Abstract
Two chlorophosphites, the biphenyl-based BIFOP–Cl and the diphenyl ether-based O–BIFOP–Cl, exhibit striking differences regarding their reaction with water. While BIFOP–Cl is nearly completely unreactive, its oxo-derivative O–BIFOP–Cl reacts instantly with water, yielding a tricyclic hydrocarbon unit after rearrangement. The analysis of the crystal structure of O–BIFOP–Cl and BIFOP–Cl revealed that the large steric demand of encapsulating fenchane units renders the phosphorus atom nearly inaccessible by nucleophilic reagents, but only for BIFOP–Cl. In addition to the steric effect, a hypervalent P(III)–O interaction as well as an electronic conjugation effect causes the high reactivity of O–BIFOP–Cl. A DFT study of the hydrolysis in BIFOP–Cl verifies a higher repulsive interaction to water and a decreased leaving tendency of the chloride nucleofuge, which is caused by the fenchane units. This high stability of BIFOP–Cl against nucleophiles supports its application as a chiral ligand, for example, in Pd catalysts.

Graphical Abstract
Introduction
Phosphorus halides are highly reactive intermediates for the synthesis of phosphites and phosphoramidites [1-5], which are widely used, for example, as ligands in catalysts [6-9]. There are also some applications of phosphine halides used as ligands in catalytic reactions, for example, in cross-coupling reactions and hydroformylations [10-12]. We recently reported the application of the fenchole-based, phosphine halide BIFOP–Hal (Hal = F, Cl, Br) (Scheme 1) in an intramolecular palladium-catalyzed alkyl–aryl cross-coupling reaction [13] and in Pd-catalyzed allylic substitutions [14]. Several of the highly sterically hindered BIFOP derivatives were employed as ligands in Cu-catalyzed 1,4-additions [15]. Similar chelating fencholates [16-22] (Scheme 1) were employed in enantioselective organozinc catalysis reactions [23-26], umpolung catalysis [27] and in organoaluminum [17] and chiral n-butyllithium aggregates [28-33].
![[1860-5397-11-36-i1]](/bjoc/content/inline/1860-5397-11-36-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Fenchyl-based ligands used as building blocks for phosphorous ligands or organoaluminum reagents.
Scheme 1: Fenchyl-based ligands used as building blocks for phosphorous ligands or organoaluminum reagents.
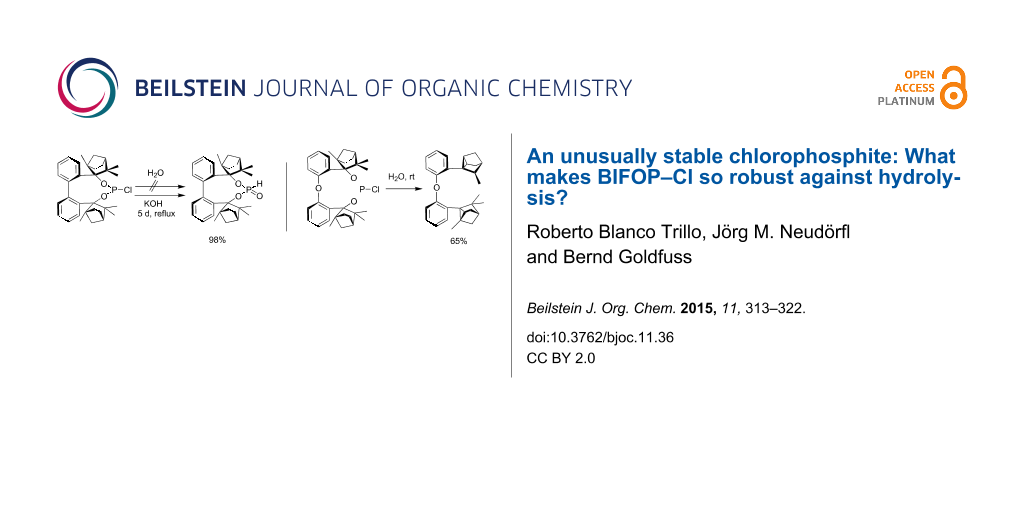
The chlorophosphite BIFOP-Cl (1) is air-stable and very resistant to hydrolysis (Scheme 2) [13,15]. The low reactivity of 1 to O- and C-nucleophiles is explained by the tight encapsulation of the P–Cl unit of the endo-fenchane moieties [15]. This unusual stability of the BIFOP–halides prompted the comparison of BIFOP–Cl (1) with its diphenyl ether derivative O–BIFOP–Cl (3). Despite similar encapsulation by two fencholate moieties, O–BIFOP–Cl 3 exhibits a significantly higher reactivity with nucleophiles (e.g., with water). Here we rationalize the different reactivities of 1 and 3.
![[1860-5397-11-36-i2]](/bjoc/content/inline/1860-5397-11-36-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction of BIFOP–Cl (1) to BIFOP–(O)H (2) and of O–BIFOP–Cl (3) yielding O–BIFOP–H (4), O–BIFOP–(O)H (6) as well as diphenyl ether-2,2’-biscyclofenchene 7.
Scheme 2: Reaction of BIFOP–Cl (1) to BIFOP–(O)H (2) and of O–BIFOP–Cl (3) yielding O–BIFOP–H (4), O–BIFOP–(O...
Results and Discussion
In contrast to BIFOP–Cl (1), the diphenyl ether analogue O–BIFOP–Cl (3) exhibits the expected halophosphite reactivity and instantly reacts with water (Scheme 2, Figure 1). NMR monitoring of the hydrolysis showed that O–BIFOP–Cl (3, 31P NMR, δ = 161.9, Figure 1) is immediately hydrolyzed, yielding O–BIFOP–(O)H (6, 31P NMR, δ = −8.2, Figure 1). After 37 min, the amount of starting O–BIFOP–Cl (3) as well as the primary hydrolysis product 6 (Figure 1) is nearly completely depleted. The details of the reaction mixture that yielded diphenyl ether-2,2’-biscyclofenchene 7 (Figure 2) are shown in Scheme 2.
![[1860-5397-11-36-1]](/bjoc/content/figures/1860-5397-11-36-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 31P NMR (125 MHz, CDCl3) of O–BIFOP–Cl (3, δ = 161.9) after the addition of 1 equiv H2O and formation of O–BIFOP–(O)H (6, δ = −8.2), which vanished after 37 min forming 7 (cf. Scheme 2 and Scheme 3). The integration values are shown below the signals.
Figure 1: 31P NMR (125 MHz, CDCl3) of O–BIFOP–Cl (3, δ = 161.9) after the addition of 1 equiv H2O and formati...
![[1860-5397-11-36-2]](/bjoc/content/figures/1860-5397-11-36-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray crystal structure of diphenyl ether-2,2’-biscyclofenchene 7. Ellipsoids are shown with 50% probability.
Figure 2: X-ray crystal structure of diphenyl ether-2,2’-biscyclofenchene 7. Ellipsoids are shown with 50% pr...
The formation of a cyclopropane ring in 7 can be rationalized to proceed through a fenchyl carbocation (Scheme 3) [34-40]. Intramolecular cyclopropanation reactions are often characterized by prolonged treatment with an acid [41-46]. Stabilization of the intermediate carbocation by the lone pair of the oxygen atom is enabled by lone-pair conjugation (O-lp conjugation) of the benzyl cation and supports elimination of the oxido unit (Scheme 3).
![[1860-5397-11-36-i3]](/bjoc/content/inline/1860-5397-11-36-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed mechanism for the formation of diphenyl ether-2,2’-biscyclofenchene 7 through stabilization of the intermediate carbocation by O-lp conjugation and cyclopropane formation starting from O–BIFOP–Cl (3).
Scheme 3: Proposed mechanism for the formation of diphenyl ether-2,2’-biscyclofenchene 7 through stabilizatio...
To assess whether this rearrangement, formaing 7, is mediated by HCl originating from 3, chlorine-free O–BIFOP–H (4) was treated with O2, yielding 6 (79%, Scheme 2). While O–BIFOP–H (4) readily reacts with water, O–BIFOP–(O)H (6) was found to be stable in air and water (Figure 3d). However, addition of HCl to O–BIFOP–(O)H (6) gave diphenyl ether-2,2’-biscyclofenchene 7 in 65% yield. Hence, acidic conditions (HCl) are necessary to form 7 from 6, which is generated by hydrolysis of 3 (Scheme 2 and Scheme 3).
![[1860-5397-11-36-3]](/bjoc/content/figures/1860-5397-11-36-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: 31P NMR (125 MHz, CDCl3) of O–BIFOP–H (4, δ = 152.5) adding O2 after a) 5 min; b) 15 min; c) 120 min; d) adding 1 equiv H2O forming O-BIFOP-(O)H (6, δ = −8.2).
Figure 3: 31P NMR (125 MHz, CDCl3) of O–BIFOP–H (4, δ = 152.5) adding O2 after a) 5 min; b) 15 min; c) 120 mi...
The analysis of the crystal structure of BIFOP–Cl (1) reveals the large steric demand of the fenchane units, which embed the phosphorus atom, thus making it inaccessible to nucleophilic reagents (Figure 4, Table 1) [14,15]. In contrast, the reduced protection of the phosphorus atom in O-BIFOP-Cl (3), which is primarily caused by the relatively large H13b–P distance (4.08 Å to 3.01 Å), provides an explanation for the higher reactivity of the >P–Cl moiety in O–BIFOP–Cl (3, Scheme 4, Figure 5, Table 1).
![[1860-5397-11-36-4]](/bjoc/content/figures/1860-5397-11-36-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: X-ray crystal structure of BIFOP-Cl 1. Ellipsoids are shown with 50% probability [15].
Figure 4: X-ray crystal structure of BIFOP-Cl 1. Ellipsoids are shown with 50% probability [15].
Table 1: Geometries bases on the X-ray structure of BIFOP–Cla and O–BIFOP–Cl.
| BIFOP–Cl (1) | O–BIFOP–Cl (3) | |
|---|---|---|
| Angle sum at P (°)b | 305.2 | 290.7 |
| FAA–lp (°)c | 38.9 | −26,6 |
| FAA (°)c | 37.1 | −52.5 |
| H13b–P (Å) | 3.01 | 4.08 |
| H15b–P (Å) | 3.24 | 3.42 |
| H28a–P (Å) | 3.10 | 3.20 |
| H30a–P (Å) | 2.69 | 3.06 |
aPublished in reference [14]. bAngle sum at phosphorous atom (pyramidality). cFenchyl–aryl dihedral angles (FAA, C1–C2–C3–O1) on the lone-pair side of phosphorus (FAA–lp) and at the substituent side (FAA) biaryl axis.
![[1860-5397-11-36-i4]](/bjoc/content/inline/1860-5397-11-36-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The different backbones provoke different reactivities due to tighter encapsulation of the P–Cl unit by the fenchane moieties in BIFOP–Cl (1) relative to O–BIFOP–Cl (3).
Scheme 4: The different backbones provoke different reactivities due to tighter encapsulation of the P–Cl uni...
![[1860-5397-11-36-5]](/bjoc/content/figures/1860-5397-11-36-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: X-ray crystal structure of O-BIFOP-Cl (3). Ellipsoids are shown with 50% probability.
Figure 5: X-ray crystal structure of O-BIFOP-Cl (3). Ellipsoids are shown with 50% probability.
Moreover, the shorter P–O distance (2.4 Å) in O–BIFOP–Cl (3) and the nearly linear (176.5°) O–P–Cl arrangement (Scheme 4) suggest a neighbor-group effect through an O-lp donor to σ*P–Cl acceptor interaction, supporting chloride substitution (Figure 5, Scheme 4). Hypervalent P(III)–O interactions with similar P–O distances are documented for five membered rings [47,48] as well as for acyclic systems [49].
The computational analysis of the hydrolysis of the chlorophosphites BIFOP–Cl (1) and O–BIFOP–Cl (3, as well as the smaller model system 2-chloro-1,3,2-dioxaphospholane 8) provides further comparison of the >P–Cl reactivity. The nucleophilic substitution reaction takes place at a triple-coordinated chlorophosphite (in R2PCl) due to a single-well potential energy surface [50,51]. The initial step of the water addition proceeds through the formation of the transition state (TS1) in which the oxygen atom of the water molecule binds to the phosphorous atom (Scheme 3, Table 2) and chloride substitution forms the product (G2). Here, chloride is replaced at the phosphorus center with the hydroxide nucleophile (Table 2).
Table 2: Computed relative energies (Erel, kcal/mol) for the reaction of 2, 4 or 8 with water.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-36-i5.svg?max-width=637&scale=1.0)
|
||||
| Entry | Chlorophosphane | Erel (G1) | Erel (TS1) | Erel (G2) |
|---|---|---|---|---|
| 1a | BIFOP–Cl (1) | 0.0 | 31.2 | −4.7 |
| 2a | O–BIFOP–Cl (3) | 0.0 | 22.5 | −3.1 |
| 3b | [CH2O]2P–Cl (8) | 0.0 | 18.3 | −5.3 |
aBP86/SVP + ZPE. bMP2/cc-p-VQZ//BP86/SVP + ZPE.
The relatively high hydrolyzation barrier of BIFOP–Cl (1, 31.2 kcal/mol) in comparison to O–BIFOP–Cl (3, 22.5 kcal/mol) and the smaller, glycol-based, chlorophosphite [CH2O]2P–Cl (8, 18.3 kcal/mol, Table 2) agrees with the experimental finding that BIFOP–Cl (1) is unusually robust against hydrolysis (Figure 1 and Figure 3). The lower hydrolysis barriers of 3 and 8 agree with the expected high reactivity of the >P–Cl in water [52-57].
A comparison of the transition state structures of chlorophosphites 1 (Figure 6) and 3 (Figure 7) reveals a higher steric congestion of the P–Cl unit by the fenchane moiety in BIFOP–Cl (1) relative to O–BIFOP–Cl (3). In BIFOP–Cl (1), the shorter distances of the endo-oriented hydrogen atoms of the fenchane moiety (H35 and H75) to the Cl atom of the P–Cl unit and to the O atom of water (Table 3) prevent both the elimination of the chloride nucleofuge and the attack of the water nucleophile. This steric congestion of the transition state structures in reactions with water explains the surprisingly low reactivity of BIFOP–Cl (1, Figure 6) relative to the much more reactive O–BIFOP–Cl (3, Figure 7).
![[1860-5397-11-36-6]](/bjoc/content/figures/1860-5397-11-36-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Transition state structure for the reaction of BIFOP–Cl (1) with water (BP86/def-SV(P)).
Figure 6: Transition state structure for the reaction of BIFOP–Cl (1) with water (BP86/def-SV(P)).
![[1860-5397-11-36-7]](/bjoc/content/figures/1860-5397-11-36-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Transition state structure for the reaction of O-BIFOP–Cl (3) with water (BP86/def-SV(P)).
Figure 7: Transition state structure for the reaction of O-BIFOP–Cl (3) with water (BP86/def-SV(P)).
Table 3: Selected, computed distances in the transition state structures for the addition of water to chlorophosphites 1 and 3.a
| Distance | BIFOP–Cl (1) | O–BIFOP–Cl (3) |
|---|---|---|
| Cl–H35 (Å) | 2.65 | 3.05 |
| Cl–H75 (Å) | 4.70 | 5.21 |
| O2–H35 (Å) | 2.68 | 2.98 |
| O2–H75 (Å) | 2.60 | 3.36 |
aBP86/def-SV(P) optimized transition state structures, cf. Figure 6 and Figure 7.
Conclusion
Two fenchole-based chlorophosphites, BIFOP–Cl (1) and O–BIFOP–Cl (3), were studied with respect to their striking differences in regards to their reaction with water. While BIFOP–Cl (1) exhibits a surprisingly high stability against hydrolysis, O–BIFOP–Cl (3) reacts instantly with water, leading to cyclofenchene 6. X-ray studies revealed that the increased reactivity of the intermediate carbenium ion and cyclopropane formation is due to a steric effect caused by the shielding of the fenchane groups and a hypervalent P(III)–O interaction. Formation of the cyclofenchene derivative 7 is explained by rearrangement via a 2-fenchyl carbocation. The DFT computations of the hydrolysis revealed a higher degree of steric congestion in BIFOP–Cl (1) caused by the fenchane units, relative to the less-shielded and hence much more reactive O–BIFOP–Cl (3). This result demonstrates that steric and electronic effects can be used to render the inherently highly reactive and electrophilic phosphorus–halogen units essentially inert against nucleophilic reagents. The stability of BIFOP–Cl (and other phosphorus–halogen systems) against nucleophiles promotes its application as a chiral ligand to be used in, for example, Pd catalysis [13-15].
Experimental
All reactions were carried out under an inert argon atmosphere and in heated glassware using standard Schlenk techniques. Anhydrous solvents were obtained by distillation from sodium benzophenone ketyl. The NMR spectra were measured with Bruker instruments (Avance II 600, Avance II 300 and DPX Acance 300). Deuterated chloroform was used as solvent. The proton shifts are reported in ppm (δ) downfield from TMS and are referenced to residual signals of the solvent (CHCl3 7.24 ppm for hydrogen, 77.0 ppm for carbon atoms). The coupling constants (J) are given in Hz. As an external standard, 85% phosphoric acid was used for the 31P NMR spectra. The infrared spectra were recorded on a Shimadzu, IRAffinity-1 instrument. The wavenumbers (ν) of the recorded IR signals are given in cm−1. The GC–MS spectra were recorded using an Agilent Technologies, Model GC 6890N gas chromatograph coupled with an HP 5973N series mass selective detector and an HP 7683 GC autosampler. Optical rotation was measured with an IBZ, Messtechnik POLAR LµP-WR polarimeter, using a 1 dm path length cell. The reactions were carried out under dry argon. X-ray analysis was performed with a Nonius, Kappa CCD diffractometer (Mo Kα, λ = 0.71073). The starting material, O–BIFOL, was obtained in an analogous manner to a procedure previously described [15].
Diphenyl ether-2,2'-bisfencholphosphane chloride (O–BIFOP–Cl, 3)
The O–BIFOP–Cl compound was prepared in a manner analogous to the procedure described in [15]. 1.48 mL (3 mmol) of n-butyllithium in hexane (1.6 M) was slowly added to a 200 mg solution (0.42 mmol) of O–BIFOL in 1.4 mL abs. THF at −20 °C. The mixture was stirred for 30 min at −20 °C, then for 1 h at rt. After again cooling to −20 °C, 0.06 mL (0.46 mmol) of freshly distilled PCl3 was slowly added and the reaction mixture was stirred for 6 h at rt. Recrystallization from Et2O/CH2Cl2 resulted in 111 mg (0.21 mmol, 50%) of compound 3 as colorless crystals. [α]D20 +46.47 (c 4.5, hexane); 31P NMR (125.5 MHz, CDCl3) δ 161.9; 1H NMR (300 MHz, CDCl3) δ 0.11 (s, 3H), 0.42 (s, 3H), 0.77 (s, 3H), 0.82 (s, 3H), 1.22–1.58 (m, 8H), 2.37 (d, J = 9 Hz, 1H), 2.49 (m, 3H), 2.45 (m, 3H), 2.76 (m, 4H), 6.75 (d, J = 6 Hz, 1H), 6.96 (t, J = 9 Hz, 1H), 7.17–7.22 (m, 2H), 7.54 (d, J = 6 Hz, 1H), 7.62 (d, J = 6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 18.3, 21.1, 22.2, 22.7, 32.6, 38.6, 42.4, 42.9, 49.8, 49.4, 51.3, 52.7, 96.3, 115.2, 121.9, 122.8, 125.6, 128.4, 148.8; X-ray crystal data: C32H41O4P; Mr = 539.1 g·mol−1; space group: P212121; a = 12.2504(6), b = 14.9267(9), c = 30.6807(12) Å; V = 5610.2(5) Å3; Z = 8; ρ = 1.276 g·mL−3; T = 100(2) K; λ = 0.71073; μ = 0.123 mm−1; total reflections: 22204; unique reflections: 11385; observed: 5685 [I>2s(I)]; parameters refined: 679; R1 = 0.0611, wR2 = 0.0981; GOF = 0.924; H atoms bound to oxygen were refined, the positions of the H atoms bound to carbon were calculated.
Diphenyl ether-2,2'-bisfencholphosphane hydride (O–BIFOP–H, 4)
The O–BIFOP–H compound was prepared in a manner analogous to the procedure as described in [15]. 8.7 mg (0.23 mmol) of LiAlH4 was added to 100 mg (0.19 mmol) of O–BIFOP–Cl (3) in 1 mL of THF and the mixture was stirred for 3 h at rt. The solvent was removed in vacuum and the residue was taken up in 10 mL of toluene and stirred for 30 min at rt. After filtration through celite to remove LiCl and other salts, the resulting solution was concentrated in vacuum until precipitation. The recrystallization from toluene yielded 83 mg (0.16 mmol, 87%) of 4 as a white solid. [α]D20 +53.74 (c 2.8, hexane); 31P NMR (125.5 MHz, toluene-d8) δ 153.5 (1JP–H = 190 Hz); 1H NMR (300 MHz, toluene-d8) δ 0.35 (s, 3H), 0.55 (s, 3H), 0.73 (s, 3H), 0.96 (t, J = 6 Hz, 1H), 1.04–1.11 (m, 6H), 1.22 (s, 3H), 1.31 (t, J = 6 Hz, 1H), 1.52 (d, J = 6 Hz, 2H), 1.68 (s, 1H), 1.72 (s, 1H), 6.62 (d, 1JP–H = 190 Hz, 1H), 6.84–7.03 (m, 2H), 7.53 (d, J = 6 Hz, 1H); 13C NMR (75 MHz, toluene-d8) δ 18.3, 22.8, 23.8, 24.1, 24.8, 23.2, 34.4, 42.4, 43.5, 49.5, 52.4, 54.6, 97.9, 99.3, 116.9, 118.2, 122.5, 122.8, 123.9, 124.5, 125.0, 126.1, 136.8, 138.8, 145.2, 149.7.
Diphenyl ether-2,2'-bisfencholphosphate (O-BIFOPH(O), 6)
O2 was supplied to 83 mg (0.16 mmol) of O–BIFOP–H (4) for 5 min. The recrystallization from toluene yielded 38 mg (79%) of 6 as a colorless solid. [α]D20 +55.7 (c 4.5; hexane); 31P NMR (125.5 MHz, toluene-d8) δ −8.2 (1JP–H = 710.8 Hz); 1H NMR (300 MHz, CDCl3) δ 0.41 (s, 3H), 0.52 (s, 3H), 0.57 (s, 3H), 0.82 (s, 3H), 1.04 (s, 1H), 1.20 (s, 1H), 1.28 (s, 6H), 1.34 (m, 6H), 1.37 (m, 6H), 1.49 (s, 4H), 1.65 (d, J = 9 Hz, 2H), 1.75 (m, 4H), 6.75 (d, 1JP–H = 710.8 Hz, 1H), 6.96 (d, J = 6 Hz, 1H), 7.17 (t, J = 9 Hz, 2H), 6.64 (d, J = 6 Hz, 1H), 7.71 (d, J = 6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 18.03, 18.17, 21.90, 22.97, 23.63, 23.73, 28.62, 29.69, 34.31, 35.60, 41.02, 42.33, 43.06, 48.73, 49.04, 49.31, 50.30, 55.60, 118.05, 119.87, 121.08, 121.69, 122.93, 123.36, 126.58, 127.82, 128.20, 129.97, 130.55, 131.46.
Diphenyl ether-2,2’-biscyclofenchene-1,3,3-trimethyltricyclo[2.2.1.0]heptane (7)
2.9 mL (0.16 mmol) of H2O was slowly added to 83 mg of O–BIFOP–Cl (3, 0.16 mmol) in 2 mL of THF, and the mixture was stirred for 20 min at rt. The solvent was removed in vacuum, and the residue was taken up in 10 mL of toluene and filtered through celite. The resulting solution was concentrated in vacuum until precipitation. The recrystallization from toluene yielded 44 mg (65%) of 7 as colorless crystals. 1H NMR (300 MHz, CDCl3) δ 0.74 (s, 1H), 0.90 (s, 3H), 0.94 (s, 3H), 1.07 (s, 1H), 1.12 (s, 3H), 1.24–1.29 (m, 3H), 1.46 (s, 1H), 1.53 (d, J = 10 Hz, 2H), 1.84 (d, J = 10 Hz, 2H), 6.89 (d, J = 6 Hz, 1H), 6.99 (t, J = 9 Hz, 1H), 7.09 (d, J = 6 Hz, 1H), 7.19 (d, J = 6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 15.63, 15.90, 21.76, 22.23, 22.61, 25.86, 27.09, 32.72, 33.04, 35.68, 38.20, 38.33, 42.61, 42.72, 47.65, 47.81, 118.62, 120.29, 121.47, 121.86, 126.86, 127.40, 127.91, 134.34, 134.69; IR (KBr) ν: 3334 (s), 2987 (vs), 1503 (m), 1434 (m); ESIMS (%) m/z: [M]+ 438.3; Anal. calcd for C32H38O (438.3 g·mol−1): C, 87.62; H, 8.73; found: C, 87.60; H, 9.19. X-ray crystal data: C32H38O; Mr = 438.6 g·mol−1; space group: P21; a = 7.2593(6), b = 16.903(2), c = 20.472(2) Å; V = 2508.4(4) Å3; Z = 4; ρ = 1.161 g·mL−3; T = 100(2) K; λ = 0.71073; μ = 0.068 mm−1; total reflections: 10449; unique reflections: 8172; observed: 3859 [I>2s(I)]; parameters refined: 578; R1 = 0.1649, wR2 = 0.3899; GOF = 1.264; H atoms bound to oxygen were refined, the positions of the H atoms bound to carbon were calculated.
Computational details
The computations were performed with the program package TURBOMOLE-5.10 [58-60]. The employed functional was BP86 with an m3 grid size combined with the contracted, SVP basis set from Ahlrichs et al. The resolution-of-identity approximation for a two-electron integral evaluation was used. All stationary points were fully optimized and confirmed by separate analytical frequency calculations. The transition state structures were optimized with quasi-Newton–Raphson methods by using the Powell update algorithm for Hessian matrix approximation (analytical frequency calculation subsequent). The absolute energies were zero-point-corrected with the vibrational information resulting from the harmonic analytical frequency calculations.
Acknowledgements
We are grateful to the Funds der Chemischen Industrie for financial support. We also thank the Regionales Rechenzentrum zu Köln (RRZK) for the maintenance of the HPC systems, and Bayer AG, BASF AG, Wacker AG, Evonic AG, Raschig GmbH, Symrise GmbH, Solvay GmbH and the OMG group for their generous support.
References
-
Brunel, J. M. Chem. Rev. 2005, 105, 857. doi:10.1021/cr040079g
Return to citation in text: [1] -
van Leeuwen, P. W. N. M.; Kamer, P. C. J.; Claver, C.; Pàmies, O.; Dièguez, M. Chem. Rev. 2011, 111, 2077. doi:10.1021/cr1002497
Return to citation in text: [1] -
Fernàndez-Pèrez, H.; Etayo, P.; Panossian, A.; Vidal-Ferran, A. Chem. Rev. 2011, 111, 2119. doi:10.1021/cr100244e
Return to citation in text: [1] -
Seebach, D.; Beck, A. K.; Heckel, A. Angew. Chem. 2001, 113, 96. doi:10.1002/1521-3757(20010105)113:1<96::AID-ANGE96>3.0.CO;2-B
Angew. Chem., Int. Ed. 2001, 40, 92. doi:10.1002/1521-3773(20010105)40:1<92::AID-ANIE92>3.0.CO;2-K
Return to citation in text: [1] -
Teichert, J. F.; Feringa, B. L. Angew. Chem. 2010, 122, 2538. doi:10.1002/ange.200904948
Angew. Chem., Int. Ed. 2010, 49, 2486. doi:10.1002/anie.200904948
Return to citation in text: [1] -
Schober, K.; Hartmann, E.; Zhang, H.; Gschwind, R. M. Angew. Chem. 2010, 122, 2855. doi:10.1002/ange.200907247
Angew. Chem., Int. Ed. 2010, 49, 2794. doi:10.1002/anie.200907247
Return to citation in text: [1] -
Shibasaki, M.; Sasai, H.; Arai, T. Angew. Chem. 1997, 109, 1290. doi:10.1002/ange.19971091204
Angew. Chem., Int. Ed. Engl. 1997, 36, 1236. doi:10.1002/anie.199712361
Return to citation in text: [1] -
Sasai, H.; Arai, T.; Satow, Y.; Houk, K. N.; Shibasaki, M. J. Am. Chem. Soc. 1995, 117, 6194. doi:10.1021/ja00128a005
Return to citation in text: [1] -
d'Augustin, M.; Palais, L.; Alexakis, A. Angew. Chem. 2005, 117, 1400. doi:10.1002/ange.200462137
Angew. Chem., Int. Ed. 2005, 44, 1376. doi:10.1002/anie.200462137
Return to citation in text: [1] -
Ackermann, L.; Kapdi, A. R.; Schulzke, C. Org. Lett. 2010, 12, 2298. doi:10.1021/ol100658y
Return to citation in text: [1] -
Puckette, T. A. Chem. Ind. 2007, 115, 31.
Return to citation in text: [1] -
Timosheva, N. V.; Chandrasekaran, A.; Holmes, R. R. J. Am. Chem. Soc. 2005, 127, 12474. doi:10.1021/ja053422n
Return to citation in text: [1] -
Blanco Trillo, R.; Leven, M.; Neudörfl, J. M.; Goldfuss, B. Adv. Synth. Catal. 2012, 354, 1451. doi:10.1002/adsc.201100924
Return to citation in text: [1] [2] [3] -
Goldfuss, B.; Löschmann, T.; Kop-Weiershausen, T.; Neudörfl, J.; Rominger, F. Beilstein J. Org. Chem. 2006, 2, No. 7. doi:10.1186/1860-5397-2-7
Return to citation in text: [1] [2] [3] [4] -
Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Lange, D. A.; Neudörfl, J.-M.; Goldfuss, B. Tetrahedron 2006, 62, 3704. doi:10.1016/j.tet.2006.01.060
Return to citation in text: [1] -
Soki, F.; Neudörfl, J.-M.; Goldfuss, B. J. Organomet. Chem. 2008, 693, 2139. doi:10.1016/j.jorganchem.2008.03.013
Return to citation in text: [1] [2] -
Goldfuss, B.; Löschmann, T.; Rominger, F. Chem. – Eur. J. 2001, 7, 2028. doi:10.1002/1521-3765(20010504)7:9<2028::AID-CHEM2028>3.0.CO;2-Y
Return to citation in text: [1] -
Goldfuss, B.; Rominger, F. Tetrahedron 2000, 56, 881. doi:10.1016/S0040-4020(99)01077-7
Return to citation in text: [1] -
Goldfuss, B.; Eisenträger, E. Aust. J. Chem. 2000, 53, 209. doi:10.1071/CH99184
Return to citation in text: [1] -
Goldfuss, B.; Löschmann, T.; Rominger, F. Chem. – Eur. J. 2004, 10, 5422. doi:10.1002/chem.200400273
Return to citation in text: [1] -
Soki, F. Synthese und Charakterisierung neuartiger Fencholate und deren Einsatz als Chiralitätsvermittler in enantioselktiven C–C Knüpfungsreaktionen. Ph.D. Thesis, University of Cologne, Germany, 2008.
Return to citation in text: [1] -
Steigelmann, M.; Nisar, Y.; Rominger, F.; Goldfuss, B. Chem. – Eur. J. 2002, 8, 5211. doi:10.1002/1521-3765(20021115)8:22<5211::AID-CHEM5211>3.0.CO;2-S
Return to citation in text: [1] -
Goldfuss, B.; Steigelmann, M.; Rominger, F. Eur. J. Org. Chem. 2000, 1785. doi:10.1002/(SICI)1099-0690(200005)2000:9<1785::AID-EJOC1785>3.0.CO;2-0
Return to citation in text: [1] -
Goldfuss, B.; Steigelmann, M. J. Mol. Model. 2000, 6, 166. doi:10.1007/s0089400060166
Return to citation in text: [1] -
Leven, M.; Schlörer, N. E.; Neudörfl, J. M.; Goldfuss, B. Chem. – Eur. J. 2010, 16, 13443. doi:10.1002/chem.201001106
Return to citation in text: [1] -
Gliga, A.; Klare, H.; Schumacher, M.; Soki, F.; Neudörfl, J. M.; Goldfuss, B. Eur. J. Org. Chem. 2011, 256–263. doi:10.1002/ejoc.201001295
Return to citation in text: [1] -
Goldfuss, B.; Steigelmann, M.; Khan, S. I.; Houk, K. N. J. Org. Chem. 2000, 65, 77. doi:10.1021/jo991070v
Return to citation in text: [1] -
Goldfuss, B.; Khan, S. I.; Houk, K. N. Organometallics 1999, 18, 2927. doi:10.1021/om990184u
Return to citation in text: [1] -
Goldfuss, B. Synthesis 2005, 2271. doi:10.1055/s-2005-872107
Return to citation in text: [1] -
Goldfuss, B.; Steigelmann, M.; Löschmann, T.; Schilling, G.; Rominger, F. Chem. – Eur. J. 2005, 11, 4019. doi:10.1002/chem.200500158
Return to citation in text: [1] -
Goldfuss, B.; Steigelmann, M.; Rominger, F.; Urtel, H. Chem. – Eur. J. 2001, 7, 4456. doi:10.1002/1521-3765(20011015)7:20<4456::AID-CHEM4456>3.0.CO;2-S
Return to citation in text: [1] -
Goldfuss, B.; Steigelmann, M.; Rominger, F. Angew. Chem. 2000, 112, 4299. doi:10.1002/1521-3757(20001117)112:22<4299::AID-ANGE4299>3.0.CO;2-O
Angew. Chem., Int. Ed. 2000, 39, 4133. doi:10.1002/1521-3773(20001117)39:22<4133::AID-ANIE4133>3.0.CO;2-X
Return to citation in text: [1] -
Huang, E.; Ranganayakulu, K.; Sorensen, T. S. J. Am. Chem. Soc. 1972, 94, 1779. doi:10.1021/ja00760a079
Return to citation in text: [1] -
Sorensen, T. S. Acc. Chem. Res. 1976, 9, 257. doi:10.1021/ar50103a003
Return to citation in text: [1] -
Starling, S. M.; Vonwiller, S. C.; Reek, J. N. H. J. Org. Chem. 1998, 63, 2262. doi:10.1021/jo972025v
Return to citation in text: [1] -
Brown, H. C.; Takeuchi, K. J. Am. Chem. Soc. 1968, 90, 2693. doi:10.1021/ja01012a043
Return to citation in text: [1] -
Farnum, D. G.; Mehta, G. J. Am. Chem. Soc. 1969, 91, 3256. doi:10.1021/ja01040a028
Return to citation in text: [1] -
Brown, H. C.; Takeuchi, K.; Ravindranathan, M. J. Am. Chem. Soc. 1977, 99, 2684. doi:10.1021/ja00450a047
Return to citation in text: [1] -
Farnum, D. G.; Wolf, A. D. J. Am. Chem. Soc. 1974, 96, 5166. doi:10.1021/ja00823a025
Return to citation in text: [1] -
von Ragué Schleyer, P.; Lam, L. K. M.; Raber, D. J.; Fry, J. L.; McKervey, M. A.; Alford, J. R.; Cuddy, B. D.; Keizer, V. G.; Geluk, H. W.; Schlatmann, J. L. M. A. J. Am. Chem. Soc. 1970, 92, 5246. doi:10.1021/ja00720a056
Return to citation in text: [1] -
Majerski, Z.; von Ragué Schleyer, P.; Wolf, A. P. J. Am. Chem. Soc. 1970, 92, 5731. doi:10.1021/ja00722a034
Return to citation in text: [1] -
Nickon, A.; Weglein, R. C. J. Am. Chem. Soc. 1975, 97, 1271. doi:10.1021/ja00838a067
Return to citation in text: [1] -
Paquette, L. A.; Waykole, L.; Jendralla, H.; Cottrell, C. E. J. Am. Chem. Soc. 1986, 108, 3739. doi:10.1021/ja00273a031
Return to citation in text: [1] -
Paquette, L. A.; Lanter, J. C.; Johnston, J. N. J. Org. Chem. 1997, 62, 1702. doi:10.1021/jo962019j
Return to citation in text: [1] -
Lee, O.-S.; Yang, K.; Kang, K. D.; Koo, I. S.; Kim, C.-K.; Lee, I. J. Comput. Chem. 2004, 25, 1740. doi:10.1002/jcc.20104
Return to citation in text: [1] -
Milov, A. A.; Minyaev, R. M.; Minkin, V. I. J. Phys. Chem. A 2011, 115, 12973. doi:10.1021/jp2042119
Return to citation in text: [1] -
Kubo, K.; Nakazawa, H.; Kawamura, K.; Mizuta, T.; Miyoshi, K. J. Am. Chem. Soc. 1998, 120, 6715. doi:10.1021/ja980146m
Return to citation in text: [1] -
Dobado, J. A.; Martinez-Garcia, H.; Molina, J.; Sundberg, M. R. J. Am. Chem. Soc. 2000, 122, 1144. doi:10.1021/ja992672z
Return to citation in text: [1] -
van Bochove, M. A.; Swart, M.; Bickelhaupt, F. M. Phys. Chem. Chem. Phys. 2009, 11, 259. doi:10.1039/b813152j
Return to citation in text: [1] -
van Bochove, M. A.; Swart, M.; Bickelhaupt, F. M. ChemPhysChem 2007, 8, 2452. doi:10.1002/cphc.200700488
Return to citation in text: [1] -
Koh, H. J.; Kang, S. J.; Kevill, D. N. Phosphorus, Sulfur Silicon Relat. Elem. 2010, 185, 1404. doi:10.1080/10426500903061525
Return to citation in text: [1] -
Koh, H. J.; Kevill, D. N. Phosphorus, Sulfur Silicon Relat. Elem. 2010, 185, 865. doi:10.1080/10426500903012478
Return to citation in text: [1] -
Koh, H. J.; Kang, S. J.; Kevill, D. N. Phosphorus, Sulfur Silicon Relat. Elem. 2008, 183, 364. doi:10.1080/10426500701734943
Return to citation in text: [1] -
Kevill, D. N.; Koh, H. J. J. Phys. Org. Chem. 2007, 20, 88. doi:10.1002/poc.1124
Return to citation in text: [1] -
Kevill, D. N.; Carver, J. S. Org. Biomol. Chem. 2004, 2, 2040. doi:10.1039/b402093f
Return to citation in text: [1] -
Kevill, D. N.; Miller, B. J. Org. Chem. 2002, 67, 7399. doi:10.1021/jo020467n
Return to citation in text: [1] -
Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Chem. Phys. Lett. 1989, 162, 165. doi:10.1016/0009-2614(89)85118-8
Return to citation in text: [1] -
Staroverov, V. N.; Scuseria, G. E.; Tao, J.; Perdew, J. P. J. Chem. Phys. 2003, 119, 12129. doi:10.1063/1.1626543
Return to citation in text: [1] -
Schäfer, A.; Horn, H.; Ahlrichs, R. J. Chem. Phys. 1992, 97, 2571. doi:10.1063/1.463096
Return to citation in text: [1]
| 1. | Brunel, J. M. Chem. Rev. 2005, 105, 857. doi:10.1021/cr040079g |
| 2. | van Leeuwen, P. W. N. M.; Kamer, P. C. J.; Claver, C.; Pàmies, O.; Dièguez, M. Chem. Rev. 2011, 111, 2077. doi:10.1021/cr1002497 |
| 3. | Fernàndez-Pèrez, H.; Etayo, P.; Panossian, A.; Vidal-Ferran, A. Chem. Rev. 2011, 111, 2119. doi:10.1021/cr100244e |
| 4. |
Seebach, D.; Beck, A. K.; Heckel, A. Angew. Chem. 2001, 113, 96. doi:10.1002/1521-3757(20010105)113:1<96::AID-ANGE96>3.0.CO;2-B
Angew. Chem., Int. Ed. 2001, 40, 92. doi:10.1002/1521-3773(20010105)40:1<92::AID-ANIE92>3.0.CO;2-K |
| 5. |
Teichert, J. F.; Feringa, B. L. Angew. Chem. 2010, 122, 2538. doi:10.1002/ange.200904948
Angew. Chem., Int. Ed. 2010, 49, 2486. doi:10.1002/anie.200904948 |
| 14. | Goldfuss, B.; Löschmann, T.; Kop-Weiershausen, T.; Neudörfl, J.; Rominger, F. Beilstein J. Org. Chem. 2006, 2, No. 7. doi:10.1186/1860-5397-2-7 |
| 41. | von Ragué Schleyer, P.; Lam, L. K. M.; Raber, D. J.; Fry, J. L.; McKervey, M. A.; Alford, J. R.; Cuddy, B. D.; Keizer, V. G.; Geluk, H. W.; Schlatmann, J. L. M. A. J. Am. Chem. Soc. 1970, 92, 5246. doi:10.1021/ja00720a056 |
| 42. | Majerski, Z.; von Ragué Schleyer, P.; Wolf, A. P. J. Am. Chem. Soc. 1970, 92, 5731. doi:10.1021/ja00722a034 |
| 43. | Nickon, A.; Weglein, R. C. J. Am. Chem. Soc. 1975, 97, 1271. doi:10.1021/ja00838a067 |
| 44. | Paquette, L. A.; Waykole, L.; Jendralla, H.; Cottrell, C. E. J. Am. Chem. Soc. 1986, 108, 3739. doi:10.1021/ja00273a031 |
| 45. | Paquette, L. A.; Lanter, J. C.; Johnston, J. N. J. Org. Chem. 1997, 62, 1702. doi:10.1021/jo962019j |
| 46. | Lee, O.-S.; Yang, K.; Kang, K. D.; Koo, I. S.; Kim, C.-K.; Lee, I. J. Comput. Chem. 2004, 25, 1740. doi:10.1002/jcc.20104 |
| 13. | Blanco Trillo, R.; Leven, M.; Neudörfl, J. M.; Goldfuss, B. Adv. Synth. Catal. 2012, 354, 1451. doi:10.1002/adsc.201100924 |
| 14. | Goldfuss, B.; Löschmann, T.; Kop-Weiershausen, T.; Neudörfl, J.; Rominger, F. Beilstein J. Org. Chem. 2006, 2, No. 7. doi:10.1186/1860-5397-2-7 |
| 15. | Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6 |
| 10. | Ackermann, L.; Kapdi, A. R.; Schulzke, C. Org. Lett. 2010, 12, 2298. doi:10.1021/ol100658y |
| 11. | Puckette, T. A. Chem. Ind. 2007, 115, 31. |
| 12. | Timosheva, N. V.; Chandrasekaran, A.; Holmes, R. R. J. Am. Chem. Soc. 2005, 127, 12474. doi:10.1021/ja053422n |
| 15. | Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6 |
| 6. |
Schober, K.; Hartmann, E.; Zhang, H.; Gschwind, R. M. Angew. Chem. 2010, 122, 2855. doi:10.1002/ange.200907247
Angew. Chem., Int. Ed. 2010, 49, 2794. doi:10.1002/anie.200907247 |
| 7. |
Shibasaki, M.; Sasai, H.; Arai, T. Angew. Chem. 1997, 109, 1290. doi:10.1002/ange.19971091204
Angew. Chem., Int. Ed. Engl. 1997, 36, 1236. doi:10.1002/anie.199712361 |
| 8. | Sasai, H.; Arai, T.; Satow, Y.; Houk, K. N.; Shibasaki, M. J. Am. Chem. Soc. 1995, 117, 6194. doi:10.1021/ja00128a005 |
| 9. |
d'Augustin, M.; Palais, L.; Alexakis, A. Angew. Chem. 2005, 117, 1400. doi:10.1002/ange.200462137
Angew. Chem., Int. Ed. 2005, 44, 1376. doi:10.1002/anie.200462137 |
| 34. | Huang, E.; Ranganayakulu, K.; Sorensen, T. S. J. Am. Chem. Soc. 1972, 94, 1779. doi:10.1021/ja00760a079 |
| 35. | Sorensen, T. S. Acc. Chem. Res. 1976, 9, 257. doi:10.1021/ar50103a003 |
| 36. | Starling, S. M.; Vonwiller, S. C.; Reek, J. N. H. J. Org. Chem. 1998, 63, 2262. doi:10.1021/jo972025v |
| 37. | Brown, H. C.; Takeuchi, K. J. Am. Chem. Soc. 1968, 90, 2693. doi:10.1021/ja01012a043 |
| 38. | Farnum, D. G.; Mehta, G. J. Am. Chem. Soc. 1969, 91, 3256. doi:10.1021/ja01040a028 |
| 39. | Brown, H. C.; Takeuchi, K.; Ravindranathan, M. J. Am. Chem. Soc. 1977, 99, 2684. doi:10.1021/ja00450a047 |
| 40. | Farnum, D. G.; Wolf, A. D. J. Am. Chem. Soc. 1974, 96, 5166. doi:10.1021/ja00823a025 |
| 27. | Gliga, A.; Klare, H.; Schumacher, M.; Soki, F.; Neudörfl, J. M.; Goldfuss, B. Eur. J. Org. Chem. 2011, 256–263. doi:10.1002/ejoc.201001295 |
| 28. | Goldfuss, B.; Steigelmann, M.; Khan, S. I.; Houk, K. N. J. Org. Chem. 2000, 65, 77. doi:10.1021/jo991070v |
| 29. | Goldfuss, B.; Khan, S. I.; Houk, K. N. Organometallics 1999, 18, 2927. doi:10.1021/om990184u |
| 30. | Goldfuss, B. Synthesis 2005, 2271. doi:10.1055/s-2005-872107 |
| 31. | Goldfuss, B.; Steigelmann, M.; Löschmann, T.; Schilling, G.; Rominger, F. Chem. – Eur. J. 2005, 11, 4019. doi:10.1002/chem.200500158 |
| 32. | Goldfuss, B.; Steigelmann, M.; Rominger, F.; Urtel, H. Chem. – Eur. J. 2001, 7, 4456. doi:10.1002/1521-3765(20011015)7:20<4456::AID-CHEM4456>3.0.CO;2-S |
| 33. |
Goldfuss, B.; Steigelmann, M.; Rominger, F. Angew. Chem. 2000, 112, 4299. doi:10.1002/1521-3757(20001117)112:22<4299::AID-ANGE4299>3.0.CO;2-O
Angew. Chem., Int. Ed. 2000, 39, 4133. doi:10.1002/1521-3773(20001117)39:22<4133::AID-ANIE4133>3.0.CO;2-X |
| 23. | Steigelmann, M.; Nisar, Y.; Rominger, F.; Goldfuss, B. Chem. – Eur. J. 2002, 8, 5211. doi:10.1002/1521-3765(20021115)8:22<5211::AID-CHEM5211>3.0.CO;2-S |
| 24. | Goldfuss, B.; Steigelmann, M.; Rominger, F. Eur. J. Org. Chem. 2000, 1785. doi:10.1002/(SICI)1099-0690(200005)2000:9<1785::AID-EJOC1785>3.0.CO;2-0 |
| 25. | Goldfuss, B.; Steigelmann, M. J. Mol. Model. 2000, 6, 166. doi:10.1007/s0089400060166 |
| 26. | Leven, M.; Schlörer, N. E.; Neudörfl, J. M.; Goldfuss, B. Chem. – Eur. J. 2010, 16, 13443. doi:10.1002/chem.201001106 |
| 13. | Blanco Trillo, R.; Leven, M.; Neudörfl, J. M.; Goldfuss, B. Adv. Synth. Catal. 2012, 354, 1451. doi:10.1002/adsc.201100924 |
| 15. | Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6 |
| 16. | Lange, D. A.; Neudörfl, J.-M.; Goldfuss, B. Tetrahedron 2006, 62, 3704. doi:10.1016/j.tet.2006.01.060 |
| 17. | Soki, F.; Neudörfl, J.-M.; Goldfuss, B. J. Organomet. Chem. 2008, 693, 2139. doi:10.1016/j.jorganchem.2008.03.013 |
| 18. | Goldfuss, B.; Löschmann, T.; Rominger, F. Chem. – Eur. J. 2001, 7, 2028. doi:10.1002/1521-3765(20010504)7:9<2028::AID-CHEM2028>3.0.CO;2-Y |
| 19. | Goldfuss, B.; Rominger, F. Tetrahedron 2000, 56, 881. doi:10.1016/S0040-4020(99)01077-7 |
| 20. | Goldfuss, B.; Eisenträger, E. Aust. J. Chem. 2000, 53, 209. doi:10.1071/CH99184 |
| 21. | Goldfuss, B.; Löschmann, T.; Rominger, F. Chem. – Eur. J. 2004, 10, 5422. doi:10.1002/chem.200400273 |
| 22. | Soki, F. Synthese und Charakterisierung neuartiger Fencholate und deren Einsatz als Chiralitätsvermittler in enantioselktiven C–C Knüpfungsreaktionen. Ph.D. Thesis, University of Cologne, Germany, 2008. |
| 15. | Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6 |
| 17. | Soki, F.; Neudörfl, J.-M.; Goldfuss, B. J. Organomet. Chem. 2008, 693, 2139. doi:10.1016/j.jorganchem.2008.03.013 |
| 47. | Milov, A. A.; Minyaev, R. M.; Minkin, V. I. J. Phys. Chem. A 2011, 115, 12973. doi:10.1021/jp2042119 |
| 48. | Kubo, K.; Nakazawa, H.; Kawamura, K.; Mizuta, T.; Miyoshi, K. J. Am. Chem. Soc. 1998, 120, 6715. doi:10.1021/ja980146m |
| 15. | Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6 |
| 14. | Goldfuss, B.; Löschmann, T.; Kop-Weiershausen, T.; Neudörfl, J.; Rominger, F. Beilstein J. Org. Chem. 2006, 2, No. 7. doi:10.1186/1860-5397-2-7 |
| 15. | Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6 |
| 58. | Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Chem. Phys. Lett. 1989, 162, 165. doi:10.1016/0009-2614(89)85118-8 |
| 59. | Staroverov, V. N.; Scuseria, G. E.; Tao, J.; Perdew, J. P. J. Chem. Phys. 2003, 119, 12129. doi:10.1063/1.1626543 |
| 60. | Schäfer, A.; Horn, H.; Ahlrichs, R. J. Chem. Phys. 1992, 97, 2571. doi:10.1063/1.463096 |
| 15. | Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6 |
| 15. | Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6 |
| 52. | Koh, H. J.; Kang, S. J.; Kevill, D. N. Phosphorus, Sulfur Silicon Relat. Elem. 2010, 185, 1404. doi:10.1080/10426500903061525 |
| 53. | Koh, H. J.; Kevill, D. N. Phosphorus, Sulfur Silicon Relat. Elem. 2010, 185, 865. doi:10.1080/10426500903012478 |
| 54. | Koh, H. J.; Kang, S. J.; Kevill, D. N. Phosphorus, Sulfur Silicon Relat. Elem. 2008, 183, 364. doi:10.1080/10426500701734943 |
| 55. | Kevill, D. N.; Koh, H. J. J. Phys. Org. Chem. 2007, 20, 88. doi:10.1002/poc.1124 |
| 56. | Kevill, D. N.; Carver, J. S. Org. Biomol. Chem. 2004, 2, 2040. doi:10.1039/b402093f |
| 57. | Kevill, D. N.; Miller, B. J. Org. Chem. 2002, 67, 7399. doi:10.1021/jo020467n |
| 13. | Blanco Trillo, R.; Leven, M.; Neudörfl, J. M.; Goldfuss, B. Adv. Synth. Catal. 2012, 354, 1451. doi:10.1002/adsc.201100924 |
| 14. | Goldfuss, B.; Löschmann, T.; Kop-Weiershausen, T.; Neudörfl, J.; Rominger, F. Beilstein J. Org. Chem. 2006, 2, No. 7. doi:10.1186/1860-5397-2-7 |
| 15. | Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6 |
| 49. | Dobado, J. A.; Martinez-Garcia, H.; Molina, J.; Sundberg, M. R. J. Am. Chem. Soc. 2000, 122, 1144. doi:10.1021/ja992672z |
| 50. | van Bochove, M. A.; Swart, M.; Bickelhaupt, F. M. Phys. Chem. Chem. Phys. 2009, 11, 259. doi:10.1039/b813152j |
| 51. | van Bochove, M. A.; Swart, M.; Bickelhaupt, F. M. ChemPhysChem 2007, 8, 2452. doi:10.1002/cphc.200700488 |
© 2015 Blanco Trillo et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)