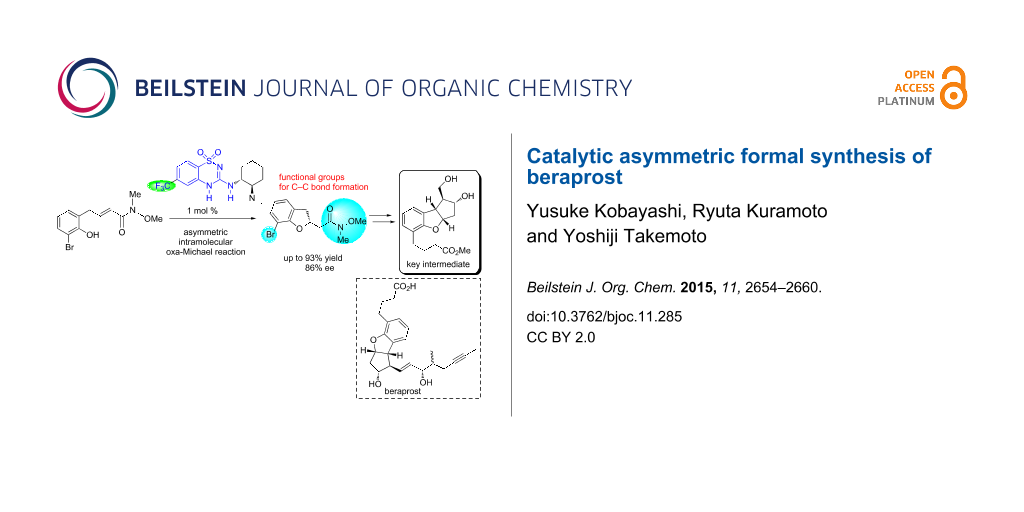
Abstract
The first catalytic asymmetric synthesis of the key intermediate for beraprost has been achieved through an enantioselective intramolecular oxa-Michael reaction of an α,β-unsaturated amide mediated by a newly developed benzothiadiazine catalyst. The Weinreb amide moiety and bromo substituent of the Michael adduct were utilized for the C–C bond formations to construct the scaffold. All four contiguous stereocenters of the tricyclic core were controlled via Rh-catalyzed stereoselective C–H insertion and the subsequent reduction from the convex face.

Graphical Abstract
Introduction
Prostacyclin (PGI2, Figure 1) is a physiologically active compound known to inhibit platelet activation and also acting as an effective vasodilator [1-3]. In addition to these properties, PGI2 derived from new vessels has attracted much attention due to its ability to promote axonal remodeling of injured neuronal networks after central nervous system disease [4,5]. However, PGI2 possesses an unstable enol ether moiety, which can be hydrolyzed even under neutral aqueous conditions, resulting in a loss of pharmacological action [6-8]. Therefore, an increasing number of more stable PGI2 derivatives have been developed. Among these, beraprost (1) has already been used as a pharmaceutical or under clinical trial in several countries for the treatment of arteriosclerosis obliterans and pulmonary hypertension [9]. Beraprost can be dosed orally as its sodium salt, and sold as a mixture of four diastereomers (1a, ent-1a, 1b, and ent-1b) [10-13], although it was reported that each of the isomers have different activities [11]. In order to reduce the adverse effects while maintaining the pharmacological activities, an effective route for the asymmetric synthesis of 1 is highly sought after, and such methodologies should also lead to the expanded clinical application of 1, as well as the development of more active derivatives.
![[1860-5397-11-285-1]](/bjoc/content/figures/1860-5397-11-285-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of PGI2 and beraprost (1).
Figure 1: Structure of PGI2 and beraprost (1).
Due to the unique tricyclic core of 1, which bears four contiguous stereocenters, various approaches for the synthesis of key intermediate 2 (Scheme 1) have been reported [14-23], including a few asymmetric syntheses relying on the optical resolution of racemic intermediates [16-18,23]. Herein we report the first catalytic asymmetric synthesis of the key intermediate 2 through organocatalyzed-enantioselective intramolecular oxa-Michael reaction [24-26].
Results and Discussion
Our retrosynthetic analysis for 2 is shown in Scheme 1, with the derivatization of 2 to beraprost (1) having already been reported. We planned to introduce the ester side chain on the aromatic ring at a later stage, utilizing radical-mediated reactions with acrylate [22] when the functional group (X) at the ortho position was methyl, or via coupling reactions with C4 units when X was a bromo substituent. The cis-fused tricyclic core of 3 was assumed to be constructed by a stereoselective C–H insertion of diazoester 4, which can be readily prepared from the Weinreb amides 5 or 6 via Claisen condensation followed by diazo-transfer reaction. The chiral dihydrobenzofuran scaffold (5 or 6) could be synthesized by asymmetric intramolecular oxa-Michael reaction (AIOM) of α,β-unsaturated amides 7 or 8. Such reactions are generally considered to be challenging due to low nucleophilicity of the oxygen nucleophile and relatively unreactive Michael acceptors [27-33]. We envisioned that our recently developed powerful hydrogen bond (HB)-donor bifunctional organocatalyst [33] could promote the desired reaction of 7 or 8, which can be synthesized from commercial sources 9 or 10. Overall, the proposed strategy offers an efficient construction of all stereocenters of tricyclic core 2, based on the initially established chiral stereocenter, as the configuration at the C1 and C2 positions of 2 would presumably be controlled by face-selective reduction of ketone 3.
![[1860-5397-11-285-i1]](/bjoc/content/inline/1860-5397-11-285-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis of beraprost (1).
Scheme 1: Retrosynthetic analysis of beraprost (1).
The Michael precursor 7 could be readily prepared from ortho-bromophenol (9, Scheme 2). O-Allylation of 9 followed by Lewis acid-mediated Claisen rearrangement afforded ortho-allylphenol 11, whose olefin moiety was ozonolyzed and subsequently treated with Wittig reagent 13 to provide amide 7 in 55% yield over four steps from 9. Amide 8 was similarly synthesized in 48% yield from 10.
![[1860-5397-11-285-i2]](/bjoc/content/inline/1860-5397-11-285-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of Michael precursors 7 and 8.
Scheme 2: Preparation of Michael precursors 7 and 8.
With the Michael precursors in hand, we next investigated the key AIOM reaction of 7 and 8 (Table 1). When the methylated substrate 7 was employed, the thiourea B [34,35] or benzothiadiazine C [33,36-38] catalysts efficiently promoted the reaction to furnish the dihydrobenzofuran 6 in 90% yield with high enantioselectivities (Table 1, entries 2 and 3). Conversely, thiourea A showed less catalytic activity, and the reaction required a much longer time for completion (Table 1, entry 1), indicating that the HB-donor moiety played an important role in facilitating the AIOM reaction. In addition, the AIOM reaction of bromo-substituted substrate 7 resulted in lower chemical yields (72–82%) and enantioselectivities (70–75% ee) even when catalysts B or C were employed for 72–120 hours (Table 1, entries 4 and 5). These results suggest that the relatively bulky bromo-substituent prevents recognition of the substrate by the catalyst. In order to improve recognition of the substrate through increased HB-donating abilities, we then tried catalyst D bearing a fluorine atom on the C6 position of the benzothiadiazine ring (Table 1, entry 6) [33]. As expected, both the chemical yield and enantioselectivity were improved, and the adduct 5 was obtained in 92% yield with 80% ee. In our preliminary DFT calculation, the HB-donor moiety would recognize an oxyanion generated from phenolic OH of substrates with tertiary amine moiety of the catalyst. It was also suggested that the SO2 moiety of benzothiadiazine catalyst would interact with the N-methyl substituent of the substrate by a non-classical hydrogen-bonding [39], improving the catalytic activities (see Supporting Information File 1 for details). The absolute configuration of 5 and 6 were assigned as (2R) by reference from the previous work [32,33]. Encouraged by this result, we next designed the new catalyst E with a stronger electron-withdrawing CF3 group on the aromatic ring, and applied it to the present AIOM reaction of 7 (Table 1, entry 7). To our delight, the enantioselectivity was improved to 85% ee while maintaining the high reactivity. Employing catalyst E we further investigated the reaction conditions and found that a scale-up synthesis can be performed using only 1 mol % of E with no loss of enantioselectivity, although gentle heating was required to ensure a high chemical yield (Table 1, entry 8).
Table 1: Optimization of asymmetric intramolecular oxa-Michael reaction.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-285-i5.svg?max-width=637&scale=1.0)
|
|||||||
| entry | X | cat. | y | temp | time (h) | yield (%)a | ee (%)b |
|---|---|---|---|---|---|---|---|
| 1 | Me | A | 10 | rt | 192 | 95 | 90 |
| 2 | Me | B | 10 | rt | 72 | 90 | 91 |
| 3 | Me | C | 10 | rt | 72 | 90 | 93 |
| 4 | Br | B | 10 | rt | 72 | 82 | 75 |
| 5 | Br | C | 10 | rt | 120 | 72 | 70 |
| 6 | Br | D | 10 | rt | 120 | 92 | 80 |
| 7 | Br | E | 10 | rt | 96 | 83 | 85 |
| 8 | Br | E | 1 | 35 | 96 | 93 | 86 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-285-i6.svg?max-width=637&scale=1.18182)
aIsolated yields. bDetermined by HPLC.
With both the AIOM adducts 5 and 6 in hand, we next investigated the construction of the tricyclic core (Scheme 3 and Scheme 4). The cross-Claisen condensation of 6 with lithium tert-butyl acetate afforded the corresponding β-ketoester, which was then treated with 2-azido-1,3-dimethylimidazolinium hexafluorophosphate (ADMP) [40-42] to give the diazoester 14. Rhodium catalysed C–H insertion [43,44] of 14 proceeded smoothly to furnish the tricyclic ketoester, which was found to be unstable to purification on column chromatography, presumably due to decomposition of the ketoester moiety. Therefore, the product was isolated as alcohol 15 after a one-pot reduction of the ketone moiety (60% in four steps from 6), along with the minor diastereomer at C1 position (dr > 18:1). These results mean that the stereochemistry at the C2 position was fully controlled, presumably due to hydride attack from the less-hindered convex face. The relative configuration of 15 was unambiguously determined by NOESY analysis (see Supporting Information File 1 for details). As all four desired stereocenters were constructed, we next investigated the introduction of the ester side chain on the aromatic ring via benzylic bromination followed by elongation of the C3 unit [22]. To this end, the ester group at the C1 position of 15 was reduced by lithium borohydride, and the resultant 1,3-diol protected to give acetal 16 [16]. After various experiments, selective bromination of the methyl group on the aromatic ring of 16, however, was found to be difficult due to competitive bromination of the electron-rich aromatic ring, and thus the desired bromide 17 was obtained in only 14% yield.
![[1860-5397-11-285-i3]](/bjoc/content/inline/1860-5397-11-285-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: First attempt at the synthesis of 2 from 6.
Scheme 3: First attempt at the synthesis of 2 from 6.
![[1860-5397-11-285-i4]](/bjoc/content/inline/1860-5397-11-285-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Achievement of a formal synthesis of 2.
Scheme 4: Achievement of a formal synthesis of 2.
We then investigated an alternative route from adduct 5, even though the enantiomeric excess of 5 (86% ee) was a little lower than that of adduct 6 (93% ee) (Scheme 4). Fortunately the diazoester 20, as similarly derived as in Scheme 3, was obtained as a crystalline solid, and one recrystallization increased the ee to 95%. The tricyclic scaffold 21 with all four stereocenters of the desired configuration was synthesized in 67% yield (dr > 10:1) via the method established in Scheme 3. After derivatization to acetal 22 in 2 steps, we then turned our attention to the introduction of the C4 ester substituents. Amongst various different conditions investigated – including Pd-catalyzed coupling reactions – a halogen-lithium exchange and subsequent addition to methyl 4-oxobutanoate was found to be the best method to introduce the C4 subunit with reproducibility in the case of scale-up synthesis. Deprotection of the resultant ester 23 followed by reduction of the benzylic OH group finally afforded the key intermediate 2.
Conclusion
We have developed the first asymmetric catalytic synthesis of the key intermediate for beraprost in 14 steps, via an organocatalyzed AIOM reaction of α,β-unsaturated amides. During the course of this study, it was revealed that a bromo substituent ortho to the phenolic OH group significantly decreased the reactivity and enantioselectivity. However, we found that the newly developed organocatalyst E, bearing increased HB-donating abilities, could improve both the reactivity and selectivity. In addition, the Weinreb amide moieties of the AIOM adduct were shown to be efficiently converted to β-ketoesters and diazoesters, a reactivity that could be further extended to various other molecular transformations. We believe that these findings could be applied to the synthesis of other biologically active oxo-heterocycles, and thus this is currently under investigation in our laboratory and will be reported in due course.
Experimental
General procedure for asymmetric oxa-Michael reaction
The benzothiadiazine catalyst E (8.6 mg, 0.022 mmol, 1 mol %) was added to a solution of 7 (661 mg, 2.20 mmol) in CH2Cl2 (20 mL), and the resulting mixture was stirred at 35 ºC for 96 h. The reaction mixture was then evaporated and the resulting crude residue purified by column chromatography on silica gel eluting with n-hexane/ethyl acetate (60/40) to give the analytically pure compound 5 (614 mg, 93%). The enantiomeric ratio was determined by HPLC on a chiral stationary phase (86% ee).
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data. | ||
| Format: PDF | Size: 1.9 MB | Download |
References
-
Moncada, S.; Gryglewski, R.; Bunting, S.; Vane, J. R. Nature 1976, 263, 663. doi:10.1038/263663a0
Return to citation in text: [1] -
Johnson, R. A.; Morton, D. R.; Kinner, J. H.; Gorman, R. R.; McGuire, J. C.; Sun, F. F.; Whittaker, N.; Bunting, S.; Salmon, J.; Moncada, S.; Vane, J. R. Prostaglandins 1976, 12, 915. doi:10.1016/0090-6980(76)90126-X
Return to citation in text: [1] -
Johnson, R. A.; Lincoln, F. H.; Nidy, E. G.; Schneider, W. P.; Thompson, J. L.; Axen, U. J. Am. Chem. Soc. 1978, 100, 7690. doi:10.1021/ja00492a043
Return to citation in text: [1] -
Muramatsu, R.; Takahashi, C.; Miyake, S.; Fujimura, H.; Mochizuki, H.; Yamashita, T. Nat. Med. 2012, 18, 1658. doi:10.1038/nm.2943
Return to citation in text: [1] -
Takahashi, C.; Muramatsu, R.; Fujimura, H.; Mochizuki, H.; Yamashita, T. Cell Death Dis. 2013, 4, e795. doi:10.1038/cddis.2013.335
Return to citation in text: [1] -
Nickolson, R. C.; Town, M. H.; Vorbrüggen, H. Med. Res. Rev. 1985, 5, 1. doi:10.1002/med.2610050102
Return to citation in text: [1] -
Ansoff, P. A. Adv. Prostaglandin, Thromboxane, Leukotriene Res. 1985, 14, 309.
Return to citation in text: [1] -
Collins, P. W.; Djuric, S. W. Chem. Rev. 1993, 93, 1533. doi:10.1021/cr00020a007
Return to citation in text: [1] -
Nagase, H.; Matsumoto, K.; Nishiyama, H. J. Synth. Org. Chem., Jpn. 1996, 54, 1055. doi:10.5059/yukigoseikyokaishi.54.1055
Return to citation in text: [1] -
Nishio, S.; Nagase, H.; Kanou, K.; Aoki, S.; Kanbayashi, Y. Yakugaku Zasshi 1997, 117, 509.
Return to citation in text: [1] -
Wakita, H.; Yoshiwara, H.; Nishiyama, H.; Nagase, H. Heterocycles 2000, 53, 1085. doi:10.3987/COM-00-8868
Return to citation in text: [1] [2] -
Das, S.; Chandrasekhar, S.; Yadav, J. S.; Grée, R. Chem. Rev. 2007, 107, 3286. doi:10.1021/cr068365a
Return to citation in text: [1] -
Jahn, U.; Galano, J.-M.; Durand, T. Angew. Chem., Int. Ed. 2008, 47, 5894. doi:10.1002/anie.200705122
Return to citation in text: [1] -
Ohno, K.; Nagase, H.; Matsumoto, K.; Nishio, S. 5,6,7-Trinor-4,8-inter-m-phenylene prostaglandin I2 derivative. Eur. Pat. Appl. EP0084856 A1, Aug 3, 1983.
Return to citation in text: [1] -
Ohno, K.; Nishiyama, H.; Nagase, H.; Matsumoto, K.; Ishikawa, M. Tetrahedron Lett. 1990, 31, 4489. doi:10.1016/S0040-4039(00)97656-1
Return to citation in text: [1] -
Nagase, H.; Matsumoto, K.; Yoshiwara, H.; Tajima, A.; Ohno, K. Tetrahedron Lett. 1990, 31, 4493. doi:10.1016/S0040-4039(00)97657-3
Return to citation in text: [1] [2] [3] -
Wakita, H.; Matsumoto, K.; Yoshiwara, H.; Hosono, Y.; Hayashi, R.; Nishiyama, H.; Nagase, H. Tetrahedron 1999, 55, 2449. doi:10.1016/S0040-4020(99)00020-4
Return to citation in text: [1] [2] -
Wakita, H.; Yoshiwara, H.; Kitano, Y.; Nishiyama, H.; Nagase, H. Tetrahedron: Asymmetry 2000, 11, 2981. doi:10.1016/S0957-4166(00)00251-2
Return to citation in text: [1] [2] -
Larock, R. C.; Lee, N. H. J. Org. Chem. 1991, 56, 6253. doi:10.1021/jo00022a002
Return to citation in text: [1] -
Yoshida, Y.; Sato, Y.; Okamoto, S.; Sato, F. J. Chem. Soc., Chem. Commun. 1995, 811. doi:10.1039/c39950000811
Return to citation in text: [1] -
Wakita, H.; Yoshiwara, H.; Tajima, A.; Kitano, Y.; Nagase, H. Tetrahedron: Asymmetry 1999, 10, 4099. doi:10.1016/S0957-4166(99)00442-5
Return to citation in text: [1] -
Higuchi, K.; Sawada, K.; Nambu, H.; Shogaki, T.; Kita, Y. Org. Lett. 2003, 5, 3703. doi:10.1021/ol035371+
Return to citation in text: [1] [2] [3] -
Reddy, N. K.; Vijaykumar, B. V. D.; Chandrasekhar, S. Org. Lett. 2012, 14, 299. doi:10.1021/ol203060v
Return to citation in text: [1] [2] -
Nising, C. F.; Bräse, S. Chem. Soc. Rev. 2008, 37, 1218. doi:10.1039/b718357g
Return to citation in text: [1] -
Nising, C. F.; Bräse, S. Chem. Soc. Rev. 2012, 41, 988. doi:10.1039/C1CS15167C
Return to citation in text: [1] -
Hintermann, L. Top. Organomet. Chem. 2010, 31, 123. doi:10.1007/978-3-642-12073-2_6
Return to citation in text: [1] -
Merschaert, A.; Delbeke, P.; Daloze, D.; Dive, G. Tetrahedron Lett. 2004, 45, 4697. doi:10.1016/j.tetlet.2004.04.090
Return to citation in text: [1] -
Saito, N.; Ryoda, A.; Nakanishi, W.; Kumamoto, T.; Ishikawa, T. Eur. J. Org. Chem. 2008, 2759. doi:10.1002/ejoc.200800089
Return to citation in text: [1] -
Gioia, C.; Fini, F.; Mazzanti, A.; Bernardi, L.; Ricci, A. J. Am. Chem. Soc. 2009, 131, 9614. doi:10.1021/ja902458m
Return to citation in text: [1] -
Tokunou, S.; Nakanishi, W.; Kagawa, N.; Kumamoto, T.; Ishikawa, T. Heterocycles 2012, 84, 1045. doi:10.3987/COM-11-S(P)82
Return to citation in text: [1] -
Hintermann, L.; Dittmer, C. Eur. J. Org. Chem. 2012, 5573. doi:10.1002/ejoc.201200838
Return to citation in text: [1] -
Hintermann, L.; Ackerstaff, J.; Boeck, F. Chem. – Eur. J. 2013, 19, 2311. doi:10.1002/chem.201203505
Return to citation in text: [1] [2] -
Kobayashi, Y.; Taniguchi, Y.; Hayama, N.; Inokuma, T.; Takemoto, Y. Angew. Chem., Int. Ed. 2013, 52, 11114. doi:10.1002/anie.201305492
Return to citation in text: [1] [2] [3] [4] [5] -
Doyle, A. G.; Jacobsen, E. N. Chem. Rev. 2007, 107, 5713. doi:10.1021/cr068373r
Return to citation in text: [1] -
Takemoto, Y. Chem. Pharm. Bull. 2010, 58, 593. doi:10.1248/cpb.58.593
Return to citation in text: [1] -
Inokuma, T.; Furukawa, M.; Uno, T.; Suzuki, Y.; Yoshida, K.; Yano, Y.; Matsuzaki, K.; Takemoto, Y. Chem. – Eur. J. 2011, 17, 10470. doi:10.1002/chem.201101338
Return to citation in text: [1] -
Inokuma, T.; Furukawa, M.; Suzuki, Y.; Kimachi, T.; Kobayashi, Y.; Takemoto, Y. ChemCatChem 2012, 4, 983. doi:10.1002/cctc.201200065
Return to citation in text: [1] -
Kobayashi, Y.; Li, S.; Takemoto, Y. Asian J. Org. Chem. 2014, 3, 403. doi:10.1002/ajoc.201400031
Return to citation in text: [1] -
Johnston, R. C.; Cheong, P. H.-Y. Org. Biomol. Chem. 2013, 11, 5057. doi:10.1039/c3ob40828k
Return to citation in text: [1] -
Kitamura, M.; Tashiro, N.; Okauchi, T. Synlett 2009, 2943. doi:10.1055/s-0029-1218269
Return to citation in text: [1] -
Kitamura, M.; Tashiro, N.; Miyagawa, S.; Okauchi, T. Synthesis 2011, 1037. doi:10.1055/s-0030-1258457
Return to citation in text: [1] -
Kitamura, M. J. Synth. Org. Chem., Jpn. 2014, 72, 14. doi:10.5059/yukigoseikyokaishi.72.14
Return to citation in text: [1] -
Davies, H. M. L.; Manning, J. R. Nature 2008, 451, 417. doi:10.1038/nature06485
Return to citation in text: [1] -
Doyle, M. P.; Duffy, R.; Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704. doi:10.1021/cr900239n
Return to citation in text: [1]
| 43. | Davies, H. M. L.; Manning, J. R. Nature 2008, 451, 417. doi:10.1038/nature06485 |
| 44. | Doyle, M. P.; Duffy, R.; Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704. doi:10.1021/cr900239n |
| 32. | Hintermann, L.; Ackerstaff, J.; Boeck, F. Chem. – Eur. J. 2013, 19, 2311. doi:10.1002/chem.201203505 |
| 33. | Kobayashi, Y.; Taniguchi, Y.; Hayama, N.; Inokuma, T.; Takemoto, Y. Angew. Chem., Int. Ed. 2013, 52, 11114. doi:10.1002/anie.201305492 |
| 40. | Kitamura, M.; Tashiro, N.; Okauchi, T. Synlett 2009, 2943. doi:10.1055/s-0029-1218269 |
| 41. | Kitamura, M.; Tashiro, N.; Miyagawa, S.; Okauchi, T. Synthesis 2011, 1037. doi:10.1055/s-0030-1258457 |
| 42. | Kitamura, M. J. Synth. Org. Chem., Jpn. 2014, 72, 14. doi:10.5059/yukigoseikyokaishi.72.14 |
| 1. | Moncada, S.; Gryglewski, R.; Bunting, S.; Vane, J. R. Nature 1976, 263, 663. doi:10.1038/263663a0 |
| 2. | Johnson, R. A.; Morton, D. R.; Kinner, J. H.; Gorman, R. R.; McGuire, J. C.; Sun, F. F.; Whittaker, N.; Bunting, S.; Salmon, J.; Moncada, S.; Vane, J. R. Prostaglandins 1976, 12, 915. doi:10.1016/0090-6980(76)90126-X |
| 3. | Johnson, R. A.; Lincoln, F. H.; Nidy, E. G.; Schneider, W. P.; Thompson, J. L.; Axen, U. J. Am. Chem. Soc. 1978, 100, 7690. doi:10.1021/ja00492a043 |
| 10. | Nishio, S.; Nagase, H.; Kanou, K.; Aoki, S.; Kanbayashi, Y. Yakugaku Zasshi 1997, 117, 509. |
| 11. | Wakita, H.; Yoshiwara, H.; Nishiyama, H.; Nagase, H. Heterocycles 2000, 53, 1085. doi:10.3987/COM-00-8868 |
| 12. | Das, S.; Chandrasekhar, S.; Yadav, J. S.; Grée, R. Chem. Rev. 2007, 107, 3286. doi:10.1021/cr068365a |
| 13. | Jahn, U.; Galano, J.-M.; Durand, T. Angew. Chem., Int. Ed. 2008, 47, 5894. doi:10.1002/anie.200705122 |
| 33. | Kobayashi, Y.; Taniguchi, Y.; Hayama, N.; Inokuma, T.; Takemoto, Y. Angew. Chem., Int. Ed. 2013, 52, 11114. doi:10.1002/anie.201305492 |
| 9. | Nagase, H.; Matsumoto, K.; Nishiyama, H. J. Synth. Org. Chem., Jpn. 1996, 54, 1055. doi:10.5059/yukigoseikyokaishi.54.1055 |
| 39. | Johnston, R. C.; Cheong, P. H.-Y. Org. Biomol. Chem. 2013, 11, 5057. doi:10.1039/c3ob40828k |
| 6. | Nickolson, R. C.; Town, M. H.; Vorbrüggen, H. Med. Res. Rev. 1985, 5, 1. doi:10.1002/med.2610050102 |
| 7. | Ansoff, P. A. Adv. Prostaglandin, Thromboxane, Leukotriene Res. 1985, 14, 309. |
| 8. | Collins, P. W.; Djuric, S. W. Chem. Rev. 1993, 93, 1533. doi:10.1021/cr00020a007 |
| 34. | Doyle, A. G.; Jacobsen, E. N. Chem. Rev. 2007, 107, 5713. doi:10.1021/cr068373r |
| 35. | Takemoto, Y. Chem. Pharm. Bull. 2010, 58, 593. doi:10.1248/cpb.58.593 |
| 4. | Muramatsu, R.; Takahashi, C.; Miyake, S.; Fujimura, H.; Mochizuki, H.; Yamashita, T. Nat. Med. 2012, 18, 1658. doi:10.1038/nm.2943 |
| 5. | Takahashi, C.; Muramatsu, R.; Fujimura, H.; Mochizuki, H.; Yamashita, T. Cell Death Dis. 2013, 4, e795. doi:10.1038/cddis.2013.335 |
| 33. | Kobayashi, Y.; Taniguchi, Y.; Hayama, N.; Inokuma, T.; Takemoto, Y. Angew. Chem., Int. Ed. 2013, 52, 11114. doi:10.1002/anie.201305492 |
| 36. | Inokuma, T.; Furukawa, M.; Uno, T.; Suzuki, Y.; Yoshida, K.; Yano, Y.; Matsuzaki, K.; Takemoto, Y. Chem. – Eur. J. 2011, 17, 10470. doi:10.1002/chem.201101338 |
| 37. | Inokuma, T.; Furukawa, M.; Suzuki, Y.; Kimachi, T.; Kobayashi, Y.; Takemoto, Y. ChemCatChem 2012, 4, 983. doi:10.1002/cctc.201200065 |
| 38. | Kobayashi, Y.; Li, S.; Takemoto, Y. Asian J. Org. Chem. 2014, 3, 403. doi:10.1002/ajoc.201400031 |
| 24. | Nising, C. F.; Bräse, S. Chem. Soc. Rev. 2008, 37, 1218. doi:10.1039/b718357g |
| 25. | Nising, C. F.; Bräse, S. Chem. Soc. Rev. 2012, 41, 988. doi:10.1039/C1CS15167C |
| 26. | Hintermann, L. Top. Organomet. Chem. 2010, 31, 123. doi:10.1007/978-3-642-12073-2_6 |
| 27. | Merschaert, A.; Delbeke, P.; Daloze, D.; Dive, G. Tetrahedron Lett. 2004, 45, 4697. doi:10.1016/j.tetlet.2004.04.090 |
| 28. | Saito, N.; Ryoda, A.; Nakanishi, W.; Kumamoto, T.; Ishikawa, T. Eur. J. Org. Chem. 2008, 2759. doi:10.1002/ejoc.200800089 |
| 29. | Gioia, C.; Fini, F.; Mazzanti, A.; Bernardi, L.; Ricci, A. J. Am. Chem. Soc. 2009, 131, 9614. doi:10.1021/ja902458m |
| 30. | Tokunou, S.; Nakanishi, W.; Kagawa, N.; Kumamoto, T.; Ishikawa, T. Heterocycles 2012, 84, 1045. doi:10.3987/COM-11-S(P)82 |
| 31. | Hintermann, L.; Dittmer, C. Eur. J. Org. Chem. 2012, 5573. doi:10.1002/ejoc.201200838 |
| 32. | Hintermann, L.; Ackerstaff, J.; Boeck, F. Chem. – Eur. J. 2013, 19, 2311. doi:10.1002/chem.201203505 |
| 33. | Kobayashi, Y.; Taniguchi, Y.; Hayama, N.; Inokuma, T.; Takemoto, Y. Angew. Chem., Int. Ed. 2013, 52, 11114. doi:10.1002/anie.201305492 |
| 16. | Nagase, H.; Matsumoto, K.; Yoshiwara, H.; Tajima, A.; Ohno, K. Tetrahedron Lett. 1990, 31, 4493. doi:10.1016/S0040-4039(00)97657-3 |
| 17. | Wakita, H.; Matsumoto, K.; Yoshiwara, H.; Hosono, Y.; Hayashi, R.; Nishiyama, H.; Nagase, H. Tetrahedron 1999, 55, 2449. doi:10.1016/S0040-4020(99)00020-4 |
| 18. | Wakita, H.; Yoshiwara, H.; Kitano, Y.; Nishiyama, H.; Nagase, H. Tetrahedron: Asymmetry 2000, 11, 2981. doi:10.1016/S0957-4166(00)00251-2 |
| 23. | Reddy, N. K.; Vijaykumar, B. V. D.; Chandrasekhar, S. Org. Lett. 2012, 14, 299. doi:10.1021/ol203060v |
| 33. | Kobayashi, Y.; Taniguchi, Y.; Hayama, N.; Inokuma, T.; Takemoto, Y. Angew. Chem., Int. Ed. 2013, 52, 11114. doi:10.1002/anie.201305492 |
| 14. | Ohno, K.; Nagase, H.; Matsumoto, K.; Nishio, S. 5,6,7-Trinor-4,8-inter-m-phenylene prostaglandin I2 derivative. Eur. Pat. Appl. EP0084856 A1, Aug 3, 1983. |
| 15. | Ohno, K.; Nishiyama, H.; Nagase, H.; Matsumoto, K.; Ishikawa, M. Tetrahedron Lett. 1990, 31, 4489. doi:10.1016/S0040-4039(00)97656-1 |
| 16. | Nagase, H.; Matsumoto, K.; Yoshiwara, H.; Tajima, A.; Ohno, K. Tetrahedron Lett. 1990, 31, 4493. doi:10.1016/S0040-4039(00)97657-3 |
| 17. | Wakita, H.; Matsumoto, K.; Yoshiwara, H.; Hosono, Y.; Hayashi, R.; Nishiyama, H.; Nagase, H. Tetrahedron 1999, 55, 2449. doi:10.1016/S0040-4020(99)00020-4 |
| 18. | Wakita, H.; Yoshiwara, H.; Kitano, Y.; Nishiyama, H.; Nagase, H. Tetrahedron: Asymmetry 2000, 11, 2981. doi:10.1016/S0957-4166(00)00251-2 |
| 19. | Larock, R. C.; Lee, N. H. J. Org. Chem. 1991, 56, 6253. doi:10.1021/jo00022a002 |
| 20. | Yoshida, Y.; Sato, Y.; Okamoto, S.; Sato, F. J. Chem. Soc., Chem. Commun. 1995, 811. doi:10.1039/c39950000811 |
| 21. | Wakita, H.; Yoshiwara, H.; Tajima, A.; Kitano, Y.; Nagase, H. Tetrahedron: Asymmetry 1999, 10, 4099. doi:10.1016/S0957-4166(99)00442-5 |
| 22. | Higuchi, K.; Sawada, K.; Nambu, H.; Shogaki, T.; Kita, Y. Org. Lett. 2003, 5, 3703. doi:10.1021/ol035371+ |
| 23. | Reddy, N. K.; Vijaykumar, B. V. D.; Chandrasekhar, S. Org. Lett. 2012, 14, 299. doi:10.1021/ol203060v |
| 22. | Higuchi, K.; Sawada, K.; Nambu, H.; Shogaki, T.; Kita, Y. Org. Lett. 2003, 5, 3703. doi:10.1021/ol035371+ |
| 11. | Wakita, H.; Yoshiwara, H.; Nishiyama, H.; Nagase, H. Heterocycles 2000, 53, 1085. doi:10.3987/COM-00-8868 |
| 22. | Higuchi, K.; Sawada, K.; Nambu, H.; Shogaki, T.; Kita, Y. Org. Lett. 2003, 5, 3703. doi:10.1021/ol035371+ |
| 16. | Nagase, H.; Matsumoto, K.; Yoshiwara, H.; Tajima, A.; Ohno, K. Tetrahedron Lett. 1990, 31, 4493. doi:10.1016/S0040-4039(00)97657-3 |
© 2015 Kobayashi et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)