Abstract
A detailed DFT study of the mechanism of metathesis of fluoroethene, 1-fluoroethene, 1,1-difluoroethene, cis- and trans-1,2-difluoroethene, tetrafluoroethene and chlorotrifluoroethene catalysed with the Hoveyda–Grubbs 2nd generation catalyst was performed. It revealed that a successful metathesis of hydrofluoroethenes is hampered by a high preference for a non-productive catalytic cycle proceeding through a ruthenacyclobutane intermediate bearing fluorines in positions 2 and 4. Moreover, the calculations showed that the cross-metathesis of perfluoro- or perhaloalkenes should be a feasible process and that the metathesis is not very sensitive to stereochemical issues.

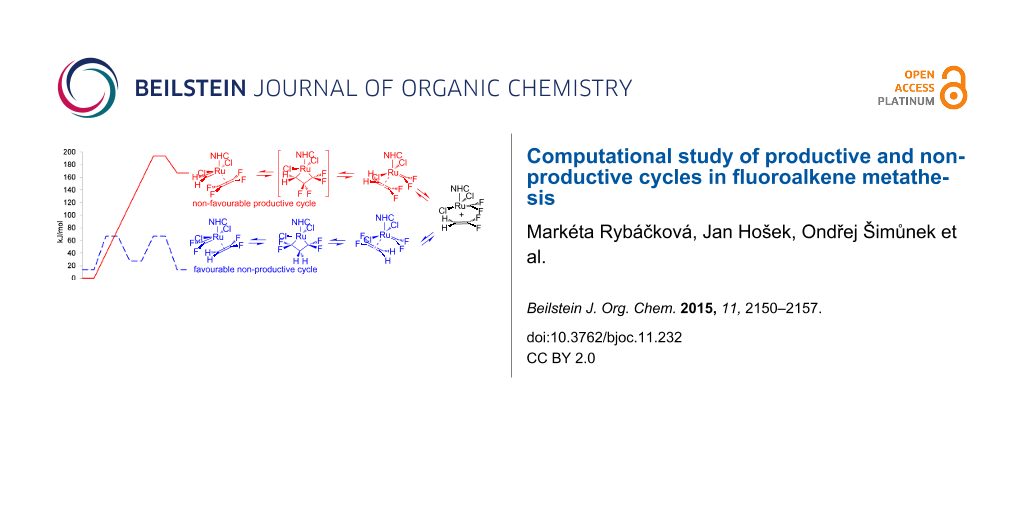
Graphical Abstract
Introduction
Over the course of the last 20 years, alkene metathesis catalysed with homogeneous transitition metal-based precatalysts evolved into a valuable tool for organic synthetic chemists mainly due to its variability and high compatibility with functional groups. It hence became the subject of multiple books [1-3] and reviews [4-8] discussing its synthetic applications, catalysts, mechanism, regio- and stereoselectivity.
Computational chemistry proved to be extremely valuable in the study of reaction mechanisms. In particular, the use of time-efficient DFT methods for the theoretical study of alkene metathesis has been extensively reviewed [9-11] and computational results have been found to agree well with recent experimental mechanistic studies based on easily initiating ruthenium precatalysts [12,13]. A theoretical approach has been also employed in attempts to gain a better insight into the complex structure of intertwined productive and non-productive catalytic cycles of alkene metathesis [14]. In contrast to the older computations, new publications also include the initiation steps starting from metathesis precatalysts as Grubbs or Hoveyda–Grubbs 2nd generation precatalysts [15-22].
In contrast to common alkenes, the metathesis of fluoroalkenes has attracted far less attention [23]. Fluorinated modifications have mostly concentrated on the side chain of the vinyl group [24-27] or applications of 2-fluoroalkenes [28-30]. As an exception, the reaction of the Grubbs 2nd generation catalyst with 1,1-difluoroethene gave an isolable difluoromethylene-containing ruthenium complex with very poor catalytic activity [31] and the analogous reaction of 1-fluoroalkene formed a fluoromethylene-containing complex with low catalytic activity [32,33]. Up to now, the only metathesis which included tetrafluoroethene and its analogues has been reported in a patent [34], describing the disproportionation of perfluoroalkenes and alkenes to hydrofluoroalkenes. Moreover, just recently a successful cross metathesis of perfluoroalkenes with vinyl ethers has been published [35].
The reported computations dealing with the metathesis of fluoroalkenes are also extremely scarce. Thus, the mechanism of the cross metathesis of norbornene with selected fluoro- and chloroalkenes has been studied and the higher stability of a ruthenium intermediate containing a difluoromethylene ligand has been emphasized [36,37].
A complete mechanism of alkene metathesis including initiation, productive and non-productive cycles represents a highly complex system [38], the understanding of which for fluoroalkenes is negligible. We hence report herein the results of a computational study dealing with the metathesis of most fluoroethenes with the emphasis on subsequent catalytic cycles, catalysed with the Hoveyda–Grubbs 2nd generation precatalyst (HG2).
Results and Discussion
In contrast to textbook pictures describing alkene metathesis as a single catalytic cycle, even a simplified system of homometathesis of 1-alkene represents a complex system, in any step of which problems can arise due to a high energetical barrier or unfavourable equilibrium. Moreover, the preference for non-productive cycles can hamper the formation of the desired product even in the absence of kinetic and thermodynamic issues (Scheme 1).
![[1860-5397-11-232-i1]](/bjoc/content/inline/1860-5397-11-232-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Initiation, productive and non-productive cycles in alkene homometathesis.
Scheme 1: Initiation, productive and non-productive cycles in alkene homometathesis.
At the beginning, the starting precomplex HG2 reacts in the initiation phase with alkene forming ruthenacyclobutane I, which releases 2-isopropoxystyrene and the first active catalytic species, alkylideneruthenium complex AC. Depending on the regioselectivity of the coordination of the second alkene molecule, the AC complex can form ruthenacyclobutane PA in the first part of productive catalytic cycle, or symmetrical ruthenacyclobutane NA in the first part of the non-productive catalytic cycle. While the first species PA reacts further to the product and methyleneruthenium complex MC, the non-productive ruthenacyclobutane NA can only return back to complex AC. Intermediary complex MC can again enter either the second part of the productive catalytic cycle closing the ring into the ruthenacyclobutane PB, or it can react in the non-productive cycle forming symmetrical ruthenacyclobutane NB. Finishing the productive cycle, the ruthenacyclobutane PB releases ethene and starts the next catalytic cycle, while the non-productive complex NB can again only return back to complex MC. It should be noted that the main aim of the development of the family of Hoveyda–Grubbs catalysts was the recycling of the catalyst. This implies a successful release-return mechanism, in which after a successful metathesis the active complexes AC or MC react with 2-isopropoxystyrene restoring the starting precatalyst HG2, a controversial issue, which some authors support [8,39] and the others contest [40]. Recent experiments have shown that the complex MC can be successfully transformed into the parent precatalyst HG2 [41].
In our computational study, we first addressed the initiation phase of possible metathesis of a highly unsymmetrical alkene, 1,1-difluoroethene, and compared its behaviour with the already reported initiation of ethene [18,19]. Among the three possible mechanisms, interchange, dissociative and associative, the first emerged as the most energetically favourable. In contrast to the symmetrical molecule of ethene, two orientations of 1,1-difluoroethene are possible, which we arbitrarily assigned as syn for the coordination of difluoromethylene to ruthenium forming 2,2-difluororuthenacyclobutane intermediate s2I and anti for the coordinaton of methylene to ruthenium forming 3,3-difluororuthenacyclobutane intermediate a2I. The computations started from an alkene weakly coordinated to the NHC ligand without any coordination to ruthenium (structures 1a and 2a), and continued with the first mechanistic step, the coordination of alkene to ruthenium with partial decoordination of alkoxybenzylidene oxygen (structures 1c and 2c). For ethene (1c) and the anti-coordinated 1,1-difluoroethene (a2c), shallow minima were observed in the Gibbs free energy profile, while for the syn structure (s2c), the minimum obtained by the calculation of electronic energy changed just to inflexion when converted to free Gibbs energy at 25 °C (see Figure 1, dotted red line). At this stage only minimal relative energy differences were observed for the structures 1c and 2c. However, in the next step forming metallacyclobutane the transition state energy was by ca. 20 kJ/mol higher for the syn-coordinated 1,1-difluoroethene (s2d) and by another ca. 20 kJ/mol higher for the anti-coordinationed 1,1-difluoroethene (a2d) compared to ethene (1d), already energetically preferring the syn-coordination. The picture changed dramatically on the formation of metallacyclobutane (1I or 2I), where 2,2-difluororuthenacyclobutane intermediate s2I was by 20 kJ/mol more stable than ruthenacyclobutane s1 and by another 40 kJ/mol more stable than 3,3-difluororuthenacyclobutane a2I. These differences further rose for the last step of initiation, the formation of methyleneruthenium 1f or a2f or difluoromethyleneruthenium s2f, where the relative stability of the latter was by 100 kJ/mol higher compared to the anti coordination. These results are in agreement with the previous computations of a norbornene derivative with fluoroalkenes [36,37], but are suprisingly contradictory to the experimental observations of the reaction of the Grubbs 2nd generation catalyst with 1,1-difluoroethene, where at room temperature the formation of both methylene- and difluoromethyleneruthenium complexes was observed [31].
![[1860-5397-11-232-1]](/bjoc/content/figures/1860-5397-11-232-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Initiation phase of the reaction of HG2 with ethene (1) and 1,1-difluoroethene (2).
Figure 1: Initiation phase of the reaction of HG2 with ethene (1) and 1,1-difluoroethene (2).
With the additional aim to obtain the information about stereoselectivity, we next analogously computed the initation phase of the reaction of precatalyst HG2 with 1-fluoroethene, where apart of the syn- and anti-approach, also cis- or trans- orientations of the alkene relatively to the alkoxyphenyl ring in the intermediary metallacyclobutane are possible. In the first step of the reaction, intermediary complex 3c with partial bonding of alkene and the alkoxy group of the alkoxybenzylidene ligand to ruthenium was detected with the exception of a syn–cis arranged alkene, for which just a weak inflexion was observed in analogy to [42]. No large differences in the energies of transition states 3d preceeding the formation of metallacyclobutane were found, but in analogy to the initiation of 1,1-difluoroethene, 2-fluorometallacyclobutanes sc3I and st3I with coordination of fluoromethylene to ruthenium were again more stable then the corresponding 3-fluorometallacyclobutanes ac3I and at3I. The difference is further augmented in the transition state 3e and in the final stage of the initiation, the formation of fluoromethyleneruthenium complex s3f or methyleneruthenium complex a3f. On the other hand, this difference in the energies reaches ca. 50 kJ/mol, about one half of the syn–anti difference for difluoromethylated complex 2AC. We also observed significant differences in the energies for cis- and trans-ruthenacyclobutanes c3I and t3I and complexes c3f and t3f, where the trans-structures were more stable by 25 to 60 kJ/mol, probably due to the repulsion of fluorine with the alkoxybenzylidene oxygen (Figure 2).
![[1860-5397-11-232-2]](/bjoc/content/figures/1860-5397-11-232-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Initiation phase of the reaction of HG2 with ethene (1) and 1-fluoroethene (3).
Figure 2: Initiation phase of the reaction of HG2 with ethene (1) and 1-fluoroethene (3).
We continued our study by the computation of the first step A of the catalytic cycle for 1,1-difluoroethene (2), which started with the coordination of the starting active catalytic form 2AC with 1,1-difluoroethene (2). The non-productive cycle started with the syn-coordination of 1,1-difluoroethene (2) to 1,1-difluoromethyleneruthenium complex 2AC, leading to symmetrical metallacyclobutane 2NA with activation energy around 65 kJ/mol and through the same transition state back to complex 2AC and 1,1-difluoroethene (2). However, for the productive anti-coordination of 1,1-difluoroethene (2), no stable metallacyclobutane structure 2PC was found (a detailed study detected only inflection on the potential energy surface), probably due to a steep rise in the energy leading through the transition state s2i to a highly unstable complex of methyleneruthenium with tetrafluoroethene. The comparison of the productive and non-productive cycle shows that the transition state energies differ by more than 120 kJ/mol, making thus the first part A of the productive cycle highly improbable and probably resulting in stopping the productive metathesis of vinylidene fluoride at all, because all active catalytic species AC move forth and back in the non-productive cycle (Figure 3).
![[1860-5397-11-232-3]](/bjoc/content/figures/1860-5397-11-232-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: First part A of the catalytic cycle of homometathesis of 1,1-difluoroethene (2).
Figure 3: First part A of the catalytic cycle of homometathesis of 1,1-difluoroethene (2).
The difference in the stability of the corresponding complexes a2j and s2j can be explained partially by the π-donation of difluoromethylene carbene in analogy to [37], but also by the electron donation of the π-bond of the 1,1-difluoroethene (2) molecule with a high negative charge on the CH2 group. This results in lowering of the positive charge on ruthenium and shortening of the CH2–Ru distance (262 pm) in the s2j structure compared to repulsive CF2–Ru interaction of tetrafluoroethene in the a2j structure with longer CF2–Ru distance (323 pm, Figure 4).
![[1860-5397-11-232-4]](/bjoc/content/figures/1860-5397-11-232-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Computed structures of complexes s2j and a2j.
Figure 4: Computed structures of complexes s2j and a2j.
We continued the calculations by the study of the second part B of the catalytic cycle of the metathesis of 1,1-difluoroethene (2), which starts by the decoordination of tetrafluoroethene from complex a2j and coordination of another molecule of 1,1-difluoroethene (2) to methyleneruthenium complex MC forming complex 2k. While both syn- and anti-coordinations of starting complex 2k and subsequent transition states 2l have nearly equal energies, the subsequent non-symmetrical productive metallacyclobutane 2PB is significantly more stable by ca. 60 kJ/mol than symmetrical non-productive metallacyclobutane 2NB. The difference in the energies was again more augmented for the subsequent transition states 2m and finally with the alkene coordinated to the alkylidene ruthenium, where the productive complex s2n, a complex of ethylene with difluororuthenium, is by ca. 80 kJ/mol more stable than the non-productive complex a2n (Figure 5). Thus, in the second part B of the catalytic cycle the non-productive cycle has no negative influence on the 1,1-difluoroethene (2) metathesis.
![[1860-5397-11-232-5]](/bjoc/content/figures/1860-5397-11-232-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Second part B of the catalytic cycle of homometathesis of 1,1-difluoroethene (2).
Figure 5: Second part B of the catalytic cycle of homometathesis of 1,1-difluoroethene (2).
To obtain the information about the stereoselectivity in the active catalytic cycle, we again decided to study the productive and non-productive cycles in the homometathesis of 1-fluoroethene (3). In the first step A of the catalytic cycle, a similar pattern, although less emphasized, could be observed for syn- and anti-coordination of the starting 1-fluoroethene (3) as in the case of 1,1-difluoroethene (2). Thus, the relative energies of the complexes of 1-fluoroethene (3) with fluoromethyleneruthenium 3g, as well as the subsequent transition states 3h, differ minimally regardless of the regio- and stereoselectivity, while the intermediary symmetrical metallacyclobutane 3NA of the non-productive cycle with the syn-coordination of 1-fluoroethene (3) shows a significantly higher stability compared to the productive anti-intermediate 3PA. These differences again increased for the transition states 3i and final complexes 3j, preferring strongly the non-productive cycle and probably significantly slowing the possible productive metathesis. The calculations also show only low stereoselectivity with the transition states of the non-productive cycle preferring trans-configuration of both fluorine atoms by ca. 10 kJ/mol, while for the productive cycle the cis-configuration is preferred (Figure 6).
![[1860-5397-11-232-6]](/bjoc/content/figures/1860-5397-11-232-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: First part A of the catalytic cycle of homometathesis of 1-fluoroethene (3).
Figure 6: First part A of the catalytic cycle of homometathesis of 1-fluoroethene (3).
For the homometathesis of 1-fluoroethene (3), we finally studied the second part B of the catalytic cycle. In analogy to the homometathesis of 1,1-difluoroethene (2), the productive cycle is energetically more favourable, indicating that in the part B the non-productive cycle does not block the catalytic cycle (Figure 7).
![[1860-5397-11-232-7]](/bjoc/content/figures/1860-5397-11-232-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Second part B of the catalytic cycle of homometathesis of 1-fluoroethene (3).
Figure 7: Second part B of the catalytic cycle of homometathesis of 1-fluoroethene (3).
The obtained results imply that the key problem in the metathesis of non-symmetrically substituted fluoroalkenes is probably not the high stability of the fluorinated methyleneruthenium complex, but the consumption of most of the active catalytic form by the non-productive cycle proceeding through symmetrical metallacyclobutane substituted with fluorines in positions 2 and 4 of the ring.
To further confirm this hypothesis, we decided to study the active catalytic cycle of the metathesis of two perhaloethenes, tetrafluoroethene (4) and chlorotrifluoroethene (5), starting from the active catalytic form 2AC. Due to symmetry, both parts A and B of the catalytic cycle for tetrafluoroethene (4) are identical and non-productive with surprisingly low transition state energy (Figure 8).
![[1860-5397-11-232-8]](/bjoc/content/figures/1860-5397-11-232-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Non-productive catalytic cycle of homometathesis of tetrafluoroethene (4).
Figure 8: Non-productive catalytic cycle of homometathesis of tetrafluoroethene (4).
For the homometathesis of chlortrifluoroethene (5), the situation again becomes more complex with two parts A and B of the catalytic cycle and both productive and non-productive cycles participating. The first part A is in analogy to the homometathesis of tetrafluoroethene (4) characteristic by the low energy of the transition state, with the preference for the non-productive cycle of ca. 20 kJ/mol, i.e., much less pronounced than in the case of 1-fluoroethene (3) complexes (Figure 6 and Figure 9).
![[1860-5397-11-232-9]](/bjoc/content/figures/1860-5397-11-232-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: First part A of the catalytic cycle of homometathesis of chlorotrifluoroethene (5).
Figure 9: First part A of the catalytic cycle of homometathesis of chlorotrifluoroethene (5).
With the second part B of the catalytic cycle starting from a non-symmetrical chlorofluoromethyleneruthenium complex, the stereochemistry of coordination in complex 5e became an issue with cis- and trans-ruthenacyclobutanes possible. In analogy to the homometathesis of tetrafluoroethene (4), the relative transition state energies were quite low with small preference for the productive cycle without any stereochemical priority (Figure 10).
![[1860-5397-11-232-10]](/bjoc/content/figures/1860-5397-11-232-10.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: Second part B of the catalytic cycle of homometathesis of chlorotrifluoroethene (5).
Figure 10: Second part B of the catalytic cycle of homometathesis of chlorotrifluoroethene (5).
Conclusion
Our computational study, which included the metathesis of both partially and fully fluorinated alkenes, showed that the formation of stable intermediary fluoro- or difluoromethyleneruthenium does not block the subsequent metathetic cycles. For partially fluorinated alkenes as 1,1-difluoroethene (2) or 1-fluoroethene (3), poor preparative results of metathesis can be caused by the overwhelming participation of the non-productive metathetic cycle proceeding through ruthenacyclobutanes substituted with fluorine atoms in positions 2 and 4. On the other hand, the results of computations of the catalytic cycles of both tetrafluoroethene (4) and chlorotrifluoroethene (5) indicate that their metathesis should proceed without any significant problems providing no reactive alkylideneruthenium complexes, e.g., methyleneruthenium, participates in the active catalytic cycles. Our results are in full agreement with the recently described surprisingly successful metathesis of perhaloalkenes with vinyl ethers [35], but contradicts the patent [34] which describes the successful synthesis of partially fluorinated alkenes from perfluorinated and non-fluorinated alkenes.
Computational Details
DFT calculations were performed using the Gaussian 09W program suite [43] using the resolution-of-identity approach [44], M06L pure functional [45] in analogy to [18], def2-SV(P) basis set [46] and universal def2 auxilliary basis set [47]. Vibrational frequencies were calculated for all structures to characterize them as minima or transition states. These computations gave their free Gibbs energies at 25 °C, which were used for the PES description in Figures 1–3 and Figures 5–10. Starting geometries were obtained by a careful series of preoptimization of structures 1a–3a (Hoveyda–Grubbs 2nd generation precatalyst HG2 with weakly coordinated alkene) and 1l–3l (metallacyclobutane) with the most stable conformation of the isopropoxy group differing from the crystal structure of the parent precatalyst HG2. For all computed structures, the corresponding pdb files are listed in Supporting Information File 1 together with a table containing the total electronic and free Gibbs energies in hartrees, and the total and relative electronic and free Gibbs energies in kJ/mol.
Supporting Information
| Supporting Information File 1: Table containing total electronic and free Gibbs energies in hartrees, total and relative electronic and free Gibbs energies in kJ/mol for all computed structures, as well as their coordinates in the pdb format. | ||
| Format: PDF | Size: 810.6 KB | Download |
Acknowledgements
We thank the Grant Agency of the Czech Republic (grant No. 207/10/1533) for financial support. Computational resources were provided by the MetaCentrum under the program LM2010005 and the CERIT-SC under the program Centre CERIT Scientific Cloud, part of the Operational Program Research and Development for Innovations, Reg. no. CZ.1.05/3.2.00/08.0144.
References
-
Grubbs, R. H.; Wenzel, A. G.; O’Leary, D. J.; Khosravi, E., Eds. Handbook of Organic Chemistry, 2nd ed.; Wiley & Sons: New York, NY, U.S.A., 2015.
Return to citation in text: [1] -
Grela, K., Ed. Olefin Metathesis: Theory and Practice; Wiley & Sons: New York, NY, U.S.A., 2014.
Return to citation in text: [1] -
Cossy, J.; Arseniyadis, S.; Meyer, C.; Grubbs, R. H., Eds. Metathesis in Natural Product Synthesis: Strategies, Substrates and Catalysts; Wiley & Sons: New York, NY, U.S.A., 2010.
Return to citation in text: [1] -
Koh, M. J.; Khan, R. K. M.; Torker, S.; Yu, M.; Mikus, M. S.; Hoveyda, A. H. Nature 2015, 517, 181–186. doi:10.1038/nature14061
Return to citation in text: [1] -
Nelson, D. J.; Manzini, S.; Urbina-Blanco, C. A.; Nolan, S. P. Chem. Commun. 2014, 50, 10355–10375. doi:10.1039/C4CC02515F
Return to citation in text: [1] -
Fürstner, A. Science 2013, 341, No. 6152. doi:10.1126/science.1229713
Return to citation in text: [1] -
Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746–1787. doi:10.1021/cr9002424
Return to citation in text: [1] -
Bieniek, M.; Michrowska, A.; Usanov, D. L.; Grela, K. Chem. – Eur. J. 2008, 14, 806–818. doi:10.1002/chem.200701340
Return to citation in text: [1] [2] -
Credendino, R.; Poater, A.; Ragone, F.; Cavallo, L. Catal. Sci. Technol. 2011, 1, 1287–1297. doi:10.1039/c1cy00052g
Return to citation in text: [1] -
Falivene, L.; Poater, A.; Cavallo, L. Tuning and Quantifying Steric and Electronic Effects of N-Heterocyclic Carbenes. In N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis; Nolan, S. P., Ed.; Wiley-VCH: New York, NY, U.S.A., 2014; pp 25–38.
Return to citation in text: [1] -
Poater, A.; Falivene, L.; Cavallo, L. Theoretical Attempts: “In Silico Olefin Metathesis”—How Can Computers Help in the Understanding of Metathesis Mechanisms and in Catalysts Development?. In Olefin Metathesis: Theory and Practice; Grela, K., Ed.; Wiley: New York, NY, U.S.A., 2014; pp 483–494.
Return to citation in text: [1] -
van der Eide, E. F.; Piers, W. E. Nat. Chem. 2010, 2, 571–576. doi:10.1038/nchem.653
Return to citation in text: [1] -
Leitao, E. M.; van der Eide, E. F.; Romero, P. E.; Piers, W. E.; McDonald, R. J. Am. Chem. Soc. 2010, 132, 2784–2794. doi:10.1021/ja910112m
Return to citation in text: [1] -
Vummaleti, S. V. C.; Cavallo, L.; Poater, A. Theor. Chem. Acc. 2015, 134, 22–27. doi:10.1007/s00214-015-1622-x
Return to citation in text: [1] -
Engle, K. M.; Lu, G.; Luo, S. X.; Henling, L. M.; Michael, K.; Takase, M. K.; Liu, P.; Houk, K. N.; Grubbs, R. H. J. Am. Chem. Soc. 2015, 137, 5782–5792. doi:10.1021/jacs.5b01144
Return to citation in text: [1] -
Manzini, S.; Urbina-Blanco, C. A.; Nelson, D. J.; Poater, A.; Lebl, T.; Meiries, S.; Slawin, A. M. Z.; Falivene, L.; Cavallo, L.; Nolan, S. P. J. Organomet. Chem. 2015, 780, 43–48. doi:10.1016/j.jorganchem.2014.12.040
Return to citation in text: [1] -
Poater, A.; Pump, E.; Vummaleti, S. V. C.; Cavallo, L. J. Chem. Theory Comput. 2014, 10, 4442–4448. doi:10.1021/ct5003863
Return to citation in text: [1] -
Ashworth, I. W.; Hillier, I. H.; Nelson, D. J.; Percy, J. M.; Vincent, M. A. ACS Catal. 2013, 3, 1929–1939. doi:10.1021/cs400164w
Return to citation in text: [1] [2] [3] -
Kvíčala, J.; Schindler, M.; Kelbichová, V.; Babuněk, M.; Rybáčková, M.; Kvíčalová, M.; Cvačka, J.; Březinová, A. J. Fluorine Chem. 2013, 153, 12–25. doi:10.1016/j.jfluchem.2013.06.001
Return to citation in text: [1] [2] -
Urbina-Blanco, C. A.; Poater, A.; Lebl, T.; Manzini, S.; Slawin, A. M. Z.; Cavallo, L.; Nolan, S. P. J. Am. Chem. Soc. 2013, 135, 7073–7079. doi:10.1021/ja402700p
Return to citation in text: [1] -
Poater, A.; Falivene, L.; Urbina-Blanco, C. A.; Manzini, S.; Nolan, S. P.; Cavallo, L. Dalton Trans. 2013, 42, 7433–7439. doi:10.1039/c3dt32980a
Return to citation in text: [1] -
Nuñez-Zarur, F.; Solans-Monfort, X.; Rodrıguez-Santiago, L.; Sodupe, M. Organometallics 2012, 31, 4203–4215. doi:10.1021/om300150d
Return to citation in text: [1] -
Fustero, S.; Simón-Fuentes, A.; Barrio, P.; Haufe, G. Chem. Rev. 2015, 115, 871–930. doi:10.1021/cr500182a
Return to citation in text: [1] -
Chatterjee, A. K.; Morgan, J. P.; Scholl, M.; Grubbs, R. H. J. Am. Chem. Soc. 2000, 122, 3783–3784. doi:10.1021/ja9939744
Return to citation in text: [1] -
Imhof, S.; Randl, S.; Blechert, S. Chem. Commun. 2001, 1692–1693. doi:10.1039/B105031C
Return to citation in text: [1] -
Cuñat, A. C.; Flores, S.; Oliver, J.; Fustero, S. Eur. J. Org. Chem. 2011, 7317–7323. doi:10.1002/ejoc.201101091
Return to citation in text: [1] -
Eignerová, B.; Slavíková, B.; Buděšínský, M.; Dračínský, M.; Klepetářová, B.; Št’astná, E.; Kotora, M. J. Med. Chem. 2009, 52, 5753–5757. doi:10.1021/jm900495f
Return to citation in text: [1] -
Marhold, M.; Stillig, C.; Fröhlich, R.; Haufe, G. Eur. J. Org. Chem. 2014, 5777–5785. doi:10.1002/ejoc.201402535
Return to citation in text: [1] -
Salim, S. S.; Bellingham, R. K.; Satcharoen, V.; Brown, R. C. D. Org. Lett. 2003, 5, 3403–3406. doi:10.1021/ol035065w
Return to citation in text: [1] -
Guérin, D.; Gaumont, A. C.; Dez, I.; Mauduit, M.; Couve-Bonnaire, S.; Pannecoucke, X. ACS Catal. 2014, 4, 2374–2378. doi:10.1021/cs500559p
Return to citation in text: [1] -
Trnka, T. M.; Day, M. W.; Grubbs, R. H. Angew. Chem., Int. Ed. 2001, 40, 3441–3444. doi:10.1002/1521-3773(20010917)40:18<3441::AID-ANIE3441>3.0.CO;2-7
Return to citation in text: [1] [2] -
Macnaughtan, M. L.; Johnson, M. J. A.; Kampf, J. W. Organometallics 2007, 26, 780–782. doi:10.1021/om061082o
Return to citation in text: [1] -
Macnaughtan, M. L.; Gary, J. B.; Gerlach, D. L.; Johnson, M. J. A.; Kampf, J. W. Organometallics 2009, 28, 2880–2887. doi:10.1021/om800463n
Return to citation in text: [1] -
Elsheikh, M. Y.; Bonnet, P. Process for the Manufacture of Hydrofluoroolefins via Metathesis. WO Patent 2009/003085, Dec 31, 2008.
Return to citation in text: [1] [2] -
Takahira, Y.; Morizawa, Y. J. Am. Chem. Soc. 2015, 137, 7031–7034. doi:10.1021/jacs.5b03342
Return to citation in text: [1] [2] -
Fomine, S.; Vargas Ortega, J.; Tlenkopatchev, M. A. J. Organomet. Chem. 2006, 691, 3343–3348. doi:10.1016/j.jorganchem.2006.04.021
Return to citation in text: [1] [2] -
Fomine, S.; Tlenkopatchev, M. A. Appl. Catal., A 2009, 355, 148–155. doi:10.1016/j.apcata.2008.12.011
Return to citation in text: [1] [2] [3] -
Stewart, I. C.; Keitz, B. K.; Kuhn, K. M.; Thomas, R. M.; Grubbs, R. H. J. Am. Chem. Soc. 2010, 132, 8534–8535. doi:10.1021/ja1029045
Return to citation in text: [1] -
Kingsbury, J. S.; Hoveyda, A. H. J. Am. Chem. Soc. 2005, 127, 4510–4517. doi:10.1021/ja042668+
Return to citation in text: [1] -
Vorfalt, T.; Wannowius, K. J.; Thiel, V.; Plenio, H. Chem. – Eur. J. 2010, 16, 12312–12315. doi:10.1002/chem.201001832
Return to citation in text: [1] -
Bates, J. M.; Lummis, J. A. M.; Bailey, G. A.; Fogg, D. E. ACS Catal. 2014, 4, 2387–2394. doi:10.1021/cs500539m
Return to citation in text: [1] -
Straub, B. F. Angew. Chem., Int. Ed. 2005, 44, 5974–5978. doi:10.1002/anie.200501114
Return to citation in text: [1] -
Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, U.S.A., 2009.
Return to citation in text: [1] -
Eichkorn, K.; Treutler, O.; Öhm, H.; Häser, M.; Ahlrichs, R. Chem. Phys. Lett. 1995, 240, 283–290. doi:10.1016/0009-2614(95)00621-A
Return to citation in text: [1] -
Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x
Return to citation in text: [1] -
Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/B508541A
Return to citation in text: [1] -
Weigend, F. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. doi:10.1039/B515623H
Return to citation in text: [1]
| 45. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 18. | Ashworth, I. W.; Hillier, I. H.; Nelson, D. J.; Percy, J. M.; Vincent, M. A. ACS Catal. 2013, 3, 1929–1939. doi:10.1021/cs400164w |
| 46. | Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/B508541A |
| 1. | Grubbs, R. H.; Wenzel, A. G.; O’Leary, D. J.; Khosravi, E., Eds. Handbook of Organic Chemistry, 2nd ed.; Wiley & Sons: New York, NY, U.S.A., 2015. |
| 2. | Grela, K., Ed. Olefin Metathesis: Theory and Practice; Wiley & Sons: New York, NY, U.S.A., 2014. |
| 3. | Cossy, J.; Arseniyadis, S.; Meyer, C.; Grubbs, R. H., Eds. Metathesis in Natural Product Synthesis: Strategies, Substrates and Catalysts; Wiley & Sons: New York, NY, U.S.A., 2010. |
| 14. | Vummaleti, S. V. C.; Cavallo, L.; Poater, A. Theor. Chem. Acc. 2015, 134, 22–27. doi:10.1007/s00214-015-1622-x |
| 38. | Stewart, I. C.; Keitz, B. K.; Kuhn, K. M.; Thomas, R. M.; Grubbs, R. H. J. Am. Chem. Soc. 2010, 132, 8534–8535. doi:10.1021/ja1029045 |
| 12. | van der Eide, E. F.; Piers, W. E. Nat. Chem. 2010, 2, 571–576. doi:10.1038/nchem.653 |
| 13. | Leitao, E. M.; van der Eide, E. F.; Romero, P. E.; Piers, W. E.; McDonald, R. J. Am. Chem. Soc. 2010, 132, 2784–2794. doi:10.1021/ja910112m |
| 8. | Bieniek, M.; Michrowska, A.; Usanov, D. L.; Grela, K. Chem. – Eur. J. 2008, 14, 806–818. doi:10.1002/chem.200701340 |
| 39. | Kingsbury, J. S.; Hoveyda, A. H. J. Am. Chem. Soc. 2005, 127, 4510–4517. doi:10.1021/ja042668+ |
| 9. | Credendino, R.; Poater, A.; Ragone, F.; Cavallo, L. Catal. Sci. Technol. 2011, 1, 1287–1297. doi:10.1039/c1cy00052g |
| 10. | Falivene, L.; Poater, A.; Cavallo, L. Tuning and Quantifying Steric and Electronic Effects of N-Heterocyclic Carbenes. In N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis; Nolan, S. P., Ed.; Wiley-VCH: New York, NY, U.S.A., 2014; pp 25–38. |
| 11. | Poater, A.; Falivene, L.; Cavallo, L. Theoretical Attempts: “In Silico Olefin Metathesis”—How Can Computers Help in the Understanding of Metathesis Mechanisms and in Catalysts Development?. In Olefin Metathesis: Theory and Practice; Grela, K., Ed.; Wiley: New York, NY, U.S.A., 2014; pp 483–494. |
| 35. | Takahira, Y.; Morizawa, Y. J. Am. Chem. Soc. 2015, 137, 7031–7034. doi:10.1021/jacs.5b03342 |
| 4. | Koh, M. J.; Khan, R. K. M.; Torker, S.; Yu, M.; Mikus, M. S.; Hoveyda, A. H. Nature 2015, 517, 181–186. doi:10.1038/nature14061 |
| 5. | Nelson, D. J.; Manzini, S.; Urbina-Blanco, C. A.; Nolan, S. P. Chem. Commun. 2014, 50, 10355–10375. doi:10.1039/C4CC02515F |
| 6. | Fürstner, A. Science 2013, 341, No. 6152. doi:10.1126/science.1229713 |
| 7. | Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746–1787. doi:10.1021/cr9002424 |
| 8. | Bieniek, M.; Michrowska, A.; Usanov, D. L.; Grela, K. Chem. – Eur. J. 2008, 14, 806–818. doi:10.1002/chem.200701340 |
| 36. | Fomine, S.; Vargas Ortega, J.; Tlenkopatchev, M. A. J. Organomet. Chem. 2006, 691, 3343–3348. doi:10.1016/j.jorganchem.2006.04.021 |
| 37. | Fomine, S.; Tlenkopatchev, M. A. Appl. Catal., A 2009, 355, 148–155. doi:10.1016/j.apcata.2008.12.011 |
| 28. | Marhold, M.; Stillig, C.; Fröhlich, R.; Haufe, G. Eur. J. Org. Chem. 2014, 5777–5785. doi:10.1002/ejoc.201402535 |
| 29. | Salim, S. S.; Bellingham, R. K.; Satcharoen, V.; Brown, R. C. D. Org. Lett. 2003, 5, 3403–3406. doi:10.1021/ol035065w |
| 30. | Guérin, D.; Gaumont, A. C.; Dez, I.; Mauduit, M.; Couve-Bonnaire, S.; Pannecoucke, X. ACS Catal. 2014, 4, 2374–2378. doi:10.1021/cs500559p |
| 32. | Macnaughtan, M. L.; Johnson, M. J. A.; Kampf, J. W. Organometallics 2007, 26, 780–782. doi:10.1021/om061082o |
| 33. | Macnaughtan, M. L.; Gary, J. B.; Gerlach, D. L.; Johnson, M. J. A.; Kampf, J. W. Organometallics 2009, 28, 2880–2887. doi:10.1021/om800463n |
| 24. | Chatterjee, A. K.; Morgan, J. P.; Scholl, M.; Grubbs, R. H. J. Am. Chem. Soc. 2000, 122, 3783–3784. doi:10.1021/ja9939744 |
| 25. | Imhof, S.; Randl, S.; Blechert, S. Chem. Commun. 2001, 1692–1693. doi:10.1039/B105031C |
| 26. | Cuñat, A. C.; Flores, S.; Oliver, J.; Fustero, S. Eur. J. Org. Chem. 2011, 7317–7323. doi:10.1002/ejoc.201101091 |
| 27. | Eignerová, B.; Slavíková, B.; Buděšínský, M.; Dračínský, M.; Klepetářová, B.; Št’astná, E.; Kotora, M. J. Med. Chem. 2009, 52, 5753–5757. doi:10.1021/jm900495f |
| 34. | Elsheikh, M. Y.; Bonnet, P. Process for the Manufacture of Hydrofluoroolefins via Metathesis. WO Patent 2009/003085, Dec 31, 2008. |
| 23. | Fustero, S.; Simón-Fuentes, A.; Barrio, P.; Haufe, G. Chem. Rev. 2015, 115, 871–930. doi:10.1021/cr500182a |
| 15. | Engle, K. M.; Lu, G.; Luo, S. X.; Henling, L. M.; Michael, K.; Takase, M. K.; Liu, P.; Houk, K. N.; Grubbs, R. H. J. Am. Chem. Soc. 2015, 137, 5782–5792. doi:10.1021/jacs.5b01144 |
| 16. | Manzini, S.; Urbina-Blanco, C. A.; Nelson, D. J.; Poater, A.; Lebl, T.; Meiries, S.; Slawin, A. M. Z.; Falivene, L.; Cavallo, L.; Nolan, S. P. J. Organomet. Chem. 2015, 780, 43–48. doi:10.1016/j.jorganchem.2014.12.040 |
| 17. | Poater, A.; Pump, E.; Vummaleti, S. V. C.; Cavallo, L. J. Chem. Theory Comput. 2014, 10, 4442–4448. doi:10.1021/ct5003863 |
| 18. | Ashworth, I. W.; Hillier, I. H.; Nelson, D. J.; Percy, J. M.; Vincent, M. A. ACS Catal. 2013, 3, 1929–1939. doi:10.1021/cs400164w |
| 19. | Kvíčala, J.; Schindler, M.; Kelbichová, V.; Babuněk, M.; Rybáčková, M.; Kvíčalová, M.; Cvačka, J.; Březinová, A. J. Fluorine Chem. 2013, 153, 12–25. doi:10.1016/j.jfluchem.2013.06.001 |
| 20. | Urbina-Blanco, C. A.; Poater, A.; Lebl, T.; Manzini, S.; Slawin, A. M. Z.; Cavallo, L.; Nolan, S. P. J. Am. Chem. Soc. 2013, 135, 7073–7079. doi:10.1021/ja402700p |
| 21. | Poater, A.; Falivene, L.; Urbina-Blanco, C. A.; Manzini, S.; Nolan, S. P.; Cavallo, L. Dalton Trans. 2013, 42, 7433–7439. doi:10.1039/c3dt32980a |
| 22. | Nuñez-Zarur, F.; Solans-Monfort, X.; Rodrıguez-Santiago, L.; Sodupe, M. Organometallics 2012, 31, 4203–4215. doi:10.1021/om300150d |
| 31. | Trnka, T. M.; Day, M. W.; Grubbs, R. H. Angew. Chem., Int. Ed. 2001, 40, 3441–3444. doi:10.1002/1521-3773(20010917)40:18<3441::AID-ANIE3441>3.0.CO;2-7 |
| 18. | Ashworth, I. W.; Hillier, I. H.; Nelson, D. J.; Percy, J. M.; Vincent, M. A. ACS Catal. 2013, 3, 1929–1939. doi:10.1021/cs400164w |
| 19. | Kvíčala, J.; Schindler, M.; Kelbichová, V.; Babuněk, M.; Rybáčková, M.; Kvíčalová, M.; Cvačka, J.; Březinová, A. J. Fluorine Chem. 2013, 153, 12–25. doi:10.1016/j.jfluchem.2013.06.001 |
| 40. | Vorfalt, T.; Wannowius, K. J.; Thiel, V.; Plenio, H. Chem. – Eur. J. 2010, 16, 12312–12315. doi:10.1002/chem.201001832 |
| 41. | Bates, J. M.; Lummis, J. A. M.; Bailey, G. A.; Fogg, D. E. ACS Catal. 2014, 4, 2387–2394. doi:10.1021/cs500539m |
| 44. | Eichkorn, K.; Treutler, O.; Öhm, H.; Häser, M.; Ahlrichs, R. Chem. Phys. Lett. 1995, 240, 283–290. doi:10.1016/0009-2614(95)00621-A |
| 35. | Takahira, Y.; Morizawa, Y. J. Am. Chem. Soc. 2015, 137, 7031–7034. doi:10.1021/jacs.5b03342 |
| 34. | Elsheikh, M. Y.; Bonnet, P. Process for the Manufacture of Hydrofluoroolefins via Metathesis. WO Patent 2009/003085, Dec 31, 2008. |
| 42. | Straub, B. F. Angew. Chem., Int. Ed. 2005, 44, 5974–5978. doi:10.1002/anie.200501114 |
| 37. | Fomine, S.; Tlenkopatchev, M. A. Appl. Catal., A 2009, 355, 148–155. doi:10.1016/j.apcata.2008.12.011 |
| 36. | Fomine, S.; Vargas Ortega, J.; Tlenkopatchev, M. A. J. Organomet. Chem. 2006, 691, 3343–3348. doi:10.1016/j.jorganchem.2006.04.021 |
| 37. | Fomine, S.; Tlenkopatchev, M. A. Appl. Catal., A 2009, 355, 148–155. doi:10.1016/j.apcata.2008.12.011 |
| 31. | Trnka, T. M.; Day, M. W.; Grubbs, R. H. Angew. Chem., Int. Ed. 2001, 40, 3441–3444. doi:10.1002/1521-3773(20010917)40:18<3441::AID-ANIE3441>3.0.CO;2-7 |
© 2015 Rybáčková et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)