Abstract
Three 2´-deoxynucleosides containing semi-flexible spin labels, namely TA, UA and UC, were prepared and incorporated into deoxyoligonucleotides using the phosphoramidite method. All three nucleosides contain 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) connected to the exocyclic amino group; TA directly and UA as well as UC through a urea linkage. TA and UC showed a minor destabilization of a DNA duplex, as registered by a small decrease in the melting temperature, while UA destabilized the duplex by more than 10 °C. Circular dichroism (CD) measurements indicated that all three labels were accommodated in B-DNA duplex. The mobility of the spin label TA varied with different base-pairing partners in duplex DNA, with the TA•T pair being the least mobile. Furthermore, TA showed decreased mobility under acidic conditions for the sequences TA•C and TA•G, to the extent that the EPR spectrum of the latter became nearly superimposable to that of TA•T. The reduced mobility of the TA•C and TA•G mismatches at pH 5 is consistent with the formation of TAH+•C and TAH+•G, in which protonation of N1 of A allows the formation of an additional hydrogen bond to N3 of C and N7 of G, respectively, with G in a syn-conformation. The urea-based spin labels UA and UC were more mobile than TA, but still showed a minor variation in their EPR spectra when paired with A, G, C or T in a DNA duplex. UA and UC had similar mobility order for the different base pairs, with the lowest mobility when paired with C and the highest when paired with T.

Graphical Abstract
Introduction
The knowledge about structures and conformational dynamics of nucleic acids, as well as other biomolecules, is essential to understand their biological functions, including interactions with other molecules. The exact atom-to-atom structural information can be obtained by X-ray crystallography [1-6] and nuclear magnetic resonance (NMR) spectroscopy [7-12]. Electron paramagnetic resonance (EPR) and fluorescence spectroscopies are nowadays routinely used to study global structures and conformational changes under biologically relevant conditions through the determination of intermediate to long-range distances [13-30]. EPR spectroscopy can also give information about the relative orientation of two rigid spin labels [31-35]. Small angle X-ray scattering is also frequently used to study global structures of large molecules and molecular assemblies [36-39].
Local structural perturbations in nucleic acids can be studied with some of the aforementioned techniques. For example, NMR has been used to study hydrogen-bonding interactions [11,40-42], non-native base-pairing properties of nucleobases [43-46] and their dynamics [42,47,48]. Fluorescence spectroscopy, using environmentally sensitive fluorescent nucleosides has been used for detection of local structural perturbations [49-57], including the investigation of single-base mismatches [51,54,56,58,59], abasic sites [60] as well as nick sites in duplex DNA [61], and ligand-induced folding of riboswitches [62,63].
Continuous wave (CW) EPR spectroscopy can be used for the determination of structure-dependent dynamics based on the line-shape analysis of the EPR spectra [64-73]. The spin labels for such experiments are attached to the nucleotide via a flexible or a semi-flexible tether, which allows some motion of the spin label independent of the nucleic acid. Spin-label motion is affected by the local surroundings of the label, which in turn is reflected in the shape of the EPR spectra. We have previously used the spin label TC [69], containing a 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) moiety conjugated to the exocyclic amino group of C (Figure 1A), to identify the base to which it is paired with in duplex DNA [69]. In other words, this label could not only distinguish between pairing with guanine and a mismatch but was also able to pinpoint the base-pairing partner. Furthermore, TC revealed a flanking-base dependent variation in the EPR spectra, showing that minor structural variations in the local surroundings of a nucleic acid groove can be detected with spin labels by EPR spectroscopy.
![[1860-5397-11-24-1]](/bjoc/content/figures/1860-5397-11-24-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Base pairing of TC with G (A), TA with T (B), UC with G (C) and UA with T (D).
Figure 1: Base pairing of TC with G (A), TA with T (B), UC with G (C) and UA with T (D).
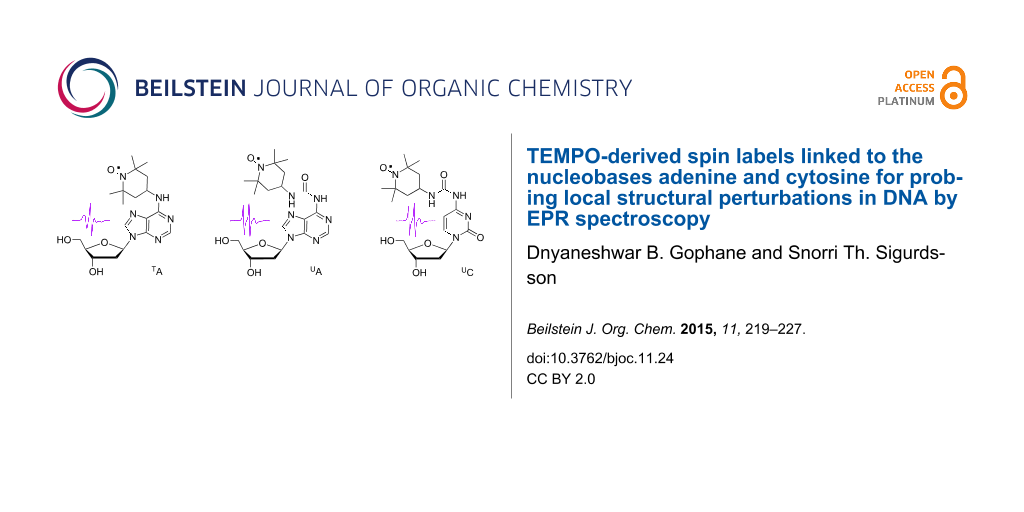
Here, we describe the use of an analogous derivative of A, namely TA, in which a TEMPO moiety is conjugated to the exocyclic amino group of 2´-deoxyadenosine (Figure 1B), to study local perturbations for a purine base in DNA. We show that TA can indeed be used to differentiate between different base-pairing partners, albeit not as clearly as TC. Lower pH causes noticeable changes in the EPR spectra for TA, in particular for the TA•G and TA•C mismatches, presumably because of protonation of the base. We have also prepared urea-linked spin-labeled derivatives of 2´-deoxycytidine (UC) and 2´-deoxyadenosine (UA) and incorporated them into DNA duplexes (Figure 1C and D). These labels provide additional possibilities for hydrogen-bonding through the urea linkage but are also more flexible than TA or TC. In spite of the increased flexibility of UC and UA, inspection of the line-shape of their CW EPR spectra reveals subtle differences between the four base-pairing partners A, T, G and C when placed in a DNA duplex.
Results and Discussion
Synthesis of TA, UA, UC and their corresponding phosphoramidites
The spin-labeled nucleoside TA and its corresponding phosphoramidite were prepared by a previously reported procedure [74] with minor modifications. The synthesis started with the reaction between 3′,5′-diacetyl-2′-deoxyinosine (1) and 2,4,6-triisopropylbenzenesulfonyl chloride to obtain compound 2 (Scheme 1). Coupling of 2 with 4-amino-TEMPO gave 3 in good yields and deprotection of the acetyl groups gave TA, which was tritylated and phosphitylated to yield compounds 4 and 5, respectively.
![[1860-5397-11-24-i1]](/bjoc/content/inline/1860-5397-11-24-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of nucleoside TA and its corresponding phosphoramidite 5. TPS = 2,4,6-triisopropylbenzenesulfonyl.
Scheme 1: Synthesis of nucleoside TA and its corresponding phosphoramidite 5. TPS = 2,4,6-triisopropylbenzene...
For the synthesis of UA, 3′,5′-TBDMS-protected 2′-deoxyadenosine [75] (6) was reacted with 4-isocyanato-TEMPO (7) [64,76], which gave compound 8 in low yields (Scheme 2). Cleavage of the TBDMS groups using TBAF gave spin-labeled nucleoside UA, which was tritylated to give compound 9 and phosphitylated to give phosphoramidite 10.
![[1860-5397-11-24-i2]](/bjoc/content/inline/1860-5397-11-24-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of nucleoside UA and its corresponding phosphoramidite 10.
Scheme 2: Synthesis of nucleoside UA and its corresponding phosphoramidite 10.
The synthesis of UC, the urea-cytidine analogue, started by reaction of 3′,5′-TBDMS-protected 2′-deoxycytidine (11) with 4-isocyanato-TEMPO (7) [64,76] to give 12 in good yields (Scheme 3). Removal of the TBDMS groups using TBAF gave spin-labeled nucleoside UC, which was tritylated to give compound 13 and subsequent phosphitylation yielded phosphoramidite 14.
![[1860-5397-11-24-i3]](/bjoc/content/inline/1860-5397-11-24-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of nucleoside UC and its corresponding phosphoramidite 14.
Scheme 3: Synthesis of nucleoside UC and its corresponding phosphoramidite 14.
Synthesis and characterization of TA-, UA- and UC-containing oligonucleotides
The phosphoramidites of TA (5), UA (10) and UC (14) were used to incorporate the spin-labeled nucleosides into DNA oligonucleotides using solid-phase synthesis [77]. The low stability of the nitroxide functional group in the TEMPO moiety towards acids lead to almost ca. 50% reduction of the nitroxide during oligonucleotide synthesis, which utilized dichloroacetic acid for the removal of the trityl groups. However, in spite of low yields, the spin-labeled oligonucleotides were readily separated from those containing the reduced spin label by denaturing polyacrylamide gel electrophoresis. The incorporation of TA, UA and UC into DNA was confirmed by MALDI–TOF mass spectrometry (Table S1, Supporting Information File 1). Circular dichroism measurements showed that the incorporation of TA, UA and UC does not alter the B-DNA conformation of DNA duplexes containing these modifications (Figure S1, Supporting Information File 1).
The thermal denaturation studies indicated only a minor destabilization of DNA the duplexes when TA was paired with T (Tm was only 2.7 °C lower), compared to the natural nucleoside A (Table S2, Supporting Information File 1). The least stable base pairing was observed with the TA•A mismatch, which showed destabilization by 8.2 °C, compared to an A•A mismatch. The duplex stability order as a function of base-paring with TA was TA•T > TA•G > TA•C > TA•A, consistent with the order of stability previously reported for A [48]. Replacing C with UC opposite G in a DNA duplex resulted in a 3.9 °C decrease in the melting temperature (Tm) and a stability order of UC•G > UC•A > UC•T > UC•C (Table S3, Supporting Information File 1). In contrast to TA and UC, UA had a large destabilizing effect on DNA duplexes (ca. 11 °C, Table S4, Supporting Information File 1). Interestingly, the melting temperatures of the DNA duplexes containing the base pairs UA•T, UA•G and UA•A were nearly identical, whereas the UA•C mismatch showed a further decrease in melting temperature of ca. 9 °C.
EPR analysis of TA-, UC-and UA-labeled DNA duplexes
To investigate the mobility of TA in duplex DNA, we analyzed the EPR spectra of the four 14-mer DNA duplexes 5′-d(GACCTCGTAATCGTG)•5′-d(CACGATYCGAGGTC), where Y is either T, C, G or A (Figure 2). The EPR spectrum of the TA•T pair was broadest, which is consistent with TA forming a Watson–Crick base pair with T, and thereby restricting the rotation around the C6–N6 bond through hydrogen bonding to N6, and consequently slowing the motion of the label (Figure 1B). On the other hand, the spectrum of TA•A was the narrowest and thereby indicating the highest mobility, while the EPR spectrum of TA•G was slightly broader than TA•A. Although base pairing schemes can be drawn for TA•G and TA•A that involve hydrogen bonding to N6 of TA (Figure S2C and S2D, Supporting Information File 1) [44,48,78-80], the increased mobility could be the result of the label being pushed further into the major groove of the DNA duplex, due to the space-demanding purine–purine pairs [81], where the spin-label mobility would be less affected by the local surroundings.
![[1860-5397-11-24-2]](/bjoc/content/figures/1860-5397-11-24-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: EPR spectra of 14-mer DNA duplexes 5′-d(GACCTCGTAATCGTG)•5′-d(CACGATYCGAGGTC), (10 mM phosphate, 100 mM NaCl, 0.1 mM Na2EDTA, pH 7.0 (A) and 5.0 (B)) at 10 °C, where TA is paired with either Y = T, C, G or A.
Figure 2: EPR spectra of 14-mer DNA duplexes 5′-d(GACCTCGTAATCGTG)•5′-d(CACGATYCGAGGTC), (10 mM phosphate, 10...
The mobility of TA•C at pH 7, as judged by its EPR spectrum (Figure 2A), was between that of TA•T and TA•G. Previous NMR studies of the TA•C mismatch at pH 8.5 [47] and 8.9 [82] showed one hydrogen bond, located between N6 of TA and N3 of cytidine (Figure S2B, Supporting Information File 1). If TA is protonated on N1 to form TAH+, it could form a wobble-pair with C [47,83] (Figure 3A), which would be expected to decrease the mobility of the spin label. The apparent pKa of the proton on N1 has been determined by NMR studies to be 7.2 [47], which means that more than half of the TA•C pairs would be protonated at pH 7. To explore if further reduction in mobility (due to conversion of the TA•C pair to the TAH+•C pair) would be detected by EPR, its spectrum was also recorded at pH 5 (Figure 2B). Indeed, comparison of the EPR spectra of TA•C at pH 5 and pH 7 clearly shows further broadening at the lower pH, almost to that of the TA•T pair, and is consistent with the formation of the TAH+•C pair.
![[1860-5397-11-24-3]](/bjoc/content/figures/1860-5397-11-24-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Possible base pairing of TAH+ with C (A) and G (B) at pH 5.
Figure 3: Possible base pairing of TAH+ with C (A) and G (B) at pH 5.
The TA•T and TA•A pairs had similar EPR spectra at pH 7 and 5 (Figure 2B). However, significant broadening was observed in the EPR spectrum of the TA•G mismatch at pH 5. In fact, the spectrum of TA•G is nearly superimposable to that of the TA•T pair, indicating reduction in mobility due to formation of another hydrogen bond at pH 5. Studies by NMR and X-ray crystallography have shown that TAH+•G pairs form when TA•G mismatches are incubated at pH below 5.6, in which the G has flipped into a syn conformation and the O6 and N7 form Hoogsteen hydrogen bonds (Figure 3B) [43,44,78]. Thus, the formation of the TAH+•G pair can be readily detected by EPR spectroscopy.
Nucleosides UA and UC, the new and readily prepared spin labels that are described here, contain a stable urea linkage that provides additional hydrogen-bonding possibilities. In particular, pairing of UA and UC to C, which has been shown by NMR to form a hydrogen bond between its N3 and the proton on N6 of A [83] as well as N4 of C [69,84], should enable the oxygen in the urea moiety of both UA and UC to pair with a N4 proton of C (Figure 4A and B). Such hydrogen bonding should have an effect on the spin label mobility and be manifested in the line width of the EPR spectra.
![[1860-5397-11-24-4]](/bjoc/content/figures/1860-5397-11-24-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Possible base pairing of UA and UC with C.
Figure 4: Possible base pairing of UA and UC with C.
Therefore, we recorded the EPR spectra of duplexes 5′-d(GACCTCGXATCGTG)•5′-d(CACGATYCGAGGTC), where X is either UA or UC, and Y is either C, A, G or T (Figure 5). Indeed, the lowest mobility, i.e., the widest spectra was observed for both UA•C and UC•C, providing circumstantial evidence for hydrogen bonding of C to the urea linkage. Interestingly, the same mobility order was observed for both UA and UC: X•C < X•A ≤ X•G < X•T. Pairing with T resulted in a high mobility for both UA and UC, whereas pairing with either A or G, caused mobility intermediate between that of C and T. As expected, the EPR spectra of the duplexes containing the urea-linked spin labels (UA and UC) were narrower than for the N6-TEMPO-dA (TA) labeled duplexes. The extended urea linker not only contains more rotatable single bonds but also projects the spin label further out of the major groove, where it is less constrained sterically by the DNA.
![[1860-5397-11-24-5]](/bjoc/content/figures/1860-5397-11-24-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: EPR spectra of 14-mer DNA duplex 5′-d(GACCTCGUAATCGTG)•5′-d(CACGATYCGAGGTC) (A), and 5′-d(GACCTCGUCATCGTG)•5′-d(CACGATYCGAGGTC) (B), (10 mM phosphate, 100 mM NaCl, 0.1 mM Na2EDTA, pH 7.0) at 10 °C, where UA or UC is paired with either Y = T, C, G or A .
Figure 5: EPR spectra of 14-mer DNA duplex 5′-d(GACCTCGUAATCGTG)•5′-d(CACGATYCGAGGTC) (A), and 5′-d(GACCTCGUC...
Conclusion
Three spin-labeled deoxynucleosides, TA, UA and UC, were prepared and incorporated into oligonucleotides by the phosphoramidite method. While UA resulted in a major decrease in the melting temperature of a DNA duplex when incorporated opposite to C, CD measurements revealed that all three spin labels were accommodated in a B-form DNA duplex. The mobility of the spin label TA was highly base-pair sensitive, allowing detection of its respective base-pairing partner in duplex DNA. Moreover, the mobility of TA was significantly reduced when paired with C or G in a DNA duplex at pH 5. This finding is consistent with protonation of TA and subsequent participation of the proton in hydrogen-bonding with C and G. The urea-linked UA and UC spin labels showed a similar mobility order when paired with A, G, C or T in a DNA duplex, with the lowest mobility when paired with C and the highest for T. These results show that the three labels, in particular TA, can report minor changes in their microenvironment, such as protonation, when placed in structured regions of nucleic acids.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 2.7 MB | Download |
References
-
Jacobo-Molina, A.; Ding, J.; Nanni, R. G.; Clark, A. D., Jr.; Lu, X.; Tantillo, C.; Williams, R. L.; Kamer, G.; Ferris, A. L.; Clark, P. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 6320–6324.
Return to citation in text: [1] -
Laughlan, G.; Murchie, A.; Norman, D. G.; Moore, M. H.; Moody, P. C.; Lilley, D. M.; Luisi, B. Science 1994, 265, 520–524. doi:10.1126/science.8036494
Return to citation in text: [1] -
Holbrook, S. R.; Holbrook, E. L.; Walukiewicz, H. E. Cell. Mol. Life Sci. 2001, 58, 234–243. doi:10.1007/pl00000851
Return to citation in text: [1] -
Vassylyev, D. G.; Sekine, S.-i.; Laptenko, O.; Lee, J.; Vassylyeva, M. N.; Borukhov, S.; Yokoyama, S. Nature 2002, 417, 712–719. doi:10.1038/nature752
Return to citation in text: [1] -
Edwards, T. E.; Klein, D. J.; Ferré-D'Amaré, A. R. Curr. Opin. Struct. Biol. 2007, 17, 273–279. doi:10.1016/j.sbi.2007.05.004
Return to citation in text: [1] -
Mooers, B. H. M. Methods 2009, 47, 168–176. doi:10.1016/j.ymeth.2008.09.006
Return to citation in text: [1] -
Clore, G. M.; Gronenborn, A. M. Curr. Opin. Chem. Biol. 1998, 2, 564–570. doi:10.1016/s1367-5931(98)80084-7
Return to citation in text: [1] -
Brutscher, B.; Boisbouvier, J.; Pardi, A.; Marion, D.; Simorre, J.-P. J. Am. Chem. Soc. 1998, 120, 11845–11851. doi:10.1021/ja982853l
Return to citation in text: [1] -
Tjandra, N. Structure 1999, 7, R205–R211.
Return to citation in text: [1] -
Riek, R.; Pervushin, K.; Wüthrich, K. Trends Biochem. Sci. 2000, 25, 462–468. doi:10.1016/S0968-0004(00)01665-0
Return to citation in text: [1] -
Latham, M. P.; Brown, D. J.; McCallum, S. A.; Pardi, A. ChemBioChem 2005, 6, 1492–1505. doi:10.1002/cbic.200500123
Return to citation in text: [1] [2] -
Campagne, S.; Gervais, V.; Milon, A. J. R. Soc., Interface 2011, 8, 1065–1078. doi:10.1098/rsif.2010.0543
Return to citation in text: [1] -
Wu, P. G.; Brand, L. Anal. Biochem. 1994, 218, 1–13. doi:10.1006/abio.1994.1134
Return to citation in text: [1] -
Prisner, T.; Rohrer, M.; MacMillan, F. Annu. Rev. Phys. Chem. 2001, 52, 279–313. doi:10.1146/annurev.physchem.52.1.279
Return to citation in text: [1] -
Schiemann, O.; Piton, N.; Mu, Y.; Stock, G.; Engels, J. W.; Prisner, T. F. J. Am. Chem. Soc. 2004, 126, 5722–5729. doi:10.1021/ja0393877
Return to citation in text: [1] -
Bowman, M. K.; Maryasov, A. G.; Kim, N.; DeRose, V. J. Appl. Magn. Reson. 2004, 26, 23–39. doi:10.1007/BF03166560
Return to citation in text: [1] -
Kim, N.-K.; Murali, A.; DeRose, V. J. Chem. Biol. 2004, 11, 939–948. doi:10.1016/j.chembiol.2004.04.013
Return to citation in text: [1] -
Piton, N.; Schiemann, O.; Mu, Y.; Stock, G.; Prisner, T.; Engels, J. W. Nucleosides, Nucleotides Nucleic Acids 2005, 24, 771–775. doi:10.1081/NCN-200060139
Return to citation in text: [1] -
Sabanayagam, C. R.; Eid, J. S.; Meller, A. J. Chem. Phys. 2005, 122, 061103. doi:10.1063/1.1854120
Return to citation in text: [1] -
Schiemann, O.; Piton, N.; Plackmeyer, J.; Bode, B. E.; Prisner, T. F.; Engels, J. W. Nat. Protoc. 2007, 2, 904–923. doi:10.1038/nprot.2007.97
Return to citation in text: [1] -
Schiemann, O.; Prisner, T. F. Q. Rev. Biophys. 2007, 40, 1–53. doi:10.1017/S003358350700460X
Return to citation in text: [1] -
Sicoli, G.; Mathis, G.; Delalande, O.; Boulard, Y.; Gasparutto, D.; Gambarelli, S. Angew. Chem. 2008, 120, 747–749. doi:10.1002/ange.200704133
Return to citation in text: [1] -
Xie, Y.; Dix, A. V.; Tor, Y. J. Am. Chem. Soc. 2009, 131, 17605–17614. doi:10.1021/ja905767g
Return to citation in text: [1] -
Sicoli, G.; Wachowius, F.; Bennati, M.; Höbartner, C. Angew. Chem., Int. Ed. 2010, 49, 6443–6447. doi:10.1002/anie.201000713
Return to citation in text: [1] -
Kim, N.-K.; Bowman, M. K.; DeRose, V. J. J. Am. Chem. Soc. 2010, 132, 8882–8884. doi:10.1021/ja101317g
Return to citation in text: [1] -
Ding, P.; Wunnicke, D.; Steinhoff, H.-J.; Seela, F. Chem. – Eur. J. 2010, 16, 14385–14396. doi:10.1002/chem.201001572
Return to citation in text: [1] -
Stoller, S.; Sicoli, G.; Baranova, T. Y.; Bennati, M.; Diederichsen, U. Angew. Chem., Int. Ed. 2011, 50, 9743–9746. doi:10.1002/anie.201103315
Return to citation in text: [1] -
Marko, A.; Denysenkov, V.; Margraf, D.; Cekan, P.; Schiemann, O.; Sigurdsson, S. T.; Prisner, T. F. J. Am. Chem. Soc. 2011, 133, 13375–13379. doi:10.1021/ja201244u
Return to citation in text: [1] -
Hengesbach, M.; Kim, N.-K.; Feigon, J.; Stone, M. D. Angew. Chem., Int. Ed. 2012, 51, 5876–5879. doi:10.1002/anie.201200526
Return to citation in text: [1] -
Pornsuwan, S.; Giller, K.; Riedel, D.; Becker, S.; Griesinger, C.; Bennati, M. Angew. Chem., Int. Ed. 2013, 52, 10290–10294. doi:10.1002/anie.201304747
Return to citation in text: [1] -
Polyhach, Y.; Godt, A.; Bauer, C.; Jeschke, G. J. Magn. Reson. 2007, 185, 118–129. doi:10.1016/j.jmr.2006.11.012
Return to citation in text: [1] -
Bode, B. E.; Plackmeyer, J.; Prisner, T. F.; Schiemann, O. J. Phys. Chem. A 2008, 112, 5064–5073. doi:10.1021/jp710504k
Return to citation in text: [1] -
Schiemann, O.; Cekan, P.; Margraf, D.; Prisner, T. F.; Sigurdsson, S. T. Angew. Chem., Int. Ed. 2009, 48, 3292–3295. doi:10.1002/anie.200805152
Return to citation in text: [1] -
Marko, A.; Margraf, D.; Cekan, P.; Sigurdsson, S. T.; Schiemann, O.; Prisner, T. F. Phys. Rev. E 2010, 81, 021911. doi:10.1103/PhysRevE.81.021911
Return to citation in text: [1] -
Tkach, I.; Pornsuwan, S.; Höbartner, C.; Wachowius, F.; Sigurdsson, S. T.; Baranova, T. Y.; Diederichsen, U.; Sicoli, G.; Bennati, M. Phys. Chem. Chem. Phys. 2013, 15, 3433–3437. doi:10.1039/C3CP44415E
Return to citation in text: [1] -
Svergun, D. I.; Koch, M. H. J. Rep. Prog. Phys. 2003, 66, 1735. doi:10.1088/0034-4885/66/10/R05
Return to citation in text: [1] -
Lipfert, J.; Doniach, S. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 307–327. doi:10.1146/annurev.biophys.36.040306.132655
Return to citation in text: [1] -
Bernadó, P.; Mylonas, E.; Petoukhov, M. V.; Blackledge, M.; Svergun, D. I. J. Am. Chem. Soc. 2007, 129, 5656–5664. doi:10.1021/ja069124n
Return to citation in text: [1] -
Mertens, H. D. T.; Svergun, D. I. J. Struct. Biol. 2010, 172, 128–141. doi:10.1016/j.jsb.2010.06.012
Return to citation in text: [1] -
Zıdek, L.; Štefl, R.; Sklenář, V. Curr. Opin. Struct. Biol. 2001, 11, 275–281. doi:10.1016/S0959-440X(00)00218-9
Return to citation in text: [1] -
Fürtig, B.; Richter, C.; Wöhnert, J.; Schwalbe, H. ChemBioChem 2003, 4, 936–962. doi:10.1002/cbic.200300700
Return to citation in text: [1] -
Al-Hashimi, H. M. J. Magn. Reson. 2013, 237, 191–204. doi:10.1016/j.jmr.2013.08.014
Return to citation in text: [1] [2] -
Gao, X.; Patel, D. J. J. Am. Chem. Soc. 1988, 110, 5178–5182. doi:10.1021/ja00223a045
Return to citation in text: [1] [2] -
Lane, A.; Jenkins, T.; Brown, D.; Brown, T. Biochem. J. 1991, 279, 269–281.
Return to citation in text: [1] [2] [3] -
Maskos, K.; Gunn, B. M.; LeBlanc, D. A.; Morden, K. M. Biochemistry 1993, 32, 3583–3595. doi:10.1021/bi00065a009
Return to citation in text: [1] -
Lane, A.; Ebel, S.; Brown, T. Eur. J. Biochem. 1994, 220, 717–727. doi:10.1111/j.1432-1033.1994.tb18672.x
Return to citation in text: [1] -
Boulard, Y.; Cognet, J. A. H.; Gabarro-Arpa, J.; Le Bret, M.; Carbonnaux, C.; Fazakerley, G. V. J. Mol. Biol. 1995, 246, 194–208. doi:10.1006/jmbi.1994.0076
Return to citation in text: [1] [2] [3] [4] -
Peyret, N.; Seneviratne, P. A.; Allawi, H. T.; SantaLucia, J., Jr. Biochemistry 1999, 38, 3468–3477. doi:10.1021/bi9825091
Return to citation in text: [1] [2] [3] -
Rist, M.; Marino, J. Nucleic Acids Res. 2001, 29, 2401–2408. doi:10.1093/nar/29.11.2401
Return to citation in text: [1] -
Liu, C.; Martin, C. T. J. Mol. Biol. 2001, 308, 465–475. doi:10.1006/jmbi.2001.4601
Return to citation in text: [1] -
Cekan, P.; Sigurdsson, S. T. Chem. Commun. 2008, 3393–3395. doi:10.1039/b801833b
Return to citation in text: [1] [2] -
Sinkeldam, R. W.; Greco, N. J.; Tor, Y. Chem. Rev. 2010, 110, 2579–2619. doi:10.1021/cr900301e
Return to citation in text: [1] -
Wilhelmsson, L. M. Q. Rev. Biophys. 2010, 43, 159–183. doi:10.1017/S0033583510000090
Return to citation in text: [1] -
Gardarsson, H.; Sigurdsson, S. T. Bioorg. Med. Chem. 2010, 18, 6121–6126. doi:10.1016/j.bmc.2010.06.060
Return to citation in text: [1] [2] -
Shin, D.; Sinkeldam, R. W.; Tor, Y. J. Am. Chem. Soc. 2011, 133, 14912–14915. doi:10.1021/ja206095a
Return to citation in text: [1] -
Gardarsson, H.; Kale, A. S.; Sigurdsson, S. T. ChemBioChem 2011, 12, 567–575. doi:10.1002/cbic.201000478
Return to citation in text: [1] [2] -
Dierckx, A.; Miannay, F.-A.; Ben Gaied, N.; Preus, S.; Björck, M.; Brown, T.; Wilhelmsson, L. M. Chem. – Eur. J. 2012, 18, 5987–5997. doi:10.1002/chem.201103419
Return to citation in text: [1] -
Saito, Y.; Miyauchi, Y.; Okamoto, A.; Saito, I. Tetrahedron Lett. 2004, 45, 7827–7831. doi:10.1016/j.tetlet.2004.09.003
Return to citation in text: [1] -
Xie, Y.; Maxson, T.; Tor, Y. Org. Biomol. Chem. 2010, 8, 5053–5055. doi:10.1039/C0OB00413H
Return to citation in text: [1] -
Sankaran, N. B.; Sato, Y.; Sato, F.; Rajendar, B.; Morita, K.; Seino, T.; Nishizawa, S.; Teramae, N. J. Phys. Chem. B 2009, 113, 1522–1529. doi:10.1021/jp808576t
Return to citation in text: [1] -
Gislason, K.; Gophane, D. B.; Sigurdsson, S. T. Org. Biomol. Chem. 2013, 11, 149–157. doi:10.1039/c2ob26536b
Return to citation in text: [1] -
Haller, A.; Rieder, U.; Aigner, M.; Blanchard, S. C.; Micura, R. Nat. Chem. Biol. 2011, 7, 393–400. doi:10.1038/nchembio.562
Return to citation in text: [1] -
Haller, A.; Altman, R. B.; Soulière, M. F.; Blanchard, S. C.; Micura, R. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 4188–4193. doi:10.1073/pnas.1218062110
Return to citation in text: [1] -
Edwards, T. E.; Okonogi, T. M.; Robinson, B. H.; Sigurdsson, S. T. J. Am. Chem. Soc. 2001, 123, 1527–1528. doi:10.1021/ja005649i
Return to citation in text: [1] [2] [3] -
Qin, P. Z.; Butcher, S. E.; Feigon, J.; Hubbell, W. L. Biochemistry 2001, 40, 6929–6936. doi:10.1021/bi010294g
Return to citation in text: [1] -
Qin, P. Z.; Hideg, K.; Feigon, J.; Hubbell, W. L. Biochemistry 2003, 42, 6772–6783. doi:10.1021/bi027222p
Return to citation in text: [1] -
Sowa, G. Z.; Qin, P. Z. Prog. Nucleic Acid Res. Mol. Biol. 2008, 82, 147–197. doi:10.1016/s0079-6603(08)00005-6
Return to citation in text: [1] -
Zhang, X.; Cekan, P.; Sigurdsson, S. T.; Qin, P. Z. Methods Enzymol. 2009, 469, 303–328. doi:10.1016/s0076-6879(09)69015-7
Return to citation in text: [1] -
Cekan, P.; Sigurdsson, S. T. J. Am. Chem. Soc. 2009, 131, 18054–18056. doi:10.1021/ja905623k
Return to citation in text: [1] [2] [3] [4] -
Ricci, A.; Marinello, J.; Bortolus, M.; Sánchez, A.; Grandas, A.; Pedroso, E.; Pommier, Y.; Capranico, G.; Maniero, A. L.; Zagotto, G. J. Med. Chem. 2011, 54, 1003–1009. doi:10.1021/jm101232t
Return to citation in text: [1] -
Höbartner, C.; Sicoli, G.; Wachowius, F.; Gophane, D. B.; Sigurdsson, S. T. J. Org. Chem. 2012, 77, 7749–7754. doi:10.1021/jo301227w
Return to citation in text: [1] -
Nguyen, P.; Qin, P. Z. Wiley Interdiscip. Rev.: RNA 2012, 3, 62–72. doi:10.1002/wrna.104
Return to citation in text: [1] -
Gophane, D. B.; Sigurdsson, S. T. Chem. Commun. 2013, 49, 999–1001. doi:10.1039/c2cc36389e
Return to citation in text: [1] -
Giordano, C.; Fratini, F.; Attanasio, D.; Cellai, L. Synthesis 2001, 565–572. doi:10.1055/s-2001-12355
Return to citation in text: [1] -
Chen, L. S.; Bahr, M. H.; Sheppard, T. L. Bioorg. Med. Chem. Lett. 2003, 13, 1509–1512. doi:10.1016/S0960-894X(03)00204-X
Return to citation in text: [1] -
Zakrzewski, J.; Krawczyk, M. Heteroat. Chem. 2006, 17, 393–401. doi:10.1002/hc.20228
Return to citation in text: [1] [2] -
Cekan, P.; Smith, A. L.; Barhate, N.; Robinson, B. H.; Sigurdsson, S. T. Nucleic Acids Res. 2008, 36, 5946–5954. doi:10.1093/nar/gkn562
Return to citation in text: [1] -
Brown, T.; Leonard, G. A.; Booth, E. D.; Chambers, J. J. Mol. Biol. 1989, 207, 455–457. doi:10.1016/0022-2836(89)90268-4
Return to citation in text: [1] [2] -
Brown, T.; Leonard, G. A.; Booth, E. D.; Kneale, G. J. Mol. Biol. 1990, 212, 437–440. doi:10.1016/0022-2836(90)90320-L
Return to citation in text: [1] -
Leonard, G. A.; Booth, E. D.; Brown, T. Nucleic Acids Res. 1990, 18, 5617–5623. doi:10.1093/nar/18.19.5617
Return to citation in text: [1] -
Brown, T. Aldrichimica Acta 1995, 28, 15–20.
Return to citation in text: [1] -
Boulard, Y.; Cognet, J. A. H.; Gabarro-Arpa, J.; Le Bret, M.; Sowers, L.; Fazakerley, G. V. Nucleic Acids Res. 1992, 20, 1933–1941. doi:10.1093/nar/20.8.1933
Return to citation in text: [1] -
Allawi, H. T.; SantaLucia, J., Jr.. Biochemistry 1998, 37, 9435–9444. doi:10.1021/bi9803729
Return to citation in text: [1] [2] -
Boulard, Y.; Cognet, J. A. H.; Fazakerley, G. V. J. Mol. Biol. 1997, 268, 331–347. doi:10.1006/jmbi.1997.0975
Return to citation in text: [1]
| 47. | Boulard, Y.; Cognet, J. A. H.; Gabarro-Arpa, J.; Le Bret, M.; Carbonnaux, C.; Fazakerley, G. V. J. Mol. Biol. 1995, 246, 194–208. doi:10.1006/jmbi.1994.0076 |
| 43. | Gao, X.; Patel, D. J. J. Am. Chem. Soc. 1988, 110, 5178–5182. doi:10.1021/ja00223a045 |
| 44. | Lane, A.; Jenkins, T.; Brown, D.; Brown, T. Biochem. J. 1991, 279, 269–281. |
| 78. | Brown, T.; Leonard, G. A.; Booth, E. D.; Chambers, J. J. Mol. Biol. 1989, 207, 455–457. doi:10.1016/0022-2836(89)90268-4 |
| 83. | Allawi, H. T.; SantaLucia, J., Jr.. Biochemistry 1998, 37, 9435–9444. doi:10.1021/bi9803729 |
| 1. | Jacobo-Molina, A.; Ding, J.; Nanni, R. G.; Clark, A. D., Jr.; Lu, X.; Tantillo, C.; Williams, R. L.; Kamer, G.; Ferris, A. L.; Clark, P. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 6320–6324. |
| 2. | Laughlan, G.; Murchie, A.; Norman, D. G.; Moore, M. H.; Moody, P. C.; Lilley, D. M.; Luisi, B. Science 1994, 265, 520–524. doi:10.1126/science.8036494 |
| 3. | Holbrook, S. R.; Holbrook, E. L.; Walukiewicz, H. E. Cell. Mol. Life Sci. 2001, 58, 234–243. doi:10.1007/pl00000851 |
| 4. | Vassylyev, D. G.; Sekine, S.-i.; Laptenko, O.; Lee, J.; Vassylyeva, M. N.; Borukhov, S.; Yokoyama, S. Nature 2002, 417, 712–719. doi:10.1038/nature752 |
| 5. | Edwards, T. E.; Klein, D. J.; Ferré-D'Amaré, A. R. Curr. Opin. Struct. Biol. 2007, 17, 273–279. doi:10.1016/j.sbi.2007.05.004 |
| 6. | Mooers, B. H. M. Methods 2009, 47, 168–176. doi:10.1016/j.ymeth.2008.09.006 |
| 36. | Svergun, D. I.; Koch, M. H. J. Rep. Prog. Phys. 2003, 66, 1735. doi:10.1088/0034-4885/66/10/R05 |
| 37. | Lipfert, J.; Doniach, S. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 307–327. doi:10.1146/annurev.biophys.36.040306.132655 |
| 38. | Bernadó, P.; Mylonas, E.; Petoukhov, M. V.; Blackledge, M.; Svergun, D. I. J. Am. Chem. Soc. 2007, 129, 5656–5664. doi:10.1021/ja069124n |
| 39. | Mertens, H. D. T.; Svergun, D. I. J. Struct. Biol. 2010, 172, 128–141. doi:10.1016/j.jsb.2010.06.012 |
| 69. | Cekan, P.; Sigurdsson, S. T. J. Am. Chem. Soc. 2009, 131, 18054–18056. doi:10.1021/ja905623k |
| 31. | Polyhach, Y.; Godt, A.; Bauer, C.; Jeschke, G. J. Magn. Reson. 2007, 185, 118–129. doi:10.1016/j.jmr.2006.11.012 |
| 32. | Bode, B. E.; Plackmeyer, J.; Prisner, T. F.; Schiemann, O. J. Phys. Chem. A 2008, 112, 5064–5073. doi:10.1021/jp710504k |
| 33. | Schiemann, O.; Cekan, P.; Margraf, D.; Prisner, T. F.; Sigurdsson, S. T. Angew. Chem., Int. Ed. 2009, 48, 3292–3295. doi:10.1002/anie.200805152 |
| 34. | Marko, A.; Margraf, D.; Cekan, P.; Sigurdsson, S. T.; Schiemann, O.; Prisner, T. F. Phys. Rev. E 2010, 81, 021911. doi:10.1103/PhysRevE.81.021911 |
| 35. | Tkach, I.; Pornsuwan, S.; Höbartner, C.; Wachowius, F.; Sigurdsson, S. T.; Baranova, T. Y.; Diederichsen, U.; Sicoli, G.; Bennati, M. Phys. Chem. Chem. Phys. 2013, 15, 3433–3437. doi:10.1039/C3CP44415E |
| 69. | Cekan, P.; Sigurdsson, S. T. J. Am. Chem. Soc. 2009, 131, 18054–18056. doi:10.1021/ja905623k |
| 13. | Wu, P. G.; Brand, L. Anal. Biochem. 1994, 218, 1–13. doi:10.1006/abio.1994.1134 |
| 14. | Prisner, T.; Rohrer, M.; MacMillan, F. Annu. Rev. Phys. Chem. 2001, 52, 279–313. doi:10.1146/annurev.physchem.52.1.279 |
| 15. | Schiemann, O.; Piton, N.; Mu, Y.; Stock, G.; Engels, J. W.; Prisner, T. F. J. Am. Chem. Soc. 2004, 126, 5722–5729. doi:10.1021/ja0393877 |
| 16. | Bowman, M. K.; Maryasov, A. G.; Kim, N.; DeRose, V. J. Appl. Magn. Reson. 2004, 26, 23–39. doi:10.1007/BF03166560 |
| 17. | Kim, N.-K.; Murali, A.; DeRose, V. J. Chem. Biol. 2004, 11, 939–948. doi:10.1016/j.chembiol.2004.04.013 |
| 18. | Piton, N.; Schiemann, O.; Mu, Y.; Stock, G.; Prisner, T.; Engels, J. W. Nucleosides, Nucleotides Nucleic Acids 2005, 24, 771–775. doi:10.1081/NCN-200060139 |
| 19. | Sabanayagam, C. R.; Eid, J. S.; Meller, A. J. Chem. Phys. 2005, 122, 061103. doi:10.1063/1.1854120 |
| 20. | Schiemann, O.; Piton, N.; Plackmeyer, J.; Bode, B. E.; Prisner, T. F.; Engels, J. W. Nat. Protoc. 2007, 2, 904–923. doi:10.1038/nprot.2007.97 |
| 21. | Schiemann, O.; Prisner, T. F. Q. Rev. Biophys. 2007, 40, 1–53. doi:10.1017/S003358350700460X |
| 22. | Sicoli, G.; Mathis, G.; Delalande, O.; Boulard, Y.; Gasparutto, D.; Gambarelli, S. Angew. Chem. 2008, 120, 747–749. doi:10.1002/ange.200704133 |
| 23. | Xie, Y.; Dix, A. V.; Tor, Y. J. Am. Chem. Soc. 2009, 131, 17605–17614. doi:10.1021/ja905767g |
| 24. | Sicoli, G.; Wachowius, F.; Bennati, M.; Höbartner, C. Angew. Chem., Int. Ed. 2010, 49, 6443–6447. doi:10.1002/anie.201000713 |
| 25. | Kim, N.-K.; Bowman, M. K.; DeRose, V. J. J. Am. Chem. Soc. 2010, 132, 8882–8884. doi:10.1021/ja101317g |
| 26. | Ding, P.; Wunnicke, D.; Steinhoff, H.-J.; Seela, F. Chem. – Eur. J. 2010, 16, 14385–14396. doi:10.1002/chem.201001572 |
| 27. | Stoller, S.; Sicoli, G.; Baranova, T. Y.; Bennati, M.; Diederichsen, U. Angew. Chem., Int. Ed. 2011, 50, 9743–9746. doi:10.1002/anie.201103315 |
| 28. | Marko, A.; Denysenkov, V.; Margraf, D.; Cekan, P.; Schiemann, O.; Sigurdsson, S. T.; Prisner, T. F. J. Am. Chem. Soc. 2011, 133, 13375–13379. doi:10.1021/ja201244u |
| 29. | Hengesbach, M.; Kim, N.-K.; Feigon, J.; Stone, M. D. Angew. Chem., Int. Ed. 2012, 51, 5876–5879. doi:10.1002/anie.201200526 |
| 30. | Pornsuwan, S.; Giller, K.; Riedel, D.; Becker, S.; Griesinger, C.; Bennati, M. Angew. Chem., Int. Ed. 2013, 52, 10290–10294. doi:10.1002/anie.201304747 |
| 62. | Haller, A.; Rieder, U.; Aigner, M.; Blanchard, S. C.; Micura, R. Nat. Chem. Biol. 2011, 7, 393–400. doi:10.1038/nchembio.562 |
| 63. | Haller, A.; Altman, R. B.; Soulière, M. F.; Blanchard, S. C.; Micura, R. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 4188–4193. doi:10.1073/pnas.1218062110 |
| 7. | Clore, G. M.; Gronenborn, A. M. Curr. Opin. Chem. Biol. 1998, 2, 564–570. doi:10.1016/s1367-5931(98)80084-7 |
| 8. | Brutscher, B.; Boisbouvier, J.; Pardi, A.; Marion, D.; Simorre, J.-P. J. Am. Chem. Soc. 1998, 120, 11845–11851. doi:10.1021/ja982853l |
| 9. | Tjandra, N. Structure 1999, 7, R205–R211. |
| 10. | Riek, R.; Pervushin, K.; Wüthrich, K. Trends Biochem. Sci. 2000, 25, 462–468. doi:10.1016/S0968-0004(00)01665-0 |
| 11. | Latham, M. P.; Brown, D. J.; McCallum, S. A.; Pardi, A. ChemBioChem 2005, 6, 1492–1505. doi:10.1002/cbic.200500123 |
| 12. | Campagne, S.; Gervais, V.; Milon, A. J. R. Soc., Interface 2011, 8, 1065–1078. doi:10.1098/rsif.2010.0543 |
| 64. | Edwards, T. E.; Okonogi, T. M.; Robinson, B. H.; Sigurdsson, S. T. J. Am. Chem. Soc. 2001, 123, 1527–1528. doi:10.1021/ja005649i |
| 65. | Qin, P. Z.; Butcher, S. E.; Feigon, J.; Hubbell, W. L. Biochemistry 2001, 40, 6929–6936. doi:10.1021/bi010294g |
| 66. | Qin, P. Z.; Hideg, K.; Feigon, J.; Hubbell, W. L. Biochemistry 2003, 42, 6772–6783. doi:10.1021/bi027222p |
| 67. | Sowa, G. Z.; Qin, P. Z. Prog. Nucleic Acid Res. Mol. Biol. 2008, 82, 147–197. doi:10.1016/s0079-6603(08)00005-6 |
| 68. | Zhang, X.; Cekan, P.; Sigurdsson, S. T.; Qin, P. Z. Methods Enzymol. 2009, 469, 303–328. doi:10.1016/s0076-6879(09)69015-7 |
| 69. | Cekan, P.; Sigurdsson, S. T. J. Am. Chem. Soc. 2009, 131, 18054–18056. doi:10.1021/ja905623k |
| 70. | Ricci, A.; Marinello, J.; Bortolus, M.; Sánchez, A.; Grandas, A.; Pedroso, E.; Pommier, Y.; Capranico, G.; Maniero, A. L.; Zagotto, G. J. Med. Chem. 2011, 54, 1003–1009. doi:10.1021/jm101232t |
| 71. | Höbartner, C.; Sicoli, G.; Wachowius, F.; Gophane, D. B.; Sigurdsson, S. T. J. Org. Chem. 2012, 77, 7749–7754. doi:10.1021/jo301227w |
| 72. | Nguyen, P.; Qin, P. Z. Wiley Interdiscip. Rev.: RNA 2012, 3, 62–72. doi:10.1002/wrna.104 |
| 73. | Gophane, D. B.; Sigurdsson, S. T. Chem. Commun. 2013, 49, 999–1001. doi:10.1039/c2cc36389e |
| 49. | Rist, M.; Marino, J. Nucleic Acids Res. 2001, 29, 2401–2408. doi:10.1093/nar/29.11.2401 |
| 50. | Liu, C.; Martin, C. T. J. Mol. Biol. 2001, 308, 465–475. doi:10.1006/jmbi.2001.4601 |
| 51. | Cekan, P.; Sigurdsson, S. T. Chem. Commun. 2008, 3393–3395. doi:10.1039/b801833b |
| 52. | Sinkeldam, R. W.; Greco, N. J.; Tor, Y. Chem. Rev. 2010, 110, 2579–2619. doi:10.1021/cr900301e |
| 53. | Wilhelmsson, L. M. Q. Rev. Biophys. 2010, 43, 159–183. doi:10.1017/S0033583510000090 |
| 54. | Gardarsson, H.; Sigurdsson, S. T. Bioorg. Med. Chem. 2010, 18, 6121–6126. doi:10.1016/j.bmc.2010.06.060 |
| 55. | Shin, D.; Sinkeldam, R. W.; Tor, Y. J. Am. Chem. Soc. 2011, 133, 14912–14915. doi:10.1021/ja206095a |
| 56. | Gardarsson, H.; Kale, A. S.; Sigurdsson, S. T. ChemBioChem 2011, 12, 567–575. doi:10.1002/cbic.201000478 |
| 57. | Dierckx, A.; Miannay, F.-A.; Ben Gaied, N.; Preus, S.; Björck, M.; Brown, T.; Wilhelmsson, L. M. Chem. – Eur. J. 2012, 18, 5987–5997. doi:10.1002/chem.201103419 |
| 60. | Sankaran, N. B.; Sato, Y.; Sato, F.; Rajendar, B.; Morita, K.; Seino, T.; Nishizawa, S.; Teramae, N. J. Phys. Chem. B 2009, 113, 1522–1529. doi:10.1021/jp808576t |
| 42. | Al-Hashimi, H. M. J. Magn. Reson. 2013, 237, 191–204. doi:10.1016/j.jmr.2013.08.014 |
| 47. | Boulard, Y.; Cognet, J. A. H.; Gabarro-Arpa, J.; Le Bret, M.; Carbonnaux, C.; Fazakerley, G. V. J. Mol. Biol. 1995, 246, 194–208. doi:10.1006/jmbi.1994.0076 |
| 48. | Peyret, N.; Seneviratne, P. A.; Allawi, H. T.; SantaLucia, J., Jr. Biochemistry 1999, 38, 3468–3477. doi:10.1021/bi9825091 |
| 61. | Gislason, K.; Gophane, D. B.; Sigurdsson, S. T. Org. Biomol. Chem. 2013, 11, 149–157. doi:10.1039/c2ob26536b |
| 43. | Gao, X.; Patel, D. J. J. Am. Chem. Soc. 1988, 110, 5178–5182. doi:10.1021/ja00223a045 |
| 44. | Lane, A.; Jenkins, T.; Brown, D.; Brown, T. Biochem. J. 1991, 279, 269–281. |
| 45. | Maskos, K.; Gunn, B. M.; LeBlanc, D. A.; Morden, K. M. Biochemistry 1993, 32, 3583–3595. doi:10.1021/bi00065a009 |
| 46. | Lane, A.; Ebel, S.; Brown, T. Eur. J. Biochem. 1994, 220, 717–727. doi:10.1111/j.1432-1033.1994.tb18672.x |
| 69. | Cekan, P.; Sigurdsson, S. T. J. Am. Chem. Soc. 2009, 131, 18054–18056. doi:10.1021/ja905623k |
| 84. | Boulard, Y.; Cognet, J. A. H.; Fazakerley, G. V. J. Mol. Biol. 1997, 268, 331–347. doi:10.1006/jmbi.1997.0975 |
| 11. | Latham, M. P.; Brown, D. J.; McCallum, S. A.; Pardi, A. ChemBioChem 2005, 6, 1492–1505. doi:10.1002/cbic.200500123 |
| 40. | Zıdek, L.; Štefl, R.; Sklenář, V. Curr. Opin. Struct. Biol. 2001, 11, 275–281. doi:10.1016/S0959-440X(00)00218-9 |
| 41. | Fürtig, B.; Richter, C.; Wöhnert, J.; Schwalbe, H. ChemBioChem 2003, 4, 936–962. doi:10.1002/cbic.200300700 |
| 42. | Al-Hashimi, H. M. J. Magn. Reson. 2013, 237, 191–204. doi:10.1016/j.jmr.2013.08.014 |
| 51. | Cekan, P.; Sigurdsson, S. T. Chem. Commun. 2008, 3393–3395. doi:10.1039/b801833b |
| 54. | Gardarsson, H.; Sigurdsson, S. T. Bioorg. Med. Chem. 2010, 18, 6121–6126. doi:10.1016/j.bmc.2010.06.060 |
| 56. | Gardarsson, H.; Kale, A. S.; Sigurdsson, S. T. ChemBioChem 2011, 12, 567–575. doi:10.1002/cbic.201000478 |
| 58. | Saito, Y.; Miyauchi, Y.; Okamoto, A.; Saito, I. Tetrahedron Lett. 2004, 45, 7827–7831. doi:10.1016/j.tetlet.2004.09.003 |
| 59. | Xie, Y.; Maxson, T.; Tor, Y. Org. Biomol. Chem. 2010, 8, 5053–5055. doi:10.1039/C0OB00413H |
| 64. | Edwards, T. E.; Okonogi, T. M.; Robinson, B. H.; Sigurdsson, S. T. J. Am. Chem. Soc. 2001, 123, 1527–1528. doi:10.1021/ja005649i |
| 76. | Zakrzewski, J.; Krawczyk, M. Heteroat. Chem. 2006, 17, 393–401. doi:10.1002/hc.20228 |
| 74. | Giordano, C.; Fratini, F.; Attanasio, D.; Cellai, L. Synthesis 2001, 565–572. doi:10.1055/s-2001-12355 |
| 75. | Chen, L. S.; Bahr, M. H.; Sheppard, T. L. Bioorg. Med. Chem. Lett. 2003, 13, 1509–1512. doi:10.1016/S0960-894X(03)00204-X |
| 82. | Boulard, Y.; Cognet, J. A. H.; Gabarro-Arpa, J.; Le Bret, M.; Sowers, L.; Fazakerley, G. V. Nucleic Acids Res. 1992, 20, 1933–1941. doi:10.1093/nar/20.8.1933 |
| 47. | Boulard, Y.; Cognet, J. A. H.; Gabarro-Arpa, J.; Le Bret, M.; Carbonnaux, C.; Fazakerley, G. V. J. Mol. Biol. 1995, 246, 194–208. doi:10.1006/jmbi.1994.0076 |
| 83. | Allawi, H. T.; SantaLucia, J., Jr.. Biochemistry 1998, 37, 9435–9444. doi:10.1021/bi9803729 |
| 47. | Boulard, Y.; Cognet, J. A. H.; Gabarro-Arpa, J.; Le Bret, M.; Carbonnaux, C.; Fazakerley, G. V. J. Mol. Biol. 1995, 246, 194–208. doi:10.1006/jmbi.1994.0076 |
| 48. | Peyret, N.; Seneviratne, P. A.; Allawi, H. T.; SantaLucia, J., Jr. Biochemistry 1999, 38, 3468–3477. doi:10.1021/bi9825091 |
| 44. | Lane, A.; Jenkins, T.; Brown, D.; Brown, T. Biochem. J. 1991, 279, 269–281. |
| 48. | Peyret, N.; Seneviratne, P. A.; Allawi, H. T.; SantaLucia, J., Jr. Biochemistry 1999, 38, 3468–3477. doi:10.1021/bi9825091 |
| 78. | Brown, T.; Leonard, G. A.; Booth, E. D.; Chambers, J. J. Mol. Biol. 1989, 207, 455–457. doi:10.1016/0022-2836(89)90268-4 |
| 79. | Brown, T.; Leonard, G. A.; Booth, E. D.; Kneale, G. J. Mol. Biol. 1990, 212, 437–440. doi:10.1016/0022-2836(90)90320-L |
| 80. | Leonard, G. A.; Booth, E. D.; Brown, T. Nucleic Acids Res. 1990, 18, 5617–5623. doi:10.1093/nar/18.19.5617 |
| 64. | Edwards, T. E.; Okonogi, T. M.; Robinson, B. H.; Sigurdsson, S. T. J. Am. Chem. Soc. 2001, 123, 1527–1528. doi:10.1021/ja005649i |
| 76. | Zakrzewski, J.; Krawczyk, M. Heteroat. Chem. 2006, 17, 393–401. doi:10.1002/hc.20228 |
| 77. | Cekan, P.; Smith, A. L.; Barhate, N.; Robinson, B. H.; Sigurdsson, S. T. Nucleic Acids Res. 2008, 36, 5946–5954. doi:10.1093/nar/gkn562 |
© 2015 Gophane and Sigurdsson; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)