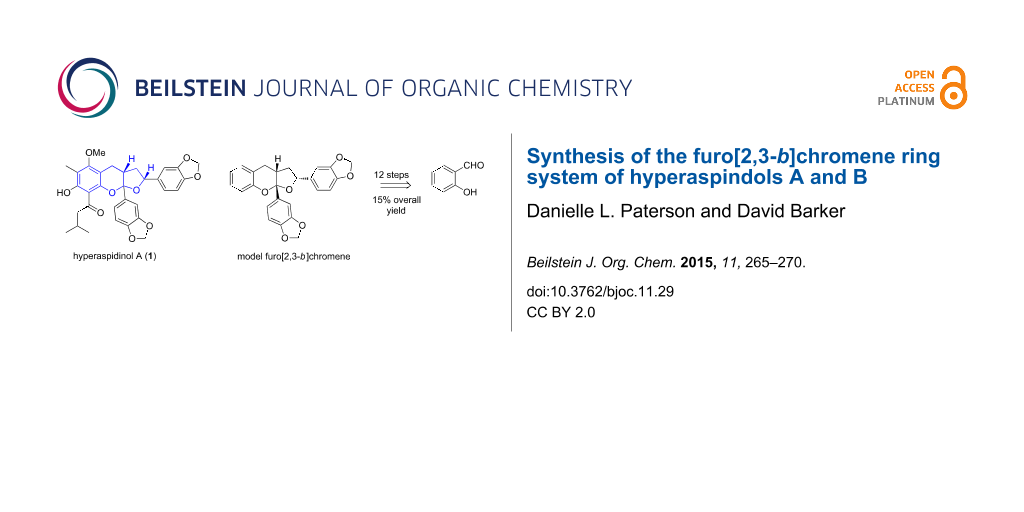
Abstract
The synthesis of the unique furo[2,3-b]chromene ring system found in hyperaspidinols A and B, acylphloroglucinols from Hypericum chinense has been achieved in twelve steps. By comparison of the NMR spectra of the synthesized compounds with those of the natural products, a relative stereochemistry is suggested, especially that of the ketal carbon.

Graphical Abstract
Introduction
Two novel racemic acylphoroglucinols, hyperaspidinols A (1) and B (2) (Figure 1) were recently isolated from the leaves of Hypericum chinense, a member of the St John’s Wort plants, which contains 490 flowering plants [1]. These plants have been used medicinally for treating illnesses such as hepatitis and depression, and as topical antimicrobials for wounds and snake bites [2-5]. There is great interest in secondary metabolites produced by plants from the Hypericum genus due to the bioactivity of many compounds that have been isolated from this source. A wide variety of compounds have been isolated from H. chinense including prenylated acylphloroglucinols such as chinesins I (3) and II (4), xanthones, flavonoids, terpenoids, naphthodianthrones, and norlignans, such as hyperione A (5) and B (6) [1,6-8]. Chinesins I (3) and II (4) are acylphloroglucinol derivatives which possess antibacterial and antiviral activities, as well as inhibitory activity on thromboxane A2 and leukotriene D4 [4]. Acylphloroglucinols are known to act as anti-oxidants, by reducing hydroperoxides and hydrogen peroxide, thereby suppressing the formation of the reactive species [9].
![[1860-5397-11-29-1]](/bjoc/content/figures/1860-5397-11-29-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Hyperaspidinols A (1) and B (2) and other compounds 3-6 from Hypericum chinense.
Figure 1: Hyperaspidinols A (1) and B (2) and other compounds 3-6 from Hypericum chinense.
The hyperaspidinols 1 and 2 both possess a highly functionalised furo[2,3-b]chromene ring system (Figure 2, highlighted in blue) and differ only with the nature of the ketone side-chain. Furo[2,3-b]chromenes have not been reported in any other natural products to date with the closest related system being the chromeno[2,3-b]chromenes. Compound which contain this motif, such as albanol A and australisine A, display potent bioactive properties including hypotensive, anticancer, antimicrobial and antimalarial activity [10-12].
![[1860-5397-11-29-2]](/bjoc/content/figures/1860-5397-11-29-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Hyperaspidinol A (1), target compound 7 and proposed precursors.
Figure 2: Hyperaspidinol A (1), target compound 7 and proposed precursors.
The unique ring system found in the hyperaspidinols combined with the potent biological activities of similar structures led us to explore routes to the carboheterocyclic skeleton of these compounds. The relative stereochemistry of the hyperaspidinols was determined using a variety of NMR spectroscopic techniques. Whilst the isolation paper reported a weak ROESY interaction between H-8 and H-10, suggesting a cis configuration between these protons, the relationship of the aryl substituents could not be determined (Figure 2) [1]. Our present aim was therefore to prepare furo[2,3-b]chromene 7 which contains the unique ring system and aryl subsitutents found in hyperaspidinols 1 and 2 and hopefully use it to determine the complete relative stereochemistry of the natural products.
Results and Discussion
The proposed route to furo[2,3-b]chromene 7 was based around the preparation of benzylic ketone 8 which was hoped to under acidic conditions would undergo cyclisation to give 7 (Figure 2). We have previously reported the formation of a number of diaryl tetrahydrofuran lignans under acidic or buffered conditions utilising the high reactivity of electron-rich benzylic alcohols to assist in the rapid and high yielding formation of the tetrahydrofuran ring [13-18]. The two methylenedioxyphenyl groups in ketone 8 would be added by sequential addition of aryllithiates to both the carbonyl and aldehyde groups, derived from the terminal alkene, of a homoallylic carboxylic acid derivative 9.
Our route to ketone 8 began from salicylaldehyde (10) which underwent a Horner–Wadsworth–Emmons reaction with triethylphosphonoacetate to give ester 11 [19] in 94% yield (Scheme 1). Hydrogenation of the alkene followed by protection of the phenol gave benzyl ether 12 in 88% yield over two steps. Initially the preparation of alkene 13 through the allylation of ester 12 was attempted, however keto-ester 14 resulting from an additional Claisen condensation was the only isolated product.
![[1860-5397-11-29-i1]](/bjoc/content/inline/1860-5397-11-29-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reagents and conditions: (i) triethylphosphonoacetate, DBU, THF, 48 h, 94%; (ii) H2, 10% Pd/C, EtOAc, 3 h, quant; (iii) BnBr, K2CO3, DMF, 3 h, 88%; (iv) LDA, THF, −78 °C, then allyl bromide, 24 h, 14%.
Scheme 1: Reagents and conditions: (i) triethylphosphonoacetate, DBU, THF, 48 h, 94%; (ii) H2, 10% Pd/C, EtOA...
It was therefore decided to functionalise the ester moiety first. This was achieved by direct conversion of ester 12 into Weinreb amide 15 in 81% yield, followed by addition of lithiate 16 (formed from 1-bromo-3,4-methylenedioxybenzene and t-butyllithium) to give ketone 17 in 84% yield (Scheme 2). Allylation of ketone 17 proved problematic with the use of strong bases such as LDA and LiHMDS, giving none of the desired product. However, the use of allyl bromide and NaH in the presence of TBAI, in THF at reflux, gave the desired alkene 18 in 95% yield. Protection of the ketone in 18 as the cyclic ketal 19 was achieved in 44% yield using excess ethylene glycol and pTSA and despite attempting a variety of alternate conditions [20-23] this yield could not be improved without degradation of both the starting material 18 and product 19. Dihydroxylation of 19, followed by oxidative cleavage of the resultant diol gave aldehyde 20 in 80% yield over two steps. Addition of lithiate 16 to aldehyde 20 gave alcohol 21 in 87% yield as an inseparable 1:1 mixture of diastereoisomers. Hydrogenolysis of the benzyl ether in 21 gave ketal-diol 22 in quantitative yield. Finally, stirring of ketal 22 in a 1:1 2 M HCl (aq):THF resulted in removal of the ketal protecting group, giving ketone 8, which under the acidic condition immediately cyclized giving furo[2,3-b]chromenes 7a and 7b in a 1.6:1 ratio, in an overall 84% yield. Separation of the diastereoisomers 7a and 7b was achieved using column chromatography and allowed characterisation of the individual isomers.
![[1860-5397-11-29-i2]](/bjoc/content/inline/1860-5397-11-29-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reagents and conditions: (i) H3C(CH3O)NH·HCl, n-BuLi, THF, −78 °C, 4 h, 81%; (ii) 1-bromo-3,4-methylenedioxybenzene, t-BuLi, THF, −78 °C, 3 h, 84%; (iii) NaH, allyl bromide, TBAI, THF, 65 °C, 20 h, 95%; (iv) ethylene glycol, pTSA, toluene, reflux, 16 h, 44%; (v) OsO4, NMO, t-BuOH, H2O, THF, 65 h, 80%; (vi) NaIO4, MeOH, H2O, 3 h, quant; (vii) 1-bromo-3,4-methylenedioxybenzene, t-BuLi, THF, −78 °C, 19 h, 87% 1:1 diastereoisomers; (viii) H2, 10% Pd/C, MeOH, 4 h, quant; (ix) 1:1 2 M HCl (aq):THF, 22 h, 7a 52%, 7b 32%.
Scheme 2: Reagents and conditions: (i) H3C(CH3O)NH·HCl, n-BuLi, THF, −78 °C, 4 h, 81%; (ii) 1-bromo-3,4-methy...
Comparison of the 1H and 13C NMR data of isomers 7a and 7b showed strong similarities between 7a and the reported data [1] for hyperaspidinols A (1) and B (2) whilst isomer 7b showed clear differences in both the chemical shift and multiplicities in the furo[2,3-b]chromene rings (see Supporting Information File 1 for a complete table of NMR data).
Extensive use of 2D NMR techniques, in particular a NOESY, allowed the complete relative stereochemistry of both 7a and 7b to be determined (Figure 3). In isomer 7a NOESY correlations between H-8 and H-10 showed the syn relationship between these two protons. An additional NOESY correlation between H-8 and H-2′ showed the syn relationship between these two groups and thus the trans relationship between the two methylenedioxyphenyl groups at C-10 and C-11.
![[1860-5397-11-29-3]](/bjoc/content/figures/1860-5397-11-29-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: NOESY correlations in isomers 7a and 7b.
Figure 3: NOESY correlations in isomers 7a and 7b.
The NOESY spectrum of 7b exhibited a NOESY correlation between H-8 and H-2′ showed the syn relationship between these two groups but no correlation between H-8 and H-10 was observed, suggesting 7b to be the C-10 epimer of 7a. A correlation of H-10 to the H-9b, which was on the opposite face to H-8 further suggested this. Further analysis of the conformations of 7a and 7b was achieved by examination of the coupling constants in the furo[2,3-b]chromene ring. The use of coupling constants to determine stereochemistry in five-membered rings is often extremely difficult, due to the large number of conformations available [24-26]. However, with the relatively fixed geometry for the five membered ring in 7a and 7b due to the fused chroman ring and the quarternary centre at C-11, analysis of the coupling constants was considered viable. In isomer 7a the coupling constant between H-9a and both H-8 and H-10 was 6.5 Hz, which corresponds to a ca. 50° dihedral angle between the protons, placing all three on the same face of the five membered ring (Figure 4). Additionally H-9b had coupling constants of 10.5 Hz and 12.6 Hz between H-8 and H-10, respectively, showing an anti-relationship between these protons [25]. In isomer 7b the coupling constant between H-8 and H-9b (9.5 Hz) represent a ca. 170° dihedral angle between these protons whilst the coupling constant between H-9b and H-10 (4.5 Hz) represent a ca. 40° dihedral angle or syn relationship between these two protons (Figure 5) [26]. Overall this represents an anti-relationship between H-8 and H-10 confirming 7b to be the C-10 epimer of 7a.
![[1860-5397-11-29-4]](/bjoc/content/figures/1860-5397-11-29-4.png?scale=1.5&max-width=1024&background=FFFFFF)
![[1860-5397-11-29-5]](/bjoc/content/figures/1860-5397-11-29-5.png?scale=1.5&max-width=1024&background=FFFFFF)
The ratio of diastereoisomers in ketal 22 was 1:1, however in the cyclised products 7 the ratio of diastereoisomers 7a to 7b was 1.6:1. This suggests partial isomerisation of the stereochemistry at C-10 during the formation of the furo[2,3-b]chromene. Indeed, one possible mechanism for the formation of isomers 7a and 7b is initial protonation of ketone 8, attack of phenol on the activated carbonyl resulting in formation of chroman-hemiketal 23, where the large C-11 aryl group and arylethyl substituent at C-8 adopt an anti relationship (Figure 6). Loss of stereochemistry at C-10 is most likely due to the formation of a quinone methide, such as 24, which upon attack by the C-11 alcohol gives a mixture of isomers with the isomer 7a being favoured over the more sterically congested 7b. We have noticed a similar process in the synthesis of other THF lignans [15,16]. To test this hypothesis we stirred a sample of isomer 7b in 1:1 2 M HCl (aq)/THF and found that after 24 hours obtained a mixture of 7a to 7b with isomer 7a being predominant. This highlights the reversibility of the ring closure and preferred formation of isomer 7a.
![[1860-5397-11-29-6]](/bjoc/content/figures/1860-5397-11-29-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Possible mechanism for the formation of furo[2,3-b]chromenes 7a and 7b.
Figure 6: Possible mechanism for the formation of furo[2,3-b]chromenes 7a and 7b.
Conclusion
In conclusion, the first synthesis of the furo[2,3-b]chromene ring system found in hyperaspidinols A (1) and B (2) has been achieved. Analysis of the NMR of synthetic furo[2,3-b]chromenes 7a and 7b and comparison to the data of 1 and 2 provides clues to the relative stereochemistry of all substituents on the natural products. The route developed allows easy introduction of alternate aryl substituents at the C-10 and C-11 positions, whilst replacing salicylaldehyde 10 with a more functionalised aldehyde could allow for the preparation of a number of highly functionalised furo[2,3-b]chromenes, including the natural products 1 and 2.
References
-
Wang, W.; Zeng, Y. H.; Osman, K.; Shinde, K.; Rahman, M.; Gibbons, S.; Mu, Q. J. Nat. Prod. 2010, 73, 1815–1820. doi:10.1021/np1004483
Return to citation in text: [1] [2] [3] [4] -
Avato, P. A survey on the hypericum genus: Secondary metabolites and bioactivity. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier B.V.: Oxford, 2005; Vol. 30, pp 603–634. doi:10.1016/S1572-5995(05)80043-2
Return to citation in text: [1] -
Tanaka, N.; Kashiwada, Y.; Kim, S. Y.; Hashida, W.; Sekiya, M.; Ikeshiro, Y.; Takaishi, Y. J. Nat. Prod. 2009, 72, 1447–1452. doi:10.1021/np900109y
Return to citation in text: [1] -
Aramaki, Y.; Chiba, K.; Tada, M. Phytochemistry 1995, 38, 1419–1421. doi:10.1016/0031-9422(94)00862-N
Return to citation in text: [1] [2] -
Abe, S.; Tanaka, N.; Kobayashi, J. J. Nat. Prod. 2012, 75, 484–488. doi:10.1021/np200741x
Return to citation in text: [1] -
Tanaka, N.; Kashiwada, Y.; Kim, S.-Y.; Sekiya, M.; Ikeshiro, Y.; Takaishi, Y. Phytochemistry 2009, 70, 1456–1461. doi:10.1016/j.phytochem.2009.08.015
Return to citation in text: [1] -
Tanaka, N.; Abe, S.; Kobayashi, J. Tetrahedron Lett. 2012, 53, 1507–1510. doi:10.1016/j.tetlet.2012.01.052
Return to citation in text: [1] -
Tanaka, N.; Abe, S.; Hasegawa, K.; Shiro, M.; Kobayashi, J. Org. Lett. 2011, 13, 5488–5491. doi:10.1021/ol2021548
Return to citation in text: [1] -
Verotta, L. Phytochem. Rev. 2002, 1, 389–407. doi:10.1023/A:1026069624278
Return to citation in text: [1] -
Osyanin, V. A.; Osipov, D. V.; Borisov, D. D.; Klimochkin, Yu. N. Chem. Heterocycl. Compd. 2013, 49, 683–688. doi:10.1007/s10593-013-1298-x
Return to citation in text: [1] -
Kikuchi, T.; Nihei, M.; Nagai, H.; Fukushi, H.; Tabata, K.; Suzuki, T.; Akihisa, T. Chem. Pharm. Bull. 2010, 58, 568–571. doi:10.1248/cpb.58.568
Return to citation in text: [1] -
Zhang, Q.-J.; Tang, Y.-B.; Chen, R.-Y.; Yu, D.-Q. Chem. Biodiversity 2007, 4, 1533–1540. doi:10.1002/cbdv.200790133
Return to citation in text: [1] -
Dickson, B. D.; Dittrich, N.; Barker, D. Tetrahedron Lett. 2012, 53, 4464–4468. doi:10.1016/j.tetlet.2012.06.088
Return to citation in text: [1] -
Barker, D.; Dickson, B. D.; Dittrich, N.; Rye, C. E. Pure Appl. Chem. 2012, 84, 1557–1565. doi:10.1351/PAC-CON-11-09-27
Return to citation in text: [1] -
Rye, C. E.; Barker, D. J. Org. Chem. 2011, 76, 6636–6648. doi:10.1021/jo200968f
Return to citation in text: [1] [2] -
Rye, C. E.; Barker, D. Synlett 2009, 3315–3319. doi:10.1055/s-0029-1218363
Return to citation in text: [1] [2] -
Pilkington, L. I.; Barker, D. J. Org. Chem. 2012, 77, 8156–8166. doi:10.1021/jo3015006
Return to citation in text: [1] -
Duhamel, N.; Rye, C. E.; Barker, D. Asian J. Org. Chem. 2013, 2, 491–493. doi:10.1002/ajoc.201300086
Return to citation in text: [1] -
Barancelli, D. A.; Salles, A. G., Jr.; Taylor, J. G.; Correia, C. R. D. Org. Lett. 2012, 14, 6036–6039. doi:10.1021/ol302923f
Return to citation in text: [1] -
Liu, G.; Lu, X. J. Am. Chem. Soc. 2006, 128, 16504–16505. doi:10.1021/ja0672425
Return to citation in text: [1] -
Sato, M.; Uchimaru, F. Chem. Pharm. Bull. 1981, 29, 3134–3144. doi:10.1248/cpb.29.3134
Return to citation in text: [1] -
Dauben, W. G.; Gerdes, J. M.; Look, G. C. J. Org. Chem. 1986, 51, 4964–4970. doi:10.1021/jo00375a039
Return to citation in text: [1] -
Firouzabadi, H.; Iranpoor, N.; Karimi, B. Synlett 1999, 321–323. doi:10.1055/s-1999-2605
Return to citation in text: [1] -
Kilpatrick, J. E.; Pitzer, K. S.; Spitzer, R. J. Am. Chem. Soc. 1947, 69, 2483–2488. doi:10.1021/ja01202a069
Return to citation in text: [1] -
Wu, A.; Cremer, D. Int. J. Mol. Sci. 2003, 4, 158–192. doi:10.3390/i4040158
Return to citation in text: [1] [2] -
Napolitano, J. G.; Gavin, J. A.; Garcia, C.; Norte, M.; Fernández, J. J.; Hernández Daranas, A. Chem. – Eur. J. 2011, 17, 6338–6347. doi:10.1002/chem.201100412
Return to citation in text: [1] [2]
| 1. | Wang, W.; Zeng, Y. H.; Osman, K.; Shinde, K.; Rahman, M.; Gibbons, S.; Mu, Q. J. Nat. Prod. 2010, 73, 1815–1820. doi:10.1021/np1004483 |
| 15. | Rye, C. E.; Barker, D. J. Org. Chem. 2011, 76, 6636–6648. doi:10.1021/jo200968f |
| 16. | Rye, C. E.; Barker, D. Synlett 2009, 3315–3319. doi:10.1055/s-0029-1218363 |
| 4. | Aramaki, Y.; Chiba, K.; Tada, M. Phytochemistry 1995, 38, 1419–1421. doi:10.1016/0031-9422(94)00862-N |
| 1. | Wang, W.; Zeng, Y. H.; Osman, K.; Shinde, K.; Rahman, M.; Gibbons, S.; Mu, Q. J. Nat. Prod. 2010, 73, 1815–1820. doi:10.1021/np1004483 |
| 6. | Tanaka, N.; Kashiwada, Y.; Kim, S.-Y.; Sekiya, M.; Ikeshiro, Y.; Takaishi, Y. Phytochemistry 2009, 70, 1456–1461. doi:10.1016/j.phytochem.2009.08.015 |
| 7. | Tanaka, N.; Abe, S.; Kobayashi, J. Tetrahedron Lett. 2012, 53, 1507–1510. doi:10.1016/j.tetlet.2012.01.052 |
| 8. | Tanaka, N.; Abe, S.; Hasegawa, K.; Shiro, M.; Kobayashi, J. Org. Lett. 2011, 13, 5488–5491. doi:10.1021/ol2021548 |
| 2. | Avato, P. A survey on the hypericum genus: Secondary metabolites and bioactivity. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier B.V.: Oxford, 2005; Vol. 30, pp 603–634. doi:10.1016/S1572-5995(05)80043-2 |
| 3. | Tanaka, N.; Kashiwada, Y.; Kim, S. Y.; Hashida, W.; Sekiya, M.; Ikeshiro, Y.; Takaishi, Y. J. Nat. Prod. 2009, 72, 1447–1452. doi:10.1021/np900109y |
| 4. | Aramaki, Y.; Chiba, K.; Tada, M. Phytochemistry 1995, 38, 1419–1421. doi:10.1016/0031-9422(94)00862-N |
| 5. | Abe, S.; Tanaka, N.; Kobayashi, J. J. Nat. Prod. 2012, 75, 484–488. doi:10.1021/np200741x |
| 26. | Napolitano, J. G.; Gavin, J. A.; Garcia, C.; Norte, M.; Fernández, J. J.; Hernández Daranas, A. Chem. – Eur. J. 2011, 17, 6338–6347. doi:10.1002/chem.201100412 |
| 19. | Barancelli, D. A.; Salles, A. G., Jr.; Taylor, J. G.; Correia, C. R. D. Org. Lett. 2012, 14, 6036–6039. doi:10.1021/ol302923f |
| 1. | Wang, W.; Zeng, Y. H.; Osman, K.; Shinde, K.; Rahman, M.; Gibbons, S.; Mu, Q. J. Nat. Prod. 2010, 73, 1815–1820. doi:10.1021/np1004483 |
| 13. | Dickson, B. D.; Dittrich, N.; Barker, D. Tetrahedron Lett. 2012, 53, 4464–4468. doi:10.1016/j.tetlet.2012.06.088 |
| 14. | Barker, D.; Dickson, B. D.; Dittrich, N.; Rye, C. E. Pure Appl. Chem. 2012, 84, 1557–1565. doi:10.1351/PAC-CON-11-09-27 |
| 15. | Rye, C. E.; Barker, D. J. Org. Chem. 2011, 76, 6636–6648. doi:10.1021/jo200968f |
| 16. | Rye, C. E.; Barker, D. Synlett 2009, 3315–3319. doi:10.1055/s-0029-1218363 |
| 17. | Pilkington, L. I.; Barker, D. J. Org. Chem. 2012, 77, 8156–8166. doi:10.1021/jo3015006 |
| 18. | Duhamel, N.; Rye, C. E.; Barker, D. Asian J. Org. Chem. 2013, 2, 491–493. doi:10.1002/ajoc.201300086 |
| 24. | Kilpatrick, J. E.; Pitzer, K. S.; Spitzer, R. J. Am. Chem. Soc. 1947, 69, 2483–2488. doi:10.1021/ja01202a069 |
| 25. | Wu, A.; Cremer, D. Int. J. Mol. Sci. 2003, 4, 158–192. doi:10.3390/i4040158 |
| 26. | Napolitano, J. G.; Gavin, J. A.; Garcia, C.; Norte, M.; Fernández, J. J.; Hernández Daranas, A. Chem. – Eur. J. 2011, 17, 6338–6347. doi:10.1002/chem.201100412 |
| 1. | Wang, W.; Zeng, Y. H.; Osman, K.; Shinde, K.; Rahman, M.; Gibbons, S.; Mu, Q. J. Nat. Prod. 2010, 73, 1815–1820. doi:10.1021/np1004483 |
| 10. | Osyanin, V. A.; Osipov, D. V.; Borisov, D. D.; Klimochkin, Yu. N. Chem. Heterocycl. Compd. 2013, 49, 683–688. doi:10.1007/s10593-013-1298-x |
| 11. | Kikuchi, T.; Nihei, M.; Nagai, H.; Fukushi, H.; Tabata, K.; Suzuki, T.; Akihisa, T. Chem. Pharm. Bull. 2010, 58, 568–571. doi:10.1248/cpb.58.568 |
| 12. | Zhang, Q.-J.; Tang, Y.-B.; Chen, R.-Y.; Yu, D.-Q. Chem. Biodiversity 2007, 4, 1533–1540. doi:10.1002/cbdv.200790133 |
| 20. | Liu, G.; Lu, X. J. Am. Chem. Soc. 2006, 128, 16504–16505. doi:10.1021/ja0672425 |
| 21. | Sato, M.; Uchimaru, F. Chem. Pharm. Bull. 1981, 29, 3134–3144. doi:10.1248/cpb.29.3134 |
| 22. | Dauben, W. G.; Gerdes, J. M.; Look, G. C. J. Org. Chem. 1986, 51, 4964–4970. doi:10.1021/jo00375a039 |
| 23. | Firouzabadi, H.; Iranpoor, N.; Karimi, B. Synlett 1999, 321–323. doi:10.1055/s-1999-2605 |
© 2015 Paterson and Barker; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)