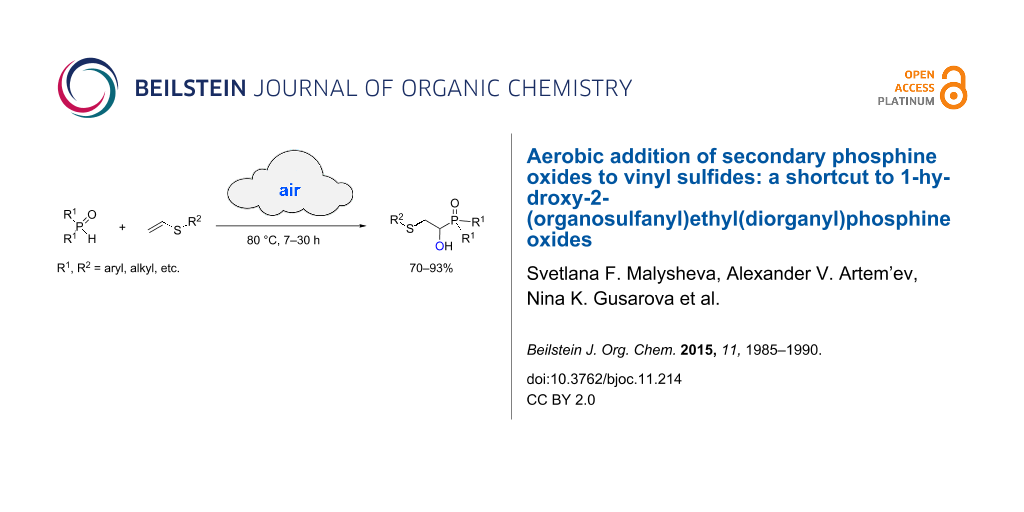
Abstract
Secondary phosphine oxides react with vinyl sulfides (both alkyl- and aryl-substituted sulfides) under aerobic and solvent-free conditions (80 °C, air, 7–30 h) to afford 1-hydroxy-2-(organosulfanyl)ethyl(diorganyl)phosphine oxides in 70–93% yields.

Graphical Abstract
Findings
Tertiary phosphines and phosphine chalcogenides are important organophosphorus compounds that are widely used in industry, organic synthesis, polymer science, medicinal and coordination chemistry [1-4]. Therefore, the synthesis of these compounds has attracted a great interest and numerous synthetic methods have been developed [5-7]. Among them, the addition of P(X)–H (X = none, O, S or Se) to diverse alkenes is one of the most powerful and 100% atom-economic approaches to construct new C–P bonds, that provide straightforward access to tertiary phosphines and their chalcogenides [8-12]. Conventionally, the activation of the P–H bonds in this reaction is achieved by using radical initiators [13-15], Brønsted/Lewis acids [16,17] and bases [18-20] as well as transition metal catalysts [21-23]. Also, examples of the microwave-assisted [24,25] and photoinduced [26] addition are described.
Recently, on example of secondary phosphines [27] as well as secondary phosphine sulfides [28] and selenides [29], it has been disclosed that the addition of P–H species to the C=C bonds readily proceeds in the absence of any catalyst or initiator (Scheme 1). The reactions occur under mild solvent-free conditions (70–80 °C, inert atmosphere, 3–15 h) to chemo- and regioselectively furnish the anti-Markovnikov adducts in excellent yields (up to 99%). The substrate scope includes both EDG- and EWG-substituted alkenes [27-29].
![[1860-5397-11-214-i1]](/bjoc/content/inline/1860-5397-11-214-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Non-catalyzed addition of P–H species to alkenes.
Scheme 1: Non-catalyzed addition of P–H species to alkenes.
In this letter, we report our serendipitous finding that secondary phosphine oxides 1a–f under aerobic conditions (air, 80 °C, 7–18 h) easily add to vinyl sulfides 2a–c to give unknown 1-hydroxy-2-(organosulfanyl)ethyl(diorganyl)phosphine oxides 3a–h in high yields (Table 1). The 10% excess of 2a–c relative to 1a–f is found to be optimal since the equimolar ratio of the reactants leads to incomplete conversion of the secondary phosphine oxides.
Table 1: The substrate scope for the aerobic addition of phosphine oxides 1a–f to vinyl sulfides 2a–c.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-214-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Phosphine oxide | Vinyl sulfide | Time, h |
Phosphine
oxide 3a–h (yield, %)b |
|---|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-214-i5.svg?max-width=637&scale=1.0)
1a |
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-214-i6.svg?max-width=637&scale=1.0)
2a |
16 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-214-i7.svg?max-width=637&scale=1.0)
3a (80%) |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-214-i8.svg?max-width=637&scale=1.0)
1a |
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-214-i9.svg?max-width=637&scale=1.0)
2b |
30 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-214-i10.svg?max-width=637&scale=1.0)
3b (78%) |
| 3 |
Ph2P(O)H
1b |
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-214-i11.svg?max-width=637&scale=1.0)
2c |
7 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-214-i12.svg?max-width=637&scale=1.0)
3c (70%) |
| 4 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-214-i13.svg?max-width=637&scale=1.0)
1a |
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-214-i14.svg?max-width=637&scale=1.0)
2c |
11 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-214-i15.svg?max-width=637&scale=1.0)
3d (91%) |
| 5 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-214-i16.svg?max-width=637&scale=1.0)
1c |
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-214-i17.svg?max-width=637&scale=1.0)
2c |
15 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-214-i18.svg?max-width=637&scale=1.0)
3e (93%) |
| 6 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-214-i19.svg?max-width=637&scale=1.0)
1d |
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-214-i20.svg?max-width=637&scale=1.0)
2c |
15 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-11-214-i21.svg?max-width=637&scale=1.0)
3f (90%) |
| 7 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-11-214-i22.svg?max-width=637&scale=1.0)
1e |
![[Graphic 20]](/bjoc/content/inline/1860-5397-11-214-i23.svg?max-width=637&scale=1.0)
2c |
15 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-11-214-i24.svg?max-width=637&scale=1.0)
3g (82%) |
| 8 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-11-214-i25.svg?max-width=637&scale=1.0)
1f |
![[Graphic 23]](/bjoc/content/inline/1860-5397-11-214-i26.svg?max-width=637&scale=1.0)
2c |
18 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-11-214-i27.svg?max-width=637&scale=1.0)
3h (89%) |
aReaction conditions: secondary phosphine oxide 1a–f (1.0 mmol), vinyl sulfide 2a–c (1.1 mmol) at 80 °C for 7–30 h under air. bIsolated yield based on 1a–f.
Importantly, under these conditions, the expected [30] anti-Markovnikov adducts are not observed in detectable amounts (31P NMR). The main byproducts are phosphinic acids, R2P(O)OH, formed by air oxidation of secondary phosphine oxides 1a–f. As seen from Table 1, the reaction is applicable to both aryl- (1b) and arylalkyl-substituted (1a,c–e) secondary phosphine oxides. The furyl-containing phosphine oxide 1f can also be reacted under these reaction conditions. On the other hand, vinyl sulfides bearing alkyl (2a,b) and aryl (2c) substituents successfully participate in the reaction to provide the corresponding phosphine oxides 3a–h. The latter were isolated as air- and moisture-stable powders (3a–f) or oils (3g,h), soluble in common organic solvents. Their structures have been established by X-ray diffraction (for 3d, Figure 1), NMR (1H, 13C, 31P, 1H,13C-HSQC) and FTIR techniques.
![[1860-5397-11-214-1]](/bjoc/content/figures/1860-5397-11-214-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: ORTEP drawing (30% thermal ellipsoid) of phosphine oxide 3d. A CIF file with the crystallographic data is available as Supporting Information File 2 and is also available on request from the Cambridge Crystallographic Data Centre as deposition 1046604.
Figure 1: ORTEP drawing (30% thermal ellipsoid) of phosphine oxide 3d. A CIF file with the crystallographic d...
The presence of an asymmetric carbon atom in the reaction products leads to non-equivalence of both heminal protons in the SCH2C* fragment and carbon signals in the arylethyl moiety. In the 1H NMR spectra of 3a–h, protons of the PCHCH2S moiety form an ABMX spin system appearing as three multiplets.
Phosphine oxide 3d crystallizes in the centrosymmetric P21/c space group. Within its extended structure, strong intermolecular H-bonding interactions between the O–H hydrogen and P=O oxygen atom of a second molecule {O(1)–H(1)···O(2), 1.80(6) Å; O–H···O angle, 174.9(7)°} leads to the formation of 1D polymeric chains along the b-axis (Figure S1, Supporting Information File 1).
In FTIR spectra of 3a–h, absorption bands of the P=O and O–H bonds appear in the regions of 1100–1150 and 3350–3450 cm−1, respectively.
Interestingly, the reaction disclosed is specific for secondary phosphine oxides. Our experiments have shown that their analogues, secondary phosphine sulfides, under similar conditions provide exclusively the anti-Markovnikov adducts (Scheme 2). On the other hand, vinyl ethers and vinyl selenides (congeners of vinyl sulfides) were found to react with phosphine oxide 1a at 80 °C for about 30 and 20 h, respectively, to deliver difficult-to-separate mixtures of organophosphorus compounds (31P NMR).
![[1860-5397-11-214-i2]](/bjoc/content/inline/1860-5397-11-214-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Addition of secondary phosphine sulfide to vinyl sulfide under aerobic catalyst-free conditions.
Scheme 2: Addition of secondary phosphine sulfide to vinyl sulfide under aerobic catalyst-free conditions.
To gain a primary insight into the reaction mechanism, several experiments were carried out. On example of phosphine oxide 1a and vinyl sulfide 2c, we have shown that the reaction proceeds in the dark with the same efficiency as in the light. Therefore, the photochemical pathway of the reaction is hardly probable. Also, the reaction was established under an argon atmosphere. Under these conditions (argon, 80 °C for 18 h, exemplified by 1a/2c pair) the formation of products 3a–h does not take place and the starting phosphine oxide remained almost intact (31P NMR). This indicates that the reaction requires the presence of oxygen. In the other experiment, when TEMPO, a widely used radical scavenger, was added (10 mol %) into the reaction system 1a/2c, the product 3d was also formed, however, a longer reaction time was required for complete conversion of secondary phosphine oxide 1a as compared to TEMPO-free conditions (15 vs 11 h). Meanwhile, this observation does not completely exclude a radical mechanism since the cross-coupling reactions between TEMPO and radical intermediates can be reversible [31]. In future, we intend to check various radical scavengers (other than TEMPO) in order to better understand the reaction mechanism.
Taking these data into account, the following mechanism is suggested (Scheme 3). The first step is assumed to be the generation of phosphinoyl (A) and hydroperoxyl (HOO•) radicals by the reaction of O2 with phosphine oxide 1. Earlier, the transfer of a hydrogen atom from the P(O)H species to molecular oxygen has been reported for example for Ph2P(O)H [30]. Then, the radical addition of A to vinyl sulfide, proceeding in an anti-Markovnikov manner, takes place. Subsequently, a 1,2-intramolecular transfer of an H atom within the radical adduct B (from PCH2 group to radical center) leads to the formation of R2P(O)-stabilized radical C. The latter recombines with a hydroperoxide radical to afford the metastable hydroperoxide D, thermal decomposition of which give rise to the final product 3.
![[1860-5397-11-214-i3]](/bjoc/content/inline/1860-5397-11-214-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Although quantum chemical computations [MP2/6-311++G(d,p)//B3LYP/6-311++G(d,p)] of the model radicals B and C (with R, R' = Me) reveals that the latter is energetically less preferred than the former, their energy difference is too small (4.38 kcal/mol) to completely prohibit the B→C transformation.
Conclusion
In summary, we have disclosed an aerobic addition of secondary phosphine oxides to vinyl sulfides under solvent- and catalyst-free conditions, which provides an efficient approach to hitherto unknown 1-hydroxy-2-(organosulfanyl)ethyl(diorganyl)phosphine oxides in one step. The synthesized phosphine oxides, bearing hydroxy and sulfide functions, represent prospective building blocks for organic synthesis and interesting ligands for metal complexes. The results obtained contribute to the basic chemistry of both phosphine oxides and vinyl sulfides.
Supporting Information
| Supporting Information File 1: General remarks, experimental procedure and characterization data; crystallographic information for 3d; 1H, 13C & 31P NMR spectra of synthesized compounds. | ||
| Format: PDF | Size: 1.1 MB | Download |
| Supporting Information File 2: CIF file of compound 3d. | ||
| Format: CIF | Size: 477.5 KB | Download |
References
-
Goldwhite, H. Introduction to Phosphorus Chemistry; Cambridge University Press: Cambridge, 1981.
Return to citation in text: [1] -
Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley and Sons: New York, USA, 2000.
Return to citation in text: [1] -
Peruzzini, M.; Gonsalvi, L. Phosphorus Compounds, Advanced Tools in Catalysis and Materials Science, Catalysis by Metal Complexes; Springer: Dordrecht, 2011. doi:10.1007/978-90-481-3817-3
Return to citation in text: [1] -
Kamer, P. C. J.; van Leeuwen, P. W. N. M. Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis; John Wiley and Sons: Chichester, U.K., 2012. doi:10.1002/9781118299715
Return to citation in text: [1] -
Hartley, F. R. Phosphine Oxides, Sulphides, Selenides and Tellurides; The Chemistry of Organophosphorus Compounds, Vol. 2; John Wiley and Sons: Chichester, U.K., 1992. doi:10.1002/0470034424
Return to citation in text: [1] -
Engel, R.; Cohen, J. I. Synthesis of carbon–phosphorus bonds, 2nd ed.; CRC Press: Boca Raton, USA, 2004.
Return to citation in text: [1] -
Kilah, N. L.; Wild, S. B. Product Class 13: Trialkylphosphine Oxides, Sulfides, Selenides, Tellurides, and Imides. In Science of Synthesis; Mathey, F., Ed.; Georg Thieme Verlag: Stuttgart, Deutschland, 2009; Vol. 42, pp 595–633.
Return to citation in text: [1] -
Delacroix, O.; Gaumont, A. C. Curr. Org. Chem. 2005, 9, 1851–1882. doi:10.2174/138527205774913079
Return to citation in text: [1] -
Arbuzova, S. N.; Gusarova, N. K.; Trofimov, B. A. ARKIVOC 2006, No. v, 12–36. doi:10.3998/ark.5550190.0007.503
Return to citation in text: [1] -
Honaker, M. T.; Hovland, J. M.; Salvatore, R. N. Curr. Org. Synth. 2007, 4, 31–45. doi:10.2174/157017907779981561
Return to citation in text: [1] -
Beletskaya, I. P.; Ananikov, V. P.; Khemchyan, L. L. Synthesis of Phosphorus Compounds via Metal-Catalyzed Addition of P–H Bond to Unsaturated Organic Molecules. In Phosphorus Compounds, Advanced Tools in Catalysis and Material Sciences; Peruzzini, M.; Gonsalvi, L., Eds.; Springer: Netherlands, 2011; Vol. 37, pp 213–264. doi:10.1007/978-90-481-3817-3_8
Return to citation in text: [1] -
Koshti, V.; Gaikwad, S.; Chikkali, S. H. Coord. Chem. Rev. 2014, 265, 52–73. doi:10.1016/j.ccr.2014.01.006
Return to citation in text: [1] -
Deprèle, S.; Montchamp, J.-L. J. Org. Chem. 2001, 66, 6745–6755. doi:10.1021/jo015876i
Return to citation in text: [1] -
Leca, D.; Fensterbank, L.; Lacôte, E.; Malacria, M. Chem. Soc. Rev. 2005, 34, 858–865. doi:10.1039/B500511F
Return to citation in text: [1] -
Oparina, L. A.; Gusarova, N. K.; Vysotskaya, O. V.; Artem’ev, A. V.; Kolyvanov, N. A.; Trofimov, B. A. Synthesis 2014, 46, 653–659. doi:10.1055/s-0033-1340497
Return to citation in text: [1] -
Dombek, B. D. J. Org. Chem. 1978, 43, 3408–3409. doi:10.1021/jo00411a038
Return to citation in text: [1] -
Routaboul, L.; Toulgoat, F.; Gatignol, J.; Lohier, J.-F.; Norah, B.; Delacroix, O.; Alayrac, C.; Taillefer, M.; Gaumont, A.-C. Chem. – Eur. J. 2013, 19, 8760–8764. doi:10.1002/chem.201301417
Return to citation in text: [1] -
Bunlaksananusorn, T.; Knochel, P. Tetrahedron Lett. 2002, 43, 5817–5819. doi:10.1016/S0040-4039(02)01177-2
Return to citation in text: [1] -
Fu, X.; Jiang, Z.; Tan, C.-H. Chem. Commun. 2007, 5058–5060. doi:10.1039/B713151H
Return to citation in text: [1] -
Perrier, A.; Comte, V.; Moïse, C.; Richard, P.; Le Gendre, P. Eur. J. Org. Chem. 2010, 1562–1568. doi:10.1002/ejoc.200901407
Return to citation in text: [1] -
Xu, Q.; Han, L.-B. J. Organomet. Chem. 2011, 696, 130–140. doi:10.1016/j.jorganchem.2010.08.043
Return to citation in text: [1] -
Xu, Q.; Zhou, Y.-B.; Zhao, C.-Q.; Yin, S.-F.; Han, L.-B. Mini-Rev. Med. Chem. 2013, 13, 824–835. doi:10.2174/1389557511313060005
Return to citation in text: [1] -
Tanaka, M. Top. Organomet. Chem. 2013, 43, 167–201. doi:10.1007/3418_2011_20
Return to citation in text: [1] -
Stockland, R. A., Jr.; Taylor, R. I.; Thompson, L. E.; Patel, P. B. Org. Lett. 2005, 7, 851–853. doi:10.1021/ol0474047
Return to citation in text: [1] -
Lenker, H. K.; Richard, M. E.; Reese, K. P.; Carter, A. F.; Zawisky, J. D.; Winter, E. F.; Bergeron, T. W.; Guydon, K. S.; Stockland, R. A., Jr. J. Org. Chem. 2012, 77, 1378–1385. doi:10.1021/jo202183u
Return to citation in text: [1] -
Kawaguchi, S.; Nomoto, A.; Sonoda, M.; Ogawa, A. Tetrahedron Lett. 2009, 50, 624–626. doi:10.1016/j.tetlet.2008.11.079
Return to citation in text: [1] -
Alonso, F.; Moglie, Y.; Radivoy, G.; Yus, M. Green Chem. 2012, 14, 2699–2702. doi:10.1039/C2GC35898K
Return to citation in text: [1] [2] -
Malysheva, S. F.; Gusarova, N. K.; Artem’ev, A. V.; Belogorlova, N. A.; Albanov, A. I.; Borodina, T. N.; Smirnov, V. I.; Trofimov, B. A. Eur. J. Org. Chem. 2014, 2516–2521. doi:10.1002/ejoc.201301786
Return to citation in text: [1] [2] -
Malysheva, S. F.; Gusarova, N. K.; Artem’ev, A. V.; Belogorlova, N. A.; Albanov, A. I.; Borodina, T. N.; Smirnov, V. I.; Trofimov, B. A. Synthesis 2014, 46, 2656–2662. doi:10.1055/s-0034-1378357
Return to citation in text: [1] [2] -
Hirai, T.; Han, L.-B. Org. Lett. 2007, 9, 53–55. doi:10.1021/ol062505l
Return to citation in text: [1] [2] -
Skene, W. G.; Belt, S. T.; Connolly, T. J.; Hahn, P.; Scaiano, J. C. Macromolecules 1998, 31, 9103–9105. doi:10.1021/ma9812229
Return to citation in text: [1]
| 1. | Goldwhite, H. Introduction to Phosphorus Chemistry; Cambridge University Press: Cambridge, 1981. |
| 2. | Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley and Sons: New York, USA, 2000. |
| 3. | Peruzzini, M.; Gonsalvi, L. Phosphorus Compounds, Advanced Tools in Catalysis and Materials Science, Catalysis by Metal Complexes; Springer: Dordrecht, 2011. doi:10.1007/978-90-481-3817-3 |
| 4. | Kamer, P. C. J.; van Leeuwen, P. W. N. M. Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis; John Wiley and Sons: Chichester, U.K., 2012. doi:10.1002/9781118299715 |
| 16. | Dombek, B. D. J. Org. Chem. 1978, 43, 3408–3409. doi:10.1021/jo00411a038 |
| 17. | Routaboul, L.; Toulgoat, F.; Gatignol, J.; Lohier, J.-F.; Norah, B.; Delacroix, O.; Alayrac, C.; Taillefer, M.; Gaumont, A.-C. Chem. – Eur. J. 2013, 19, 8760–8764. doi:10.1002/chem.201301417 |
| 31. | Skene, W. G.; Belt, S. T.; Connolly, T. J.; Hahn, P.; Scaiano, J. C. Macromolecules 1998, 31, 9103–9105. doi:10.1021/ma9812229 |
| 13. | Deprèle, S.; Montchamp, J.-L. J. Org. Chem. 2001, 66, 6745–6755. doi:10.1021/jo015876i |
| 14. | Leca, D.; Fensterbank, L.; Lacôte, E.; Malacria, M. Chem. Soc. Rev. 2005, 34, 858–865. doi:10.1039/B500511F |
| 15. | Oparina, L. A.; Gusarova, N. K.; Vysotskaya, O. V.; Artem’ev, A. V.; Kolyvanov, N. A.; Trofimov, B. A. Synthesis 2014, 46, 653–659. doi:10.1055/s-0033-1340497 |
| 8. | Delacroix, O.; Gaumont, A. C. Curr. Org. Chem. 2005, 9, 1851–1882. doi:10.2174/138527205774913079 |
| 9. | Arbuzova, S. N.; Gusarova, N. K.; Trofimov, B. A. ARKIVOC 2006, No. v, 12–36. doi:10.3998/ark.5550190.0007.503 |
| 10. | Honaker, M. T.; Hovland, J. M.; Salvatore, R. N. Curr. Org. Synth. 2007, 4, 31–45. doi:10.2174/157017907779981561 |
| 11. | Beletskaya, I. P.; Ananikov, V. P.; Khemchyan, L. L. Synthesis of Phosphorus Compounds via Metal-Catalyzed Addition of P–H Bond to Unsaturated Organic Molecules. In Phosphorus Compounds, Advanced Tools in Catalysis and Material Sciences; Peruzzini, M.; Gonsalvi, L., Eds.; Springer: Netherlands, 2011; Vol. 37, pp 213–264. doi:10.1007/978-90-481-3817-3_8 |
| 12. | Koshti, V.; Gaikwad, S.; Chikkali, S. H. Coord. Chem. Rev. 2014, 265, 52–73. doi:10.1016/j.ccr.2014.01.006 |
| 27. | Alonso, F.; Moglie, Y.; Radivoy, G.; Yus, M. Green Chem. 2012, 14, 2699–2702. doi:10.1039/C2GC35898K |
| 28. | Malysheva, S. F.; Gusarova, N. K.; Artem’ev, A. V.; Belogorlova, N. A.; Albanov, A. I.; Borodina, T. N.; Smirnov, V. I.; Trofimov, B. A. Eur. J. Org. Chem. 2014, 2516–2521. doi:10.1002/ejoc.201301786 |
| 29. | Malysheva, S. F.; Gusarova, N. K.; Artem’ev, A. V.; Belogorlova, N. A.; Albanov, A. I.; Borodina, T. N.; Smirnov, V. I.; Trofimov, B. A. Synthesis 2014, 46, 2656–2662. doi:10.1055/s-0034-1378357 |
| 5. | Hartley, F. R. Phosphine Oxides, Sulphides, Selenides and Tellurides; The Chemistry of Organophosphorus Compounds, Vol. 2; John Wiley and Sons: Chichester, U.K., 1992. doi:10.1002/0470034424 |
| 6. | Engel, R.; Cohen, J. I. Synthesis of carbon–phosphorus bonds, 2nd ed.; CRC Press: Boca Raton, USA, 2004. |
| 7. | Kilah, N. L.; Wild, S. B. Product Class 13: Trialkylphosphine Oxides, Sulfides, Selenides, Tellurides, and Imides. In Science of Synthesis; Mathey, F., Ed.; Georg Thieme Verlag: Stuttgart, Deutschland, 2009; Vol. 42, pp 595–633. |
| 26. | Kawaguchi, S.; Nomoto, A.; Sonoda, M.; Ogawa, A. Tetrahedron Lett. 2009, 50, 624–626. doi:10.1016/j.tetlet.2008.11.079 |
| 28. | Malysheva, S. F.; Gusarova, N. K.; Artem’ev, A. V.; Belogorlova, N. A.; Albanov, A. I.; Borodina, T. N.; Smirnov, V. I.; Trofimov, B. A. Eur. J. Org. Chem. 2014, 2516–2521. doi:10.1002/ejoc.201301786 |
| 24. | Stockland, R. A., Jr.; Taylor, R. I.; Thompson, L. E.; Patel, P. B. Org. Lett. 2005, 7, 851–853. doi:10.1021/ol0474047 |
| 25. | Lenker, H. K.; Richard, M. E.; Reese, K. P.; Carter, A. F.; Zawisky, J. D.; Winter, E. F.; Bergeron, T. W.; Guydon, K. S.; Stockland, R. A., Jr. J. Org. Chem. 2012, 77, 1378–1385. doi:10.1021/jo202183u |
| 29. | Malysheva, S. F.; Gusarova, N. K.; Artem’ev, A. V.; Belogorlova, N. A.; Albanov, A. I.; Borodina, T. N.; Smirnov, V. I.; Trofimov, B. A. Synthesis 2014, 46, 2656–2662. doi:10.1055/s-0034-1378357 |
| 21. | Xu, Q.; Han, L.-B. J. Organomet. Chem. 2011, 696, 130–140. doi:10.1016/j.jorganchem.2010.08.043 |
| 22. | Xu, Q.; Zhou, Y.-B.; Zhao, C.-Q.; Yin, S.-F.; Han, L.-B. Mini-Rev. Med. Chem. 2013, 13, 824–835. doi:10.2174/1389557511313060005 |
| 23. | Tanaka, M. Top. Organomet. Chem. 2013, 43, 167–201. doi:10.1007/3418_2011_20 |
| 18. | Bunlaksananusorn, T.; Knochel, P. Tetrahedron Lett. 2002, 43, 5817–5819. doi:10.1016/S0040-4039(02)01177-2 |
| 19. | Fu, X.; Jiang, Z.; Tan, C.-H. Chem. Commun. 2007, 5058–5060. doi:10.1039/B713151H |
| 20. | Perrier, A.; Comte, V.; Moïse, C.; Richard, P.; Le Gendre, P. Eur. J. Org. Chem. 2010, 1562–1568. doi:10.1002/ejoc.200901407 |
| 27. | Alonso, F.; Moglie, Y.; Radivoy, G.; Yus, M. Green Chem. 2012, 14, 2699–2702. doi:10.1039/C2GC35898K |
© 2015 Malysheva et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)