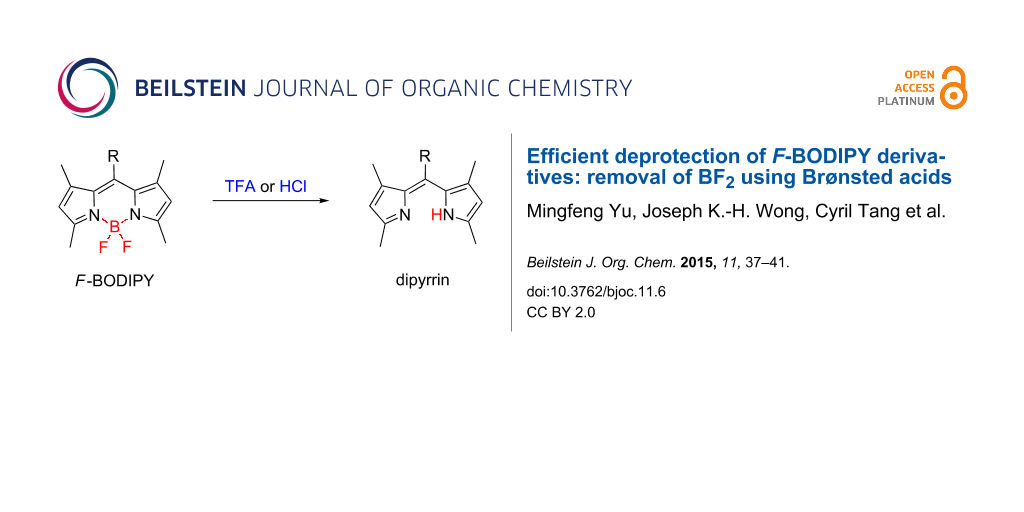
Abstract
The effective and efficient removal of the BF2 moiety from F-BODIPY derivatives has been achieved using two common Brønsted acids; treatment with trifluoroacetic acid (TFA) or methanolic hydrogen chloride (HCl) followed by work-up with Ambersep® 900 resin (hydroxide form) effects this conversion in near-quantitative yields. Compared to existing methods, these conditions are relatively mild and operationally simple, requiring only reaction at room temperature for six hours (TFA) or overnight (HCl).

Graphical Abstract
Findings
Compounds incorporating the 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (F-BODIPY) motif 1 have found widespread use in fluorescent molecular probes [1,2], photovoltaic devices [3,4] and photodynamic therapy agents [5-8]. Accordingly, there is considerable interest in extending and diversifying the F-BODIPY framework [9]. F-BODIPYs are readily prepared by condensing aldehydes, acyl chlorides or anhydrides with pyrroles and trapping the resulting dipyrrin in situ with boron trifluoride [9-11]. F-BODIPYs are generally stable and chemically robust, with photophysical properties that facilitate chromatographic purification. The parent dipyrrins are more difficult to handle but have a range of potential applications in dye and porphyrin syntheses, metal ion coordination and supramolecular chemistry [12]. Methods to enable the functionalization of dipyrrins by temporarily complexing with tin or zinc have been investigated [13,14]. More recently, the F-BODIPY motif has been envisaged as a means of protecting the dipyrrin, to enable chemical modification and purification before removal of the BF2 unit (i.e., deprotection) to reveal the functionalized dipyrrin. With this goal in mind, several recent reports have detailed methods for converting F-BODIPYs 1 to the parent dipyrrins 2 (Scheme 1) [15-19].
![[1860-5397-11-6-i1]](/bjoc/content/inline/1860-5397-11-6-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Conversion of F-BODIPYs 1 to the parent dipyrrins 2.
Scheme 1: Conversion of F-BODIPYs 1 to the parent dipyrrins 2.
Crawford and Thompson first proposed the BF2 unit as a protecting group for dipyrrins in 2010, and applied strong base under forcing conditions to effect the deprotection: potassium tert-butoxide in tert-butanol/water with microwave heating to 90–140 °C (26–98% yield) [15,16]. Kusaka et al. built on this strategy to achieve deboration of F-BODIPYs using sodium tert-butoxide in refluxing toluene (59–83% yield) en route to bis(dipyrrinato)zinc(II) complexes [17]. Two very recent reports have deployed Lewis acids to achieve this transformation: Thompson and co-workers used boron trihalides in dichloromethane under anhydrous conditions, followed by treatment with acetone/water (10:1) to achieve quantitative removal of the BF2 moiety [18]; Ravikanth and co-workers screened a range of metal-based Lewis acids (ZrCl4, TiCl4, AlCl3, Sc(OTf)3, SnCl4) and reported yields up to 96% using ZrCl4 in refluxing methanol/acetonitrile [19].
Related efforts have wrought substitution at boron in F-BODIPY analogues without removing it from the dipyrrin. For example, Lundrigan et al. have effected direct conversion of F-BODIPYs to Cl-BODIPYs using boron trichloride [20], while Jiang et al. achieved substitution of fluoride by acetate using trimethylsilyl chloride followed by acetic acid [21].
Herein we report the effective and efficient removal of the BF2 moiety from F-BODIPY derivatives using two common Brønsted acids: treatment with trifluoroacetic acid (TFA) or methanolic hydrogen chloride (HCl) at room temperature followed by work-up with Ambersep® 900 resin (hydroxide form) achieves this conversion in near-quantitative yields.
We have an ongoing interest in triazolyl-cyclam derivatives incorporating fluorescent dyes for sensing applications [22-25]. Looking to extend these systems to incorporate an F-BODIPY motif, we have synthesized the Boc-protected triazolyl-cyclam/F-BODIPY derivatives 3 and 4 from 2,4-dimethyl-1H-pyrrole (5), 4-nitrobenzaldehyde (6, Scheme 2A) and 4-bromobenzaldehyde (7, Scheme 2B) respectively (see Supporting Information File 1 for experimental data).
![[1860-5397-11-6-i2]](/bjoc/content/inline/1860-5397-11-6-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the triazolyl-cyclam/F-BODIPY conjugates 3 (A) and 4 (B). Reagents and conditions: (a) (i) 2,4-dimethyl-1H-pyrrole (5), TFA, DCM, rt, overnight; (ii) DDQ, rt, 2 h; (iii) Et3N, BF3·OEt2, rt, overnight, 27%; (b) NH2NH2·H2O, 10% Pd/C, EtOH, reflux, 2 h, 90%; (c) (i) 1 M HCl (aq), CH3OH, NaNO2, H2O, 0 °C, 1 h; (ii) NaN3, H2O, rt, 2 h, 71%; (d) propargyl-tri-Boc cyclam 12, CuSO4·5H2O, sodium ascorbate, THF/H2O (7:3), 50 °C, 12 h, 100%; (e) trimethylsilylacetylene, CuI, Pd(PPh3)4, Et3N, THF, rt, overnight, 100%; (f) K2CO3, CH3OH, rt, overnight, 71%; (g) (i) 2,4-dimethyl-1H-pyrrole (5), TFA, DCM, rt, overnight; (ii) DDQ, rt, 2 h; (iii) Et3N, BF3·OEt2, rt, overnight, 24%; (h) 2-azidoethyl-tri-Boc cyclam 13, CuSO4·5H2O, sodium ascorbate, THF/H2O (7:3), 50 °C, 12 h, 91%.
Scheme 2: Synthesis of the triazolyl-cyclam/F-BODIPY conjugates 3 (A) and 4 (B). Reagents and conditions: (a)...
Preparation of nitro-F-BODIPY 8 was initially attempted by adapting the reported synthetic method [10]. However the ethereal complex 8·Et2O was isolated by flash column chromatography, rather than 8 itself (readily evident in the 1H and 13C NMR spectra). To the best of our knowledge, the complexation of Et2O in this way has not been reported in previous syntheses of F-BODIPY derivatives. Considering that the same procedures were used to synthesize and purify 8 as we used to prepare 11 free from Et2O (vide infra), and given previous reports on the ability of nitrogen oxides (e.g., NO, N2O3 and N2O4) to complex with boron trifluoride [26-29], the nitro group is presumably the key factor in the complexation of 8 with Et2O. Washing with aqueous and organic solvents did not completely remove the complexed ether, but uncomplexed 8 was obtained after recrystallization from ethyl acetate. The structure of this F-BODIPY derivative was determined by NMR and X-ray crystallography (Figure 1), and its purity confirmed by elemental analysis. Conversion of the nitro compound 8 to the corresponding azide 9 was achieved by palladium-catalyzed reduction [10] followed by diazotization of the amine and subsequent substitution with azide [30]. 4-Bromobenzaldehyde (7) was readily converted to ethynyl-F-BODIPY 11 according to the literature procedures [11,31]. Azido-F-BODIPY 9 and ethynyl-F-BODIPY 11 were reacted respectively with the complementary propargyl-tri-Boc cyclam 12 [23,32] and 2-azidoethyl-tri-Boc cyclam 13 [24,25] under the modified click conditions we have reported previously [24] to generate the Boc-protected triazolyl-cyclam/F-BODIPY conjugates 3 and 4 in excellent yields.
![[1860-5397-11-6-1]](/bjoc/content/figures/1860-5397-11-6-1.png?scale=1.5&max-width=1024&background=FFFFFF)
Figure 1: An ORTEP plot of nitro-F-BODIPY 8 at the 50% probability level. A CIF file for the structure determination is available as Supporting Information File 2 and is also available on request from the Cambridge Crystallographic Data Centre as deposition 1018518.
Figure 1: An ORTEP plot of nitro-F-BODIPY 8 at the 50% probability level. A CIF file for the structure determ...
In attempting to remove the Boc groups from 3 and 4, we have discovered a facile method for the removal of BF2 from these F-BODIPY derivatives using Brønsted acids (Scheme 3, Table 1) (see Supporting Information File 1 for experimental data). Thus 3 was converted efficiently to 14 (96–99% yield) with the loss of three Boc groups and the BF2 moiety, using either a mixture of TFA/DCM/H2O (90:5:5) or a methanolic solution of hydrogen chloride (2.8 M) at room temperature, followed by basification with a suspension of excess Ambersep® 900 resin (hydroxide form) in methanol. Similarly, 4 afforded 15 (96–98% yield) under the same reaction conditions.
![[1860-5397-11-6-i3]](/bjoc/content/inline/1860-5397-11-6-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Conversion of F-BODIPYs to dipyrrins using Brønsted acids. Reagents and conditions: (a) (i) TFA/DCM/H2O (90:5:5), rt, 6 h; or 2.8 M HCl in CH3OH, rt, 12–48 h; (ii) Ambersep® 900 resin (hydroxide form), CH3OH, rt, 15 min (see Table 1 for yields).
Scheme 3: Conversion of F-BODIPYs to dipyrrins using Brønsted acids. Reagents and conditions: (a) (i) TFA/DCM...
Table 1: Removal of BF2 from F-BODIPYs using Brønsted acids.
| F-BODIPY | dipyrrin | yield (%) | |
|---|---|---|---|
| TFA | HCl | ||
| 3 | 14 | 99 | 96 |
| 4 | 15 | 96 | 98 |
| 8 | 16 | 100 | 92a |
| 9 | 17 | 99 | 99 |
| 11 | 18 | 53b | 53b |
aExtended reaction time (48 hours) was required. bAnalytically pure material was obtained by HPLC purification.
In an initial investigation of the scope of this transformation, each of the F-BODIPY derivatives prepared in this study was subjected to the reaction conditions that rendered BF2-removal from 3 and 4 (Scheme 3, Table 1). Nitro-F-BODIPY 8 was readily converted to dipyrrin 16 by the TFA method in quantitative yield (100%). With this substrate, the HCl method required an extended reaction time (48 hours) to give 16 in excellent yield (92%). Near-quantitative (99%) conversion of azido-F-BODIPY 9 to dipyrrin 17 was achieved using both methods. Ethynyl-F-BODIPY 11 was successfully converted to dipyrrin 18 using both Brønsted acids: LC–MS analysis (see Supporting Information File 1) revealed the desired 18 as the major product at m/z 301.2, however, the crude product also contained a minor contaminant at m/z 319.3, consistent with addition of water to the alkyne. 1H NMR analysis indicated that this impurity was present at ≤5% abundance, but HPLC purification was required to generate analytically pure 18 which compromised the final yields (53% for both methods).
We are aware of two previous reports investigating the treatment of BODIPYs with Brønsted acids. Yang et al. used 11B NMR to monitor the stability of BODIPYs in the presence of di- or trichloroacetic acid, reporting ‘partial decomposition’ of an F-BODIPY derivative without characterizing the breakdown product(s) [33]. While Liras et al. reported the synthesis of a single aminodipyrrin product from the corresponding 3-amino- and 3-acetamido-F-BODIPY precursors, using ‘HCl-catalyzed deacetylation conditions’ (HCl in ethanol) to effect both deacetylation and deboration [34].
Conclusion
In conclusion, we have serendipitously achieved efficient removal of the BF2 moiety from F-BODIPY derivatives using either the organic acid TFA or the inorganic acid HCl. These conditions are complementary to those previously reported for converting F-BODIPYs to the parent dipyrrins using either strong bases or Lewis acids. Compared to existing methods, the Brønsted acid conditions are relatively mild and operationally simple, requiring only reactions at room temperature for six hours (TFA) or overnight (HCl). Work is underway to further optimize these conditions and explore the scope of this reaction with a wider range of F-BODIPY derivatives.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data; crystallographic information for 8; 1H, 13C, 11B & 19F NMR spectra of novel compounds 3, 4, 14, 15, 16–18; LC–MS trace of crude 18. | ||
| Format: PDF | Size: 1.9 MB | Download |
| Supporting Information File 2: CIF file of 8, CCDC 1018518. | ||
| Format: CIF | Size: 21.5 KB | Download |
Acknowledgements
We thank the National Breast Cancer Foundation for a Novel Concept Grant (NC-10-69) and the Australian Research Council for funding (DP120104035). M. Yu was supported by a University of Sydney International Scholarship (USydIS) and J. K.-H. Wong by an Australian Postgraduate Award (APA) from the Australian Government.
References
-
Boens, N.; Leen, V.; Dehaen, W. Chem. Soc. Rev. 2012, 41, 1130–1172. doi:10.1039/c1cs15132k
Return to citation in text: [1] -
Boens, N.; Qin, W.; Baruah, M.; De Borggraeve, W. M.; Filarowski, A.; Smisdom, N.; Ameloot, M.; Crovetto, L.; Talavera, E. M.; Alvarez-Pez, J. M. Chem. – Eur. J. 2011, 17, 10924–10934. doi:10.1002/chem.201002280
Return to citation in text: [1] -
Bessette, A.; Hanan, G. S. Chem. Soc. Rev. 2014, 43, 3342–3405. doi:10.1039/c3cs60411j
Return to citation in text: [1] -
Kolemen, S.; Bozdemir, O. A.; Cakmak, Y.; Barin, G.; Erten-Ela, S.; Marszalek, M.; Yum, J.-H.; Zakeeruddin, S. M.; Nazeeruddin, M. K.; Grätzel, M.; Akkaya, E. U. Chem. Sci. 2011, 2, 949–954. doi:10.1039/c0sc00649a
Return to citation in text: [1] -
Cakmak, Y.; Kolemen, S.; Duman, S.; Dede, Y.; Dolen, Y.; Kilic, B.; Kostereli, Z.; Yildirim, L. T.; Dogan, A. L.; Guc, D.; Akkaya, E. U. Angew. Chem., Int. Ed. 2011, 50, 11937–11941. doi:10.1002/anie.201105736
Return to citation in text: [1] -
Erbas, S.; Gorgulu, A.; Kocakusakogullari, M.; Akkaya, E. U. Chem. Commun. 2009, 4956–4958. doi:10.1039/b908485a
Return to citation in text: [1] -
McDonnell, S. O.; Hall, M. J.; Allen, L. T.; Byrne, A.; Gallagher, W. M.; O'Shea, D. F. J. Am. Chem. Soc. 2005, 127, 16360–16361. doi:10.1021/ja0553497
Return to citation in text: [1] -
Ozlem, S.; Akkaya, E. U. J. Am. Chem. Soc. 2008, 131, 48–49. doi:10.1021/ja808389t
Return to citation in text: [1] -
Loudet, A.; Burgess, K. Chem. Rev. 2007, 107, 4891–4932. doi:10.1021/cr078381n
Return to citation in text: [1] [2] -
Cui, A.; Peng, X.; Fan, J.; Chen, X.; Wu, Y.; Guo, B. J. Photochem. Photobiol., A 2007, 186, 85–92. doi:10.1016/j.jphotochem.2006.07.015
Return to citation in text: [1] [2] [3] -
Li, Z.; Bittman, R. J. Org. Chem. 2007, 72, 8376–8382. doi:10.1021/jo701475q
Return to citation in text: [1] [2] -
Wood, T. E.; Thompson, A. Chem. Rev. 2007, 107, 1831–1861. doi:10.1021/cr050052c
Return to citation in text: [1] -
Tamaru, S.-i.; Yu, L.; Youngblood, W. J.; Muthukumaran, K.; Taniguchi, M.; Lindsey, J. S. J. Org. Chem. 2004, 69, 765–777. doi:10.1021/jo035622s
Return to citation in text: [1] -
Sáez Díaz, R. I.; Bennett, S. M.; Thompson, A. ChemMedChem 2009, 4, 742–745. doi:10.1002/cmdc.200900003
Return to citation in text: [1] -
Crawford, S. M.; Thompson, A. Org. Lett. 2010, 12, 1424–1427. doi:10.1021/ol902908j
Return to citation in text: [1] [2] -
Smithen, D. A.; Baker, A. E. G.; Offman, M.; Crawford, S. M.; Cameron, T. S.; Thompson, A. J. Org. Chem. 2012, 77, 3439–3453. doi:10.1021/jo3002003
Return to citation in text: [1] [2] -
Kusaka, S.; Sakamoto, R.; Kitagawa, Y.; Okumura, M.; Nishihara, H. Chem. – Asian J. 2012, 7, 907–910. doi:10.1002/asia.201200131
Return to citation in text: [1] [2] -
Lundrigan, T.; Cameron, T. S.; Thompson, A. Chem. Commun. 2014, 50, 7028–7031. doi:10.1039/c4cc02706j
Return to citation in text: [1] [2] -
Lakshmi, V.; Chatterjee, T.; Ravikanth, M. Eur. J. Org. Chem. 2014, 2105–2110. doi:10.1002/ejoc.201301662
Return to citation in text: [1] [2] -
Lundrigan, T.; Thompson, A. J. Org. Chem. 2012, 78, 757–761. doi:10.1021/jo302277d
Return to citation in text: [1] -
Jiang, X.-D.; Zhang, J.; Furuyama, T.; Zhao, W. Org. Lett. 2011, 14, 248–251. doi:10.1021/ol2030229
Return to citation in text: [1] -
Ast, S.; Rutledge, P. J.; Todd, M. H. Eur. J. Inorg. Chem. 2012, 5611–5615. doi:10.1002/ejic.201201072
Return to citation in text: [1] -
Yu, M.; Yu, Q.; Rutledge, P. J.; Todd, M. H. ChemBioChem 2013, 14, 224–229. doi:10.1002/cbic.201200637
Return to citation in text: [1] [2] -
Yu, M.; Ast, S.; Yu, Q.; Lo, A. T. S.; Flehr, R.; Todd, M. H.; Rutledge, P. J. PLoS One 2014, 9, e100761. doi:10.1371/journal.pone.0100761
Return to citation in text: [1] [2] [3] -
Lau, Y. H.; Price, J. R.; Todd, M. H.; Rutledge, P. J. Chem. – Eur. J. 2011, 17, 2850–2858. doi:10.1002/chem.201002477
Return to citation in text: [1] [2] -
Bachman, G. B.; Feuer, H.; Bluestein, B. R.; Vogt, C. M. J. Am. Chem. Soc. 1955, 77, 6188–6190. doi:10.1021/ja01628a026
Return to citation in text: [1] -
Pradeep, T.; Sreekanth, C. S.; Rao, C. N. R. J. Chem. Phys. 1989, 90, 4704–4708. doi:10.1063/1.456616
Return to citation in text: [1] -
Bachman, G. B.; Hokama, T. Complex of Boron Trifluoride and Dinitrogen Trioxide. U.S. Patent 2,829,029, April 1, 1958.
Return to citation in text: [1] -
Batey, H. H.; Sisler, H. H. J. Am. Chem. Soc. 1952, 74, 3408–3410. doi:10.1021/ja01133a501
Return to citation in text: [1] -
Jose, J.; Ueno, Y.; Castro, J. C.; Li, L.; Burgess, K. Tetrahedron Lett. 2009, 50, 6442–6445. doi:10.1016/j.tetlet.2009.08.130
Return to citation in text: [1] -
Wautelet, P.; Le Moigne, J.; Videva, V.; Turek, P. J. Org. Chem. 2003, 68, 8025–8036. doi:10.1021/jo034723n
Return to citation in text: [1] -
Yu, M.; Price, J. R.; Jensen, P.; Lovitt, C. J.; Shelper, T.; Duffy, S.; Windus, L. C.; Avery, V. M.; Rutledge, P. J.; Todd, M. H. Inorg. Chem. 2011, 50, 12823–12835. doi:10.1021/ic2020012
Return to citation in text: [1] -
Yang, L.; Simionescu, R.; Lough, A.; Yan, H. Dyes Pigm. 2011, 91, 264–267. doi:10.1016/j.dyepig.2011.03.027
Return to citation in text: [1] -
Liras, M.; Bañuelos Prieto, J.; Pintado-Sierra, M.; López Arbeloa, F.; García-Moreno, I.; Costela, Á.; Infantes, L.; Sastre, R.; Amat-Guerri, F. Org. Lett. 2007, 9, 4183–4186. doi:10.1021/ol701674b
Return to citation in text: [1]
| 1. | Boens, N.; Leen, V.; Dehaen, W. Chem. Soc. Rev. 2012, 41, 1130–1172. doi:10.1039/c1cs15132k |
| 2. | Boens, N.; Qin, W.; Baruah, M.; De Borggraeve, W. M.; Filarowski, A.; Smisdom, N.; Ameloot, M.; Crovetto, L.; Talavera, E. M.; Alvarez-Pez, J. M. Chem. – Eur. J. 2011, 17, 10924–10934. doi:10.1002/chem.201002280 |
| 9. | Loudet, A.; Burgess, K. Chem. Rev. 2007, 107, 4891–4932. doi:10.1021/cr078381n |
| 10. | Cui, A.; Peng, X.; Fan, J.; Chen, X.; Wu, Y.; Guo, B. J. Photochem. Photobiol., A 2007, 186, 85–92. doi:10.1016/j.jphotochem.2006.07.015 |
| 11. | Li, Z.; Bittman, R. J. Org. Chem. 2007, 72, 8376–8382. doi:10.1021/jo701475q |
| 22. | Ast, S.; Rutledge, P. J.; Todd, M. H. Eur. J. Inorg. Chem. 2012, 5611–5615. doi:10.1002/ejic.201201072 |
| 23. | Yu, M.; Yu, Q.; Rutledge, P. J.; Todd, M. H. ChemBioChem 2013, 14, 224–229. doi:10.1002/cbic.201200637 |
| 24. | Yu, M.; Ast, S.; Yu, Q.; Lo, A. T. S.; Flehr, R.; Todd, M. H.; Rutledge, P. J. PLoS One 2014, 9, e100761. doi:10.1371/journal.pone.0100761 |
| 25. | Lau, Y. H.; Price, J. R.; Todd, M. H.; Rutledge, P. J. Chem. – Eur. J. 2011, 17, 2850–2858. doi:10.1002/chem.201002477 |
| 10. | Cui, A.; Peng, X.; Fan, J.; Chen, X.; Wu, Y.; Guo, B. J. Photochem. Photobiol., A 2007, 186, 85–92. doi:10.1016/j.jphotochem.2006.07.015 |
| 5. | Cakmak, Y.; Kolemen, S.; Duman, S.; Dede, Y.; Dolen, Y.; Kilic, B.; Kostereli, Z.; Yildirim, L. T.; Dogan, A. L.; Guc, D.; Akkaya, E. U. Angew. Chem., Int. Ed. 2011, 50, 11937–11941. doi:10.1002/anie.201105736 |
| 6. | Erbas, S.; Gorgulu, A.; Kocakusakogullari, M.; Akkaya, E. U. Chem. Commun. 2009, 4956–4958. doi:10.1039/b908485a |
| 7. | McDonnell, S. O.; Hall, M. J.; Allen, L. T.; Byrne, A.; Gallagher, W. M.; O'Shea, D. F. J. Am. Chem. Soc. 2005, 127, 16360–16361. doi:10.1021/ja0553497 |
| 8. | Ozlem, S.; Akkaya, E. U. J. Am. Chem. Soc. 2008, 131, 48–49. doi:10.1021/ja808389t |
| 20. | Lundrigan, T.; Thompson, A. J. Org. Chem. 2012, 78, 757–761. doi:10.1021/jo302277d |
| 3. | Bessette, A.; Hanan, G. S. Chem. Soc. Rev. 2014, 43, 3342–3405. doi:10.1039/c3cs60411j |
| 4. | Kolemen, S.; Bozdemir, O. A.; Cakmak, Y.; Barin, G.; Erten-Ela, S.; Marszalek, M.; Yum, J.-H.; Zakeeruddin, S. M.; Nazeeruddin, M. K.; Grätzel, M.; Akkaya, E. U. Chem. Sci. 2011, 2, 949–954. doi:10.1039/c0sc00649a |
| 21. | Jiang, X.-D.; Zhang, J.; Furuyama, T.; Zhao, W. Org. Lett. 2011, 14, 248–251. doi:10.1021/ol2030229 |
| 15. | Crawford, S. M.; Thompson, A. Org. Lett. 2010, 12, 1424–1427. doi:10.1021/ol902908j |
| 16. | Smithen, D. A.; Baker, A. E. G.; Offman, M.; Crawford, S. M.; Cameron, T. S.; Thompson, A. J. Org. Chem. 2012, 77, 3439–3453. doi:10.1021/jo3002003 |
| 18. | Lundrigan, T.; Cameron, T. S.; Thompson, A. Chem. Commun. 2014, 50, 7028–7031. doi:10.1039/c4cc02706j |
| 15. | Crawford, S. M.; Thompson, A. Org. Lett. 2010, 12, 1424–1427. doi:10.1021/ol902908j |
| 16. | Smithen, D. A.; Baker, A. E. G.; Offman, M.; Crawford, S. M.; Cameron, T. S.; Thompson, A. J. Org. Chem. 2012, 77, 3439–3453. doi:10.1021/jo3002003 |
| 17. | Kusaka, S.; Sakamoto, R.; Kitagawa, Y.; Okumura, M.; Nishihara, H. Chem. – Asian J. 2012, 7, 907–910. doi:10.1002/asia.201200131 |
| 18. | Lundrigan, T.; Cameron, T. S.; Thompson, A. Chem. Commun. 2014, 50, 7028–7031. doi:10.1039/c4cc02706j |
| 19. | Lakshmi, V.; Chatterjee, T.; Ravikanth, M. Eur. J. Org. Chem. 2014, 2105–2110. doi:10.1002/ejoc.201301662 |
| 19. | Lakshmi, V.; Chatterjee, T.; Ravikanth, M. Eur. J. Org. Chem. 2014, 2105–2110. doi:10.1002/ejoc.201301662 |
| 13. | Tamaru, S.-i.; Yu, L.; Youngblood, W. J.; Muthukumaran, K.; Taniguchi, M.; Lindsey, J. S. J. Org. Chem. 2004, 69, 765–777. doi:10.1021/jo035622s |
| 14. | Sáez Díaz, R. I.; Bennett, S. M.; Thompson, A. ChemMedChem 2009, 4, 742–745. doi:10.1002/cmdc.200900003 |
| 12. | Wood, T. E.; Thompson, A. Chem. Rev. 2007, 107, 1831–1861. doi:10.1021/cr050052c |
| 17. | Kusaka, S.; Sakamoto, R.; Kitagawa, Y.; Okumura, M.; Nishihara, H. Chem. – Asian J. 2012, 7, 907–910. doi:10.1002/asia.201200131 |
| 30. | Jose, J.; Ueno, Y.; Castro, J. C.; Li, L.; Burgess, K. Tetrahedron Lett. 2009, 50, 6442–6445. doi:10.1016/j.tetlet.2009.08.130 |
| 26. | Bachman, G. B.; Feuer, H.; Bluestein, B. R.; Vogt, C. M. J. Am. Chem. Soc. 1955, 77, 6188–6190. doi:10.1021/ja01628a026 |
| 27. | Pradeep, T.; Sreekanth, C. S.; Rao, C. N. R. J. Chem. Phys. 1989, 90, 4704–4708. doi:10.1063/1.456616 |
| 28. | Bachman, G. B.; Hokama, T. Complex of Boron Trifluoride and Dinitrogen Trioxide. U.S. Patent 2,829,029, April 1, 1958. |
| 29. | Batey, H. H.; Sisler, H. H. J. Am. Chem. Soc. 1952, 74, 3408–3410. doi:10.1021/ja01133a501 |
| 10. | Cui, A.; Peng, X.; Fan, J.; Chen, X.; Wu, Y.; Guo, B. J. Photochem. Photobiol., A 2007, 186, 85–92. doi:10.1016/j.jphotochem.2006.07.015 |
| 33. | Yang, L.; Simionescu, R.; Lough, A.; Yan, H. Dyes Pigm. 2011, 91, 264–267. doi:10.1016/j.dyepig.2011.03.027 |
| 34. | Liras, M.; Bañuelos Prieto, J.; Pintado-Sierra, M.; López Arbeloa, F.; García-Moreno, I.; Costela, Á.; Infantes, L.; Sastre, R.; Amat-Guerri, F. Org. Lett. 2007, 9, 4183–4186. doi:10.1021/ol701674b |
| 24. | Yu, M.; Ast, S.; Yu, Q.; Lo, A. T. S.; Flehr, R.; Todd, M. H.; Rutledge, P. J. PLoS One 2014, 9, e100761. doi:10.1371/journal.pone.0100761 |
| 25. | Lau, Y. H.; Price, J. R.; Todd, M. H.; Rutledge, P. J. Chem. – Eur. J. 2011, 17, 2850–2858. doi:10.1002/chem.201002477 |
| 24. | Yu, M.; Ast, S.; Yu, Q.; Lo, A. T. S.; Flehr, R.; Todd, M. H.; Rutledge, P. J. PLoS One 2014, 9, e100761. doi:10.1371/journal.pone.0100761 |
| 11. | Li, Z.; Bittman, R. J. Org. Chem. 2007, 72, 8376–8382. doi:10.1021/jo701475q |
| 31. | Wautelet, P.; Le Moigne, J.; Videva, V.; Turek, P. J. Org. Chem. 2003, 68, 8025–8036. doi:10.1021/jo034723n |
| 23. | Yu, M.; Yu, Q.; Rutledge, P. J.; Todd, M. H. ChemBioChem 2013, 14, 224–229. doi:10.1002/cbic.201200637 |
| 32. | Yu, M.; Price, J. R.; Jensen, P.; Lovitt, C. J.; Shelper, T.; Duffy, S.; Windus, L. C.; Avery, V. M.; Rutledge, P. J.; Todd, M. H. Inorg. Chem. 2011, 50, 12823–12835. doi:10.1021/ic2020012 |
© 2015 Yu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)