Abstract
Kinetic experiments based on the measurement of nitrogen evolution in the reaction of ethyl diazoacetate (N2CHCO2Et, EDA) and styrene or methanol catalyzed by the [IPrAu]+ core (IPr = 1,3-bis(diisopropylphenyl)imidazole-2-ylidene) have provided evidence that the transfer of the carbene group CHCO2Et to the substrate (styrene or methanol) takes place in the coordination sphere of Au(I) by means of an inner-sphere mechanism, in contrast to the generally accepted proposal of outer-sphere mechanisms for Au(I)-catalyzed reactions.

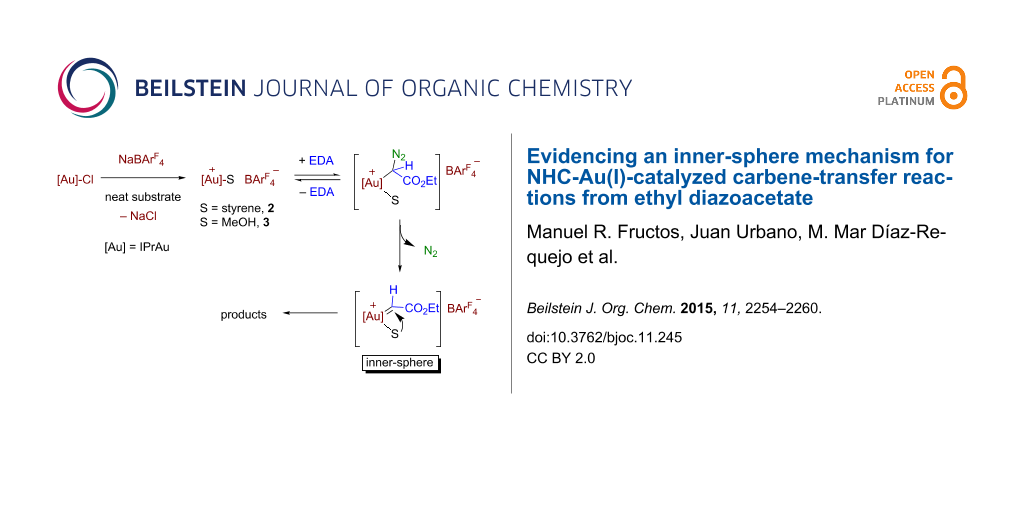
Graphical Abstract
Introduction
The discovery of the catalytic capabilities of soluble gold(I) species toward the hydration of alkynes by Teles and co-workers [1] is considered as the rising of the golden era for the use of this metal in homogeneous catalysis [2,3]. A number of transformations have been reported to date [4-13], most of them based on a particular feature of gold: a singular carbophilicity that enhances the electrophilic activation of multiple bonds upon coordination followed by subsequent inter- or intramolecular reaction with nucleophiles. Most of the reported systems contain an unsaturated fragment that is activated upon coordination to the gold center, thus triggering further transformations, the formation of very reactive gold–carbene intermediates being proposed [4-14]. As a representative example, the skeletal rearrangement of the [2 + 2] cycloaddition of 1,6-enynes [4] is shown in Scheme 1, where three different gold–carbene intermediates are involved in the possible transformations.
![[1860-5397-11-245-i1]](/bjoc/content/inline/1860-5397-11-245-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The Au(I)-catalyzed skeletal rearrangement of the [2 + 2] cycloaddition of 1,6-enynes that involves gold–carbene intermediates.
Scheme 1: The Au(I)-catalyzed skeletal rearrangement of the [2 + 2] cycloaddition of 1,6-enynes that involves...
A different reaction in which the formation of gold–carbene intermediates has been proposed arises from the interaction of a gold(I) source and a diazo compound. It was not until 2005 that the first example of this transformation was reported by our group [15,16], when the complex IPrAuCl (1) (IPr = 1,3-bis(diisopropylphenyl)imidazole-2-ylidene), in the presence of NaBArF4 (BArF4− = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate) as a halide scavenger, induced the incorporation of the :CHCO2Et group from N2CHCO2Et to styrene (Scheme 2a) or methanol (Scheme 2b), among others. With the former, in addition to the formation of the expected cyclopropanes, a second type of product was observed, derived from the incorporation of the carbene :CHCO2Et unit to the C(sp2)–H bonds. For methanol as the reactant, the ether derived from the functionalization of the O–H bond was obtained.
![[1860-5397-11-245-i2]](/bjoc/content/inline/1860-5397-11-245-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The catalytic activity of IPrAuCl + NaBArF4 in the carbene-transfer reaction to styrene or methanol.
Scheme 2: The catalytic activity of IPrAuCl + NaBArF4 in the carbene-transfer reaction to styrene or methanol....
A transformation related to this contribution is the reaction published by Echavarren (Scheme 3). In this reaction, the complex [LAu(NCR)]SbF6 (L = tertiary phosphine ligand) abstracted the carbene group :CHPh from a tropilium derivative, and further transferred the gold-bonded carbene group to an olefin [17,18]. This contribution constituted a breakthrough in gold-mediated carbene-transfer reactions since the carbene source lacks of the well-known instability of some of the diazo reagents.
![[1860-5397-11-245-i3]](/bjoc/content/inline/1860-5397-11-245-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The gold-promoted decarbenation reaction described by Echavarren and co-workers.
Scheme 3: The gold-promoted decarbenation reaction described by Echavarren and co-workers.
The reactions shown in Scheme 2 and Scheme 3 have been explained through the appearance of a [LAu=CR1R2]+ intermediate, not detected nor isolated, that further reacts with a non-coordinated nucleophile (i.e., by means of an outer-sphere mechanism). Those intermediates are quite reactive, in contrast to similar but low reactive gold–carbene complexes recently and independently described by the groups of Fürstner [19] and Straub [20]. Scheme 4a shows a generally accepted mechanism for the metal-catalyzed carbene transfer from diazo compounds to nucleophiles [21-27], where the formation of C–C or C–X bonds takes place throughout a transition state in which no interaction of the substrate and the metal center exists (styrene is shown as an example). In many cases, a non-desired side reaction is also observed, in which two molecules of the diazo compound convert into an olefin and/or an azine (Scheme 4b), the second acting as a nucleophile attacking the metallocarbene intermediate [28]. However, our previous studies have shown that this gold-based catalytic system does not induce such non-desired carbene homocoupling [15,16].
![[1860-5397-11-245-i4]](/bjoc/content/inline/1860-5397-11-245-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: (a) General representation of the metal-catalyzed carbene-transfer reaction (olefin cyclopropanation). (b) Side-reactions of carbene or diazo coupling commonly observed.
Scheme 4: (a) General representation of the metal-catalyzed carbene-transfer reaction (olefin cyclopropanatio...
In this contribution we report the results obtained from a kinetic study carried out with the IPrAuCl complex that allows proposing a plausible inner-sphere mechanism in which the substrate (styrene or methanol) seems to remain coordinated to the gold center, a feature that could be extended to many of the reported catalytic systems involving Au(I) complexes.
Results and Discussion
The probe reactions
As mentioned above, we have already described the potential of the system IPrAuCl + NaBArF4 to promote the catalytic decomposition of ethyl diazoacetate (N2CHCO2Et, EDA) and functionalize styrene or methanol (see Scheme 2). Since molecular nitrogen is evolved in these transformations, we have monitored the pressure above the reaction mixtures of EDA and styrene or methanol in the presence of catalytic amounts (5 mol %) of IPrAuCl + NaBArF4. It is worth noting that the catalyst precursors were dissolved in the neat substrate (5 mL) and stirred for 30 min to ensure halide abstraction prior to EDA addition. The experiments have been carried out using a flask connected to a pressure gauge that provides the variation in the increase of the internal pressure (see Experimental). Figure 1 shows the plots of the N2 concentration (mmol) vs time, from which kobs for N2 liberation have been obtained as 2.17(0.05) × 10−3 s−1 and 1.34(0.01) × 10−3 s−1 for styrene and methanol, respectively. Thus, both reactions decomposed EDA with similar rates within the same order of magnitude. Given the experimental fact that this catalytic system does not promote the carbene coupling side reaction, the amount of measured N2 exclusively corresponds to that evolved from the formation of the gold–carbene intermediate in the path to the products.
![[1860-5397-11-245-1]](/bjoc/content/figures/1860-5397-11-245-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Plot of evolved nitrogen with time for the reactions of EDA with styrene or methanol.
Figure 1: Plot of evolved nitrogen with time for the reactions of EDA with styrene or methanol.
The effect of substrate concentration
A second set of experiments has been performed varying the catalyst to substrate ratio, but maintaining the concentration of the catalyst as a constant. Thus, 100:0, 80:20 and 60:40 v/v mixtures of styrene and cyclohexane, respectively, have been reacted with EDA (0.285 mmol) with the same catalyst precursor (see Experimental). Under these conditions, cyclohexane is much less reactive than styrene and acts as an inert solvent [29]. The kinetic curves for these three experiments are shown in Figure 2. The plot of kobs vs substrate concentration evidences a direct correlation between styrene concentration and kobs. The same effect is observed in an array of experiments carried out with mixtures of methanol and methylene chloride (Figure 2). These results unambiguously indicate that the release of nitrogen takes place at a faster rate when increasing the amount of the reactant, styrene or methanol.
![[1860-5397-11-245-2]](/bjoc/content/figures/1860-5397-11-245-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Top: Plots of evolved nitrogen with time for the reactions of EDA with styrene (left) or methanol (right) varying the relative substrate:catalyst ratio, at a constant catalyst value. Bottom: Plots of kobs vs substrate concentration showing a direct correlation between both magnitudes.
Figure 2: Top: Plots of evolved nitrogen with time for the reactions of EDA with styrene (left) or methanol (...
It is worth mentioning that the addition of 5 equiv of NaBArF4 did not induce any change in the reaction rate compared with that of one equiv, assessing that the halide-free catalytically-active gold species is available in the latter case. Also, the fact that the same behavior regarding the “dilution effect” shown in Figure 2 is observed with cyclohexane or dichloromethane as dilution agents must be interpreted as the result of their independent behavior in the process, not being involved in any effect relative to polarity or solubility issues.
Mechanistic interpretation
It has been reported by several authors that the use of LAuX (X = halide) as catalyst precursor requires the addition of a halide scavenger with a weekly coordinating ligand (such as NTf2, BArF4, SbF6) [30,31]. Cationic complexes of type [LAu(NCR)]+ have also been isolated previously [32,33]. One way or the other, the incorporation of the reactant has been proposed to be associative in most cases, the reaction then being triggered from there, usually through an outer-sphere mechanism. To the best of our knowledge, there are no evidences which support the alternative route, the inner-sphere mechanism.
The kinetic data available from the previous section have provided the following statements: (i) there is only a slight effect of the nature of the substrate, styrene or methanol, in the rate of evolution of N2, and (ii) the reaction rate is affected by the relative substrate:catalyst ratio, both magnitudes being directly correlated. Scheme 5 displays a feasible catalytic cycle for these transformations. The mixture of IPrAuCl and NaBArF4 in styrene or methanol ensures the formation of the cationic species [IPrAu(sty)]BArF4 (2) or [IPrAu(MeOH)]BArF4 (3) similar to structurally characterized [IPrAu(NCMe)]BF4 [32,33]. The addition of EDA originates the immediate evolution of N2, the gold–carbene intermediate must form from 2 or 3. Also, this must be the rate determining step, efforts to detect such intermediates having proven unsuccessful. The commonly accepted pathway (route A, Scheme 5) would suppose an exchange of the substrate ligand with the diazo compound and then evolution of nitrogen. However, that first step of substrate dissociation would be in disagreement with the observation of the enhancement of the reaction rate of nitrogen evolution when increasing the substrate:catalyst ratio, the dissociation of the substrate should be disfavoured at larger substrate concentration. Thus, there is only a plausible explanation for the experimental data (route B, Scheme 5): the substrate remains coordinated while the diazo compound coordinates and eliminates nitrogen, the carbene transfer taking place between two ligands, the carbene and the substrate. This approach would also explain the lack of formation of the olefins derived from carbene coupling with this Au(I)-based system.
![[1860-5397-11-245-i5]](/bjoc/content/inline/1860-5397-11-245-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: The outer- and inner-sphere routes for this transformation.
Scheme 5: The outer- and inner-sphere routes for this transformation.
Conclusion
The experimental results obtained from the measurement of N2 evolution in [IPrAu]+-catalyzed carbene transfer from ethyl diazoacetate have allowed proposing that both the carbene and the substrate (styrene or methanol) are bonded to the Au(I) center previously to the corresponding coupling, that is, the functionalization reaction takes place throughout an inner-sphere mechanism. This is not the commonly proposed mechanism for most of the different Au(I)-catalyzed reactions, for which an outer-sphere mechanism has been frequently assumed. We hope that the findings reported herein could be verified for other Au(I)-catalyzed transformations.
Experimental
General methods
All preparations and manipulations were carried out under an oxygen-free nitrogen atmosphere using conventional Schlenk techniques. Solvents were rigorously dried previously to their use. The substrates were purchased from Aldrich. The complex IPrAuCl and NaBArF4 were prepared according to the literature [34,35]. NMR spectra were performed on Agilent 400 MR and 500 DD2 spectrometers. GC data were collected with a Varian GC-3900 spectrometer with a FID detector.
Kinetic experiments
The kinetic experiments have been carried out similarly to previous procedures from our laboratory [36]. For the sake of clarity, the methodology is briefly described herein. A ManontheMoonTech X201 (http://www.manonthemoontech.com) device consisting of a stainless-steel gas reservoir doubly connected to a pressure transmitter has been employed for the measurement of nitrogen evolution. The variation of the inner pressure inside the reaction flask (Figure 3) is measured with an electronic pressure meter/controller (EL-Press,Bronkhorst HI-TEC).
![[1860-5397-11-245-3]](/bjoc/content/figures/1860-5397-11-245-3.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: The experimental device for the measurement of N2 evolution.
Figure 3: The experimental device for the measurement of N2 evolution.
In a typical experiment, the N2 pressure change was measured after the addition of EDA (0.285 mmol) to a stirred solution of substrate (0. 5 mmol), and a mixture of IPrAuCl and NaBArF4 (5 mol %) in cyclohexane/CH2Cl2 (5 mL) at room temperature. kobs was obtained from experimental curves upon fitting to an exponential growth equation using ORIGIN software.
References
-
Teles, J. H.; Brode, S.; Chabanas, M. Angew. Chem., Int. Ed. 1998, 37, 1415–1418. doi:10.1002/(SICI)1521-3773(19980605)37:10<1415::AID-ANIE1415>3.0.CO;2-N
Return to citation in text: [1] -
Norman, R. O. C.; Parr, W. J. E.; Thomas, C. B. J. Chem. Soc., Perkin Trans. 1 1976, 1983–1987. doi:10.1039/p19760001983
Return to citation in text: [1] -
Fukuda, Y.; Utimoto, K. J. Org. Chem. 1991, 56, 3729–3731. doi:10.1021/jo00011a058
Return to citation in text: [1] -
Obradors, C.; Echavarren, A. M. Chem. Commun. 2014, 50, 16–28. doi:10.1039/C3CC45518A
Return to citation in text: [1] [2] [3] -
Xie, J.; Pan, C.; Abdukader, A.; Zhu, C. Chem. Soc. Rev. 2014, 43, 5245–5256. doi:10.1039/C4CS00004H
Return to citation in text: [1] [2] -
Shapiro, N. D.; Toste, F. D. Synlett 2010, 675–691. doi:10.1055/s-0029-1219369
Return to citation in text: [1] [2] -
Fürstner, A. Chem. Soc. Rev. 2009, 38, 3208–3221. doi:10.1039/b816696j
Return to citation in text: [1] [2] -
Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395–3442. doi:10.1021/cr050041j
Return to citation in text: [1] [2] -
Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g
Return to citation in text: [1] [2] -
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319
Return to citation in text: [1] [2] -
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Commun. 2007, 333–346. doi:10.1039/B612008C
Return to citation in text: [1] [2] -
Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410–3449. doi:10.1002/anie.200604335
Return to citation in text: [1] [2] -
Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x
Return to citation in text: [1] [2] -
Lauterbach, T.; Asiri, A. M.; Hashmi, A. S. K. Adv. Organomet. Chem. 2014, 62, 261–297. doi:10.1016/B978-0-12-800976-5.00005-9
Return to citation in text: [1] -
Fructos, M. R.; Belderrain, T. R.; de Fremont, P.; Scott, N. M.; Nolan, S. P.; Díaz-Requejo, M. M.; Pérez, P. J. Angew. Chem., Int. Ed. 2005, 44, 5284–5288. doi:10.1002/anie.200501056
Return to citation in text: [1] [2] -
Rivilla, I.; Gómez-Emeterio, B. P.; Fructos, M. R.; Díaz-Requejo, M. M.; Pérez, P. J. Organometallics 2011, 30, 2855–2860. doi:10.1021/om200206m
Return to citation in text: [1] [2] -
Solorio-Alvarado, C. R.; Wang, Y.; Echavarren, A. M. J. Am. Chem. Soc. 2012, 133, 11952–11955. doi:10.1021/ja205046h
Return to citation in text: [1] -
Wang, Y.; McGonigal, P. R.; Herlé, B.; Besora, M.; Echavarren, A. M. J. Am. Chem. Soc. 2014, 136, 801–809. doi:10.1021/ja411626v
Return to citation in text: [1] -
Seidel, G.; Fürstner, A. Angew. Chem., Int. Ed. 2014, 53, 4807–4811. doi:10.1002/anie.201402080
Return to citation in text: [1] -
Hussong, M. W.; Rominger, F.; Krämer, P.; Straub, B. F. Angew. Chem., Int. Ed. 2014, 53, 9372–9375. doi:10.1002/anie.201404032
Return to citation in text: [1] -
Doyle, M. P.; Duffy, R.; Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704–724. doi:10.1021/cr900239n
Return to citation in text: [1] -
Doyle, M. P.; McKervey, M. A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds; John Wiley & Sons: New York, 1998.
Return to citation in text: [1] -
Davies, H. M. L.; Dick, A. R. Top. Curr. Chem. 2010, 292, 303–345. doi:10.1007/128_2009_11
Return to citation in text: [1] -
Davies, H. M. L.; Manning, J. R. Nature 2008, 451, 417–424. doi:10.1038/nature06485
Return to citation in text: [1] -
Díaz-Requejo, M. M.; Pérez, P. J. Chem. Rev. 2008, 108, 3379–3394. doi:10.1021/cr078364y
Return to citation in text: [1] -
Díaz-Requejo, M. M.; Belderraín, T. R.; Nicasio, M. C.; Pérez, P. J. Dalton Trans. 2006, 5559–5566. doi:10.1039/b610183f
Return to citation in text: [1] -
Davies, H. M. L.; Beckwith, R. E. J. Chem. Rev. 2003, 103, 2861–2904. doi:10.1021/cr0200217
Return to citation in text: [1] -
Rivilla, I.; Sameera, W. M. C.; Alvarez, E.; Díaz-Requejo, M. M.; Maseras, F.; Pérez, P. J. Dalton Trans. 2013, 42, 4132–4138. doi:10.1039/c2dt32439c
Return to citation in text: [1] -
Fructos, M. R.; de Frémont, P.; Nolan, S. P.; Díaz-Requejo, M. M.; Pérez, P. J. Organometallics 2006, 25, 2237–2241. doi:10.1021/om0507474
See for the IPrAu-catalyzed alkane functionalization with EDA.
Return to citation in text: [1] -
Istrate, F. M.; Gagosz, F. Org. Lett. 2007, 9, 3181–3184. doi:10.1021/ol0713032
Return to citation in text: [1] -
Qian, J.; Liu, Y.; Cui, J.; Xu, Z. J. Org. Chem. 2012, 77, 4484–4490. doi:10.1021/jo300543n
Return to citation in text: [1] -
Nieto-Oberhuber, C.; López, S.; Muñoz, M. P.; Cárdenas, D. J.; Buñuel, E.; Nevado, C.; Echavarren, A. M. Angew. Chem., Int. Ed. 2005, 44, 6146–6148. doi:10.1002/anie.200501937
Return to citation in text: [1] [2] -
de Frémont, P.; Stevens, E. D.; Fructos, M. R.; Díaz-Requejo, M. M.; Pérez, P. J.; Nolan, S. P. Chem. Commun. 2006, 2045–2047. doi:10.1039/b601547f
Return to citation in text: [1] [2] -
de Frémont, P.; Scott, N. M.; Stevens, E. D.; Nolan, S. P. Organometallics 2005, 24, 2411–2418. doi:10.1021/om050111c
Return to citation in text: [1] -
Brookhart, M.; Grant, B.; Volpe, A. F., Jr. Organometallics 1992, 11, 3920–3922. doi:10.1021/om00059a071
Return to citation in text: [1] -
Pereira, A.; Champouret, Y.; Martín, C.; Álvarez, E.; Etienne, M.; Belderraín, T. R.; Pérez, P. J. Chem. – Eur. J. 2015, 21, 9769–9775. doi:10.1002/chem.201500776
Return to citation in text: [1]
| 34. | de Frémont, P.; Scott, N. M.; Stevens, E. D.; Nolan, S. P. Organometallics 2005, 24, 2411–2418. doi:10.1021/om050111c |
| 35. | Brookhart, M.; Grant, B.; Volpe, A. F., Jr. Organometallics 1992, 11, 3920–3922. doi:10.1021/om00059a071 |
| 36. | Pereira, A.; Champouret, Y.; Martín, C.; Álvarez, E.; Etienne, M.; Belderraín, T. R.; Pérez, P. J. Chem. – Eur. J. 2015, 21, 9769–9775. doi:10.1002/chem.201500776 |
| 1. | Teles, J. H.; Brode, S.; Chabanas, M. Angew. Chem., Int. Ed. 1998, 37, 1415–1418. doi:10.1002/(SICI)1521-3773(19980605)37:10<1415::AID-ANIE1415>3.0.CO;2-N |
| 4. | Obradors, C.; Echavarren, A. M. Chem. Commun. 2014, 50, 16–28. doi:10.1039/C3CC45518A |
| 32. | Nieto-Oberhuber, C.; López, S.; Muñoz, M. P.; Cárdenas, D. J.; Buñuel, E.; Nevado, C.; Echavarren, A. M. Angew. Chem., Int. Ed. 2005, 44, 6146–6148. doi:10.1002/anie.200501937 |
| 33. | de Frémont, P.; Stevens, E. D.; Fructos, M. R.; Díaz-Requejo, M. M.; Pérez, P. J.; Nolan, S. P. Chem. Commun. 2006, 2045–2047. doi:10.1039/b601547f |
| 4. | Obradors, C.; Echavarren, A. M. Chem. Commun. 2014, 50, 16–28. doi:10.1039/C3CC45518A |
| 5. | Xie, J.; Pan, C.; Abdukader, A.; Zhu, C. Chem. Soc. Rev. 2014, 43, 5245–5256. doi:10.1039/C4CS00004H |
| 6. | Shapiro, N. D.; Toste, F. D. Synlett 2010, 675–691. doi:10.1055/s-0029-1219369 |
| 7. | Fürstner, A. Chem. Soc. Rev. 2009, 38, 3208–3221. doi:10.1039/b816696j |
| 8. | Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395–3442. doi:10.1021/cr050041j |
| 9. | Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g |
| 10. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319 |
| 11. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Commun. 2007, 333–346. doi:10.1039/B612008C |
| 12. | Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410–3449. doi:10.1002/anie.200604335 |
| 13. | Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x |
| 14. | Lauterbach, T.; Asiri, A. M.; Hashmi, A. S. K. Adv. Organomet. Chem. 2014, 62, 261–297. doi:10.1016/B978-0-12-800976-5.00005-9 |
| 32. | Nieto-Oberhuber, C.; López, S.; Muñoz, M. P.; Cárdenas, D. J.; Buñuel, E.; Nevado, C.; Echavarren, A. M. Angew. Chem., Int. Ed. 2005, 44, 6146–6148. doi:10.1002/anie.200501937 |
| 33. | de Frémont, P.; Stevens, E. D.; Fructos, M. R.; Díaz-Requejo, M. M.; Pérez, P. J.; Nolan, S. P. Chem. Commun. 2006, 2045–2047. doi:10.1039/b601547f |
| 4. | Obradors, C.; Echavarren, A. M. Chem. Commun. 2014, 50, 16–28. doi:10.1039/C3CC45518A |
| 5. | Xie, J.; Pan, C.; Abdukader, A.; Zhu, C. Chem. Soc. Rev. 2014, 43, 5245–5256. doi:10.1039/C4CS00004H |
| 6. | Shapiro, N. D.; Toste, F. D. Synlett 2010, 675–691. doi:10.1055/s-0029-1219369 |
| 7. | Fürstner, A. Chem. Soc. Rev. 2009, 38, 3208–3221. doi:10.1039/b816696j |
| 8. | Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395–3442. doi:10.1021/cr050041j |
| 9. | Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g |
| 10. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319 |
| 11. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Commun. 2007, 333–346. doi:10.1039/B612008C |
| 12. | Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410–3449. doi:10.1002/anie.200604335 |
| 13. | Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x |
| 29. |
Fructos, M. R.; de Frémont, P.; Nolan, S. P.; Díaz-Requejo, M. M.; Pérez, P. J. Organometallics 2006, 25, 2237–2241. doi:10.1021/om0507474
See for the IPrAu-catalyzed alkane functionalization with EDA. |
| 2. | Norman, R. O. C.; Parr, W. J. E.; Thomas, C. B. J. Chem. Soc., Perkin Trans. 1 1976, 1983–1987. doi:10.1039/p19760001983 |
| 3. | Fukuda, Y.; Utimoto, K. J. Org. Chem. 1991, 56, 3729–3731. doi:10.1021/jo00011a058 |
| 30. | Istrate, F. M.; Gagosz, F. Org. Lett. 2007, 9, 3181–3184. doi:10.1021/ol0713032 |
| 31. | Qian, J.; Liu, Y.; Cui, J.; Xu, Z. J. Org. Chem. 2012, 77, 4484–4490. doi:10.1021/jo300543n |
| 20. | Hussong, M. W.; Rominger, F.; Krämer, P.; Straub, B. F. Angew. Chem., Int. Ed. 2014, 53, 9372–9375. doi:10.1002/anie.201404032 |
| 28. | Rivilla, I.; Sameera, W. M. C.; Alvarez, E.; Díaz-Requejo, M. M.; Maseras, F.; Pérez, P. J. Dalton Trans. 2013, 42, 4132–4138. doi:10.1039/c2dt32439c |
| 19. | Seidel, G.; Fürstner, A. Angew. Chem., Int. Ed. 2014, 53, 4807–4811. doi:10.1002/anie.201402080 |
| 15. | Fructos, M. R.; Belderrain, T. R.; de Fremont, P.; Scott, N. M.; Nolan, S. P.; Díaz-Requejo, M. M.; Pérez, P. J. Angew. Chem., Int. Ed. 2005, 44, 5284–5288. doi:10.1002/anie.200501056 |
| 16. | Rivilla, I.; Gómez-Emeterio, B. P.; Fructos, M. R.; Díaz-Requejo, M. M.; Pérez, P. J. Organometallics 2011, 30, 2855–2860. doi:10.1021/om200206m |
| 17. | Solorio-Alvarado, C. R.; Wang, Y.; Echavarren, A. M. J. Am. Chem. Soc. 2012, 133, 11952–11955. doi:10.1021/ja205046h |
| 18. | Wang, Y.; McGonigal, P. R.; Herlé, B.; Besora, M.; Echavarren, A. M. J. Am. Chem. Soc. 2014, 136, 801–809. doi:10.1021/ja411626v |
| 15. | Fructos, M. R.; Belderrain, T. R.; de Fremont, P.; Scott, N. M.; Nolan, S. P.; Díaz-Requejo, M. M.; Pérez, P. J. Angew. Chem., Int. Ed. 2005, 44, 5284–5288. doi:10.1002/anie.200501056 |
| 16. | Rivilla, I.; Gómez-Emeterio, B. P.; Fructos, M. R.; Díaz-Requejo, M. M.; Pérez, P. J. Organometallics 2011, 30, 2855–2860. doi:10.1021/om200206m |
| 21. | Doyle, M. P.; Duffy, R.; Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704–724. doi:10.1021/cr900239n |
| 22. | Doyle, M. P.; McKervey, M. A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds; John Wiley & Sons: New York, 1998. |
| 23. | Davies, H. M. L.; Dick, A. R. Top. Curr. Chem. 2010, 292, 303–345. doi:10.1007/128_2009_11 |
| 24. | Davies, H. M. L.; Manning, J. R. Nature 2008, 451, 417–424. doi:10.1038/nature06485 |
| 25. | Díaz-Requejo, M. M.; Pérez, P. J. Chem. Rev. 2008, 108, 3379–3394. doi:10.1021/cr078364y |
| 26. | Díaz-Requejo, M. M.; Belderraín, T. R.; Nicasio, M. C.; Pérez, P. J. Dalton Trans. 2006, 5559–5566. doi:10.1039/b610183f |
| 27. | Davies, H. M. L.; Beckwith, R. E. J. Chem. Rev. 2003, 103, 2861–2904. doi:10.1021/cr0200217 |
© 2015 Fructos et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)