; Hu, J. J. Mol. Struct.: THEOCHEM 2005, 732, 21–32. doi:10.1016/j.theochem.2005.06.042
Return to citation in text: [1] [2] -
Caronna, T.; Castiglione, F.; Famulari, A.; Fontana, F.; Malpezzi, L.; Mele, A.; Mendola, D.; Sora, I. N. Molecules 2012, 17, 463–479. doi:10.3390/molecules17010463
Return to citation in text: [1] -
Porter, J. J.; Murray, J. L. J. Am. Chem. Soc. 1965, 87, 1628–1630. doi:10.1021/ja01085a050
Return to citation in text: [1] -
Ellgen, P. C.; McMullin, S. L. Inorg. Chem. 1973, 12, 2004–2007. doi:10.1021/ic50127a010
Return to citation in text: [1] -
Nixon, J. F.; Kooti, M. J. Organomet. Chem. 1978, 149, 71–79. doi:10.1016/S0022-328X(00)90379-6
Return to citation in text: [1] -
Cumming, W. M.; Ferrier, G. S. J. Chem. Soc. 1924, 125, 1108–1111. doi:10.1039/ct9242501108
Return to citation in text: [1] -
Bellaart, A. C. Tetrahedron 1965, 21, 3285–3288. doi:10.1016/S0040-4020(01)96949-2
Return to citation in text: [1] -
Braithwaite, R. S. W.; Holt, P. F. J. Chem. Soc. 1959, 3025–3031. doi:10.1039/jr9590003025
Return to citation in text: [1] -
Corbett, J. F.; Holt, P. F. J. Chem. Soc. 1960, 3646–3653. doi:10.1039/jr9600003646
Return to citation in text: [1] -
Holt, P. F.; Went, C. W. J. Chem. Soc. 1963, 4099–4102. doi:10.1039/jr9630004099
Return to citation in text: [1] -
Corbett, J. F.; Holt, P. F. J. Chem. Soc. 1961, 3695–3699. doi:10.1039/jr9610003695
Return to citation in text: [1] -
Caronna, T.; Fontana, F.; Mele, A.; Sora, I. N.; Panzeri, W.; Viganò, L. Synthesis 2008, 413–416. doi:10.1055/s-2008-1032021
Return to citation in text: [1] -
De, C. K.; Pesciaioli, F.; List, B. Angew. Chem. 2013, 125, 9463–9465. doi:10.1002/ange.201304039
Angew. Chem., Int. Ed. 2013, 52, 9293–9295. doi:10.1002/anie.201304039
Return to citation in text: [1] -
Li, G.-Q.; Gao, H.; Keene, C.; Devonas, M.; Ess, D. H.; Kürti, L. J. Am. Chem. Soc. 2013, 135, 7414–7417. doi:10.1021/ja401709k
Return to citation in text: [1] -
Takeda, Y.; Okumura, S.; Minakata, S. Angew. Chem. 2012, 124, 7924–7928. doi:10.1002/ange.201202786
Angew. Chem., Int. Ed. 2012, 51, 7804–7808. doi:10.1002/anie.201202786
Return to citation in text: [1] [2] [3] -
Takeda, Y.; Okumura, S.; Minakata, S. Synthesis 2013, 45, 1029–1033. doi:10.1055/s-0032-1318388
Return to citation in text: [1] [2] [3] -
Okumura, S.; Takeda, Y.; Kiyokawa, K.; Minakata, S. Chem. Commun. 2013, 49, 9266–9268. doi:10.1039/c3cc45469j
Return to citation in text: [1] [2] -
Okumura, S.; Lin, C.-H.; Takeda, Y.; Minakata, S. J. Org. Chem. 2013, 78, 12090–12105. doi:10.1021/jo402120w
Return to citation in text: [1] [2] [3] -
Takeda, Y.; Okazaki, M.; Minakata, S. Chem. Commun. 2014, 50, 10291–10294. doi:10.1039/C4CC04911J
Return to citation in text: [1] [2] [3] [4] -
Benoit, R. L.; Lefebvre, D.; Fréchette, M. Can. J. Chem. 1987, 65, 996–1001. doi:10.1139/v87-170
Return to citation in text: [1] -
Leffek, K. T.; Pruszynski, P.; Thanapaalasingham, K. Can. J. Chem. 1989, 67, 590–595. doi:10.1139/v89-089
Return to citation in text: [1] -
Andon, R. J. L.; Cox, J. D.; Herington, E. F. G. Trans. Faraday Soc. 1954, 50, 918–927. doi:10.1039/tf9545000918
Return to citation in text: [1] -
Burrows, P. E.; Shen, Z.; Bulovic, V.; McCarty, D. M.; Forrest, S. R.; Cronin, J. A.; Thompson, M. E. J. Appl. Phys. 1996, 79, 7991. doi:10.1063/1.362350
Return to citation in text: [1]
| 1. | Meisenheimer, J.; Witte, K. Ber. Dtsch. Chem. Ges. 1903, 36, 4153–4164. doi:10.1002/cber.19030360481 |
| 12. | Corbett, J. F.; Holt, P. F.; Hughes, A. N. J. Chem. Soc. 1961, 1363–1369. doi:10.1039/jr9610001363 |
| 13. | Li, W.; Cai, X.; Hu, Y.; Ye, Y.; Luo, M.; Hu, J. J. Mol. Struct.: THEOCHEM 2005, 732, 21–32. doi:10.1016/j.theochem.2005.06.042 |
| 14. | Caronna, T.; Castiglione, F.; Famulari, A.; Fontana, F.; Malpezzi, L.; Mele, A.; Mendola, D.; Sora, I. N. Molecules 2012, 17, 463–479. doi:10.3390/molecules17010463 |
| 31. | Takeda, Y.; Okazaki, M.; Minakata, S. Chem. Commun. 2014, 50, 10291–10294. doi:10.1039/C4CC04911J |
| 31. | Takeda, Y.; Okazaki, M.; Minakata, S. Chem. Commun. 2014, 50, 10291–10294. doi:10.1039/C4CC04911J |
| 25. |
De, C. K.; Pesciaioli, F.; List, B. Angew. Chem. 2013, 125, 9463–9465. doi:10.1002/ange.201304039
Angew. Chem., Int. Ed. 2013, 52, 9293–9295. doi:10.1002/anie.201304039 |
| 26. | Li, G.-Q.; Gao, H.; Keene, C.; Devonas, M.; Ess, D. H.; Kürti, L. J. Am. Chem. Soc. 2013, 135, 7414–7417. doi:10.1021/ja401709k |
| 2. |
Martin, R. H. Angew. Chem. 1974, 86, 727–738. doi:10.1002/ange.19740862003
Angew. Chem., Int. Ed. Engl. 1974, 13, 649–660. doi:10.1002/anie.197406491 |
| 3. |
Urbano, A. Angew. Chem. 2003, 115, 4116–4119. doi:10.1002/ange.200301667
Angew. Chem., Int. Ed. 2003, 42, 3986–3989. doi:10.1002/anie.200301667 |
| 4. | Collins, S. K.; Vachon, M. P. Org. Biomol. Chem. 2006, 4, 2518–2524. doi:10.1039/b603305a |
| 5. | Dumitrascu, F.; Dumitrescu, D. G.; Aron, I. ARKIVOC 2010, No. i, 1–32. |
| 6. | Shen, Y.; Chen, C.-F. Chem. Rev. 2012, 112, 1463–1535. doi:10.1021/cr200087r |
| 7. | Gingras, M. Chem. Soc. Rev. 2012, 42, 968–1006. doi:10.1039/c2cs35154d |
| 8. | Gingras, M.; Félix, G.; Peresutti, R. Chem. Soc. Rev. 2013, 42, 1007–1050. doi:10.1039/c2cs35111k |
| 9. | Gingras, M. Chem. Soc. Rev. 2012, 42, 1051–1095. doi:10.1039/c2cs35134j |
| 10. | Aillard, P.; Voituriez, A.; Marinetti, A. Dalton Trans. 2014, 43, 15263–15278. doi:10.1039/C4DT01935K |
| 11. | Saleh, N.; Shen, C.; Crassous, J. Chem. Sci. 2014, 5, 3680–3694. doi:10.1039/C4SC01404A |
| 27. |
Takeda, Y.; Okumura, S.; Minakata, S. Angew. Chem. 2012, 124, 7924–7928. doi:10.1002/ange.201202786
Angew. Chem., Int. Ed. 2012, 51, 7804–7808. doi:10.1002/anie.201202786 |
| 28. | Takeda, Y.; Okumura, S.; Minakata, S. Synthesis 2013, 45, 1029–1033. doi:10.1055/s-0032-1318388 |
| 29. | Okumura, S.; Takeda, Y.; Kiyokawa, K.; Minakata, S. Chem. Commun. 2013, 49, 9266–9268. doi:10.1039/c3cc45469j |
| 30. | Okumura, S.; Lin, C.-H.; Takeda, Y.; Minakata, S. J. Org. Chem. 2013, 78, 12090–12105. doi:10.1021/jo402120w |
| 20. | Braithwaite, R. S. W.; Holt, P. F. J. Chem. Soc. 1959, 3025–3031. doi:10.1039/jr9590003025 |
| 21. | Corbett, J. F.; Holt, P. F. J. Chem. Soc. 1960, 3646–3653. doi:10.1039/jr9600003646 |
| 23. | Corbett, J. F.; Holt, P. F. J. Chem. Soc. 1961, 3695–3699. doi:10.1039/jr9610003695 |
| 19. | Bellaart, A. C. Tetrahedron 1965, 21, 3285–3288. doi:10.1016/S0040-4020(01)96949-2 |
| 24. | Caronna, T.; Fontana, F.; Mele, A.; Sora, I. N.; Panzeri, W.; Viganò, L. Synthesis 2008, 413–416. doi:10.1055/s-2008-1032021 |
| 1. | Meisenheimer, J.; Witte, K. Ber. Dtsch. Chem. Ges. 1903, 36, 4153–4164. doi:10.1002/cber.19030360481 |
| 18. | Cumming, W. M.; Ferrier, G. S. J. Chem. Soc. 1924, 125, 1108–1111. doi:10.1039/ct9242501108 |
| 15. | Porter, J. J.; Murray, J. L. J. Am. Chem. Soc. 1965, 87, 1628–1630. doi:10.1021/ja01085a050 |
| 16. | Ellgen, P. C.; McMullin, S. L. Inorg. Chem. 1973, 12, 2004–2007. doi:10.1021/ic50127a010 |
| 17. | Nixon, J. F.; Kooti, M. J. Organomet. Chem. 1978, 149, 71–79. doi:10.1016/S0022-328X(00)90379-6 |
| 22. | Holt, P. F.; Went, C. W. J. Chem. Soc. 1963, 4099–4102. doi:10.1039/jr9630004099 |
| 32. | Benoit, R. L.; Lefebvre, D.; Fréchette, M. Can. J. Chem. 1987, 65, 996–1001. doi:10.1139/v87-170 |
| 27. |
Takeda, Y.; Okumura, S.; Minakata, S. Angew. Chem. 2012, 124, 7924–7928. doi:10.1002/ange.201202786
Angew. Chem., Int. Ed. 2012, 51, 7804–7808. doi:10.1002/anie.201202786 |
| 28. | Takeda, Y.; Okumura, S.; Minakata, S. Synthesis 2013, 45, 1029–1033. doi:10.1055/s-0032-1318388 |
| 30. | Okumura, S.; Lin, C.-H.; Takeda, Y.; Minakata, S. J. Org. Chem. 2013, 78, 12090–12105. doi:10.1021/jo402120w |
| 31. | Takeda, Y.; Okazaki, M.; Minakata, S. Chem. Commun. 2014, 50, 10291–10294. doi:10.1039/C4CC04911J |
| 31. | Takeda, Y.; Okazaki, M.; Minakata, S. Chem. Commun. 2014, 50, 10291–10294. doi:10.1039/C4CC04911J |
| 35. | Burrows, P. E.; Shen, Z.; Bulovic, V.; McCarty, D. M.; Forrest, S. R.; Cronin, J. A.; Thompson, M. E. J. Appl. Phys. 1996, 79, 7991. doi:10.1063/1.362350 |
| 27. |
Takeda, Y.; Okumura, S.; Minakata, S. Angew. Chem. 2012, 124, 7924–7928. doi:10.1002/ange.201202786
Angew. Chem., Int. Ed. 2012, 51, 7804–7808. doi:10.1002/anie.201202786 |
| 28. | Takeda, Y.; Okumura, S.; Minakata, S. Synthesis 2013, 45, 1029–1033. doi:10.1055/s-0032-1318388 |
| 29. | Okumura, S.; Takeda, Y.; Kiyokawa, K.; Minakata, S. Chem. Commun. 2013, 49, 9266–9268. doi:10.1039/c3cc45469j |
| 30. | Okumura, S.; Lin, C.-H.; Takeda, Y.; Minakata, S. J. Org. Chem. 2013, 78, 12090–12105. doi:10.1021/jo402120w |
| 13. | Li, W.; Cai, X.; Hu, Y.; Ye, Y.; Luo, M.; Hu, J. J. Mol. Struct.: THEOCHEM 2005, 732, 21–32. doi:10.1016/j.theochem.2005.06.042 |
| 33. | Leffek, K. T.; Pruszynski, P.; Thanapaalasingham, K. Can. J. Chem. 1989, 67, 590–595. doi:10.1139/v89-089 |
| 34. | Andon, R. J. L.; Cox, J. D.; Herington, E. F. G. Trans. Faraday Soc. 1954, 50, 918–927. doi:10.1039/tf9545000918 |
© 2015 Takeda et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)
