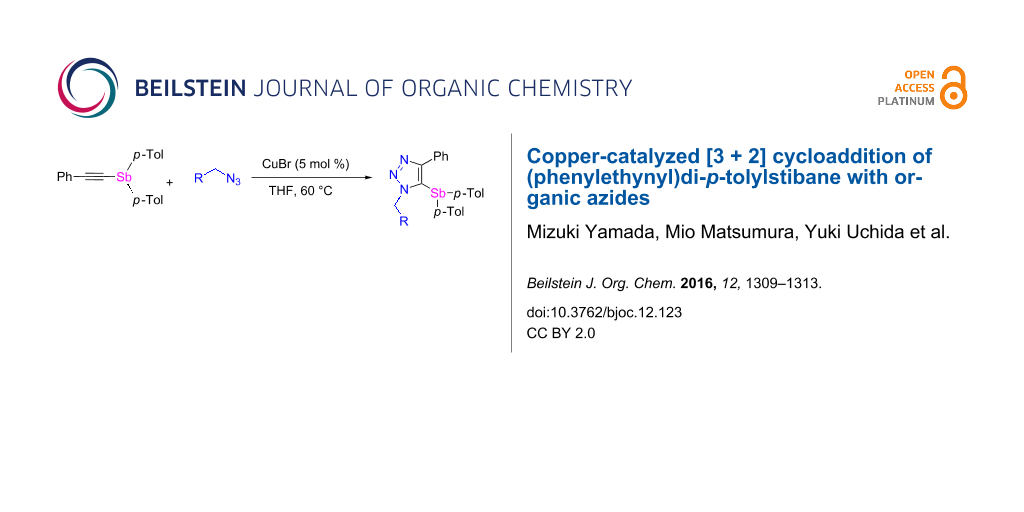
Abstract
Trisubstituted 5-stibano-1H-1,2,3-triazoles were synthesized in moderate to excellent yields by the Cu-catalyzed [3 + 2] cycloaddition of a ethynylstibane with organic azides in the presence of CuBr (5 mol %) under aerobic conditions. The reaction of 5-stibanotriazole with HCl, I2, and NOBF4 afforded 1-benzyl-4-phenyltriazole, 1-benzyl-5-iodo-4-phenyltriazole, and a pentavalent organoantimony compound, respectively.

Graphical Abstract
Introduction
The 1,3-dipolar azide–alkyne cycloaddition (AAC) has been effective for the synthesis of a wide variety of 1,2,3-triazoles [1]. However, this reaction has some limitations such as the requirement of high temperature and the generation of regioisomers. In 2002 Sharpless [2] and Meldal [3] independently reported that the addition of catalytic amounts of copper reagent in the AAC allow the reaction to proceed under milder reaction conditions, and there was also an effect on the regioselectivity for the synthesis of 1,4-disubstituted 1,2,3-triazoles. Since then, the CuAAC has been widely applied in organic synthesis [4-12], molecular biology [13-17], and materials science [18-20]. There are many reports of CuAACs by using terminal alkynes (including metal acetylides) [4-20]. But the use of internal alkynes in CuAACs for the synthesis of 1,4,5-trisubstituted 1,2,3-triazoles is a more challenging area because of the difficulty in regiocontrol based on the increased steric hindrance [21,22]. A regioselective CuAAC synthesis of fully substituted 1,2,3-triazoles having group 15 (P, Bi) elements as substituents at the C-5 position was recently attempted. Li et al. reported that the cycloaddition of alkynylphosphonate with benzyl azide did not generate 1,2,3-triazolyl-5-phosphonates, but a three-component reaction of a terminal alkyne, an organic azide, and an H-phosphate in the presence of CuCl2 (10 mol %) and triethylamine (2 equiv) afforded the desired 1,2,3-triazolyl-5-phosphonates [23]. Fokin et al. carried out the reaction of ethynylbismuthane with organic azides using CuOTf (5 mol %) and isolated 5-bismuthano-1,2,3-triazoles (Bi), which could be employed as versatile building blocks in chemical synthesis [24]. One drawback of the Cu-catalyzed cycloaddition of alkynylbismuthanes is the requirement of alkyne derivatives based on the phenothiabismuthane 5,5-dioxide framework for stabilization. The utility of organoantimony compounds in organic synthesis has attracted much interest during the last two decades [25,26]. Trivalent organoantimony compounds (stibanes) such as aryl- and ethynylstibanes are useful transmetalation agents in Pd-catalyzed cross-coupling reactions with aryl halides and acyl chlorides [27-32]. Stibanes have many advantages such as the handle ability without special care, low toxicity, and availability. Therefore, the synthesis and reactivity of novel stibanes are important for the development of effective organic reagents. However, to the best of our knowledge, there have been no reports concerning the synthesis of 1,4,5-trisubstituted 5-stibano-1,2,3-triazoles. Herein, we report a novel CuAAC of a simple alkynylstibane, (phenylethynyl)di-p-tolylstibane, with organic azides to form fully substituted 5-organostibano-1,2,3-triazoles.
Results and Discussion
We initially determined the optimal experimental conditions for the cycloaddition of (phenylethynyl)di-p-tolylstibane (1) with benzylazide (2a) under aerobic conditions. Table 1 summarizes the reaction yields obtained with various catalysts and solvents. We first examined the reaction of 1 (0.5 mmol) with 2a (0.5 mmol) using 5 mol % of various Cu catalysts under aerobic conditions in THF at 60 °C (Table 1, entries 1–9). The results showed that CuBr was the best catalyst and gave the highest yield of the expected 5-stibano-1,2,3-triazole 3a (Table 1, entry 2). The reaction without the Cu catalyst did not afford 3a (Table 1, entry 10). Screening of solvents showed that the reaction proceeds effectively in THF (93%) and 1,4-dioxane, whereas DMSO, DMF, EtOH, toluene and 1,2-DCE gave inferior results (Table 1, entries 11–17). The best result was obtained when 1 was treated with 2a using a catalytic amount of CuBr in THF at 60 °C. This cycloaddition could also be scaled up to 10 mmol and the desired product 3a was obtained in excellent yields of up to 95%, i.e., 5.11 g of the product could be generated. When Cu(OAc)2 was employed as catalyst, 1-benzyl-4-phenyltriazole 4 was isolated in 83% yield as the major product (Table 1, entry 7). Heating of 5-stibano-1,2,3-triazole 3a in the presence of Cu(OAc)2 (5 mol %) under aerobic conditions in THF at 60 °C for 3 h afforded 4 in 91% yield. Furthermore, heating of 1 without 2a under the same conditions did not give the phenylacetylene and the starting compound was recovered (84%). These two experiments indicate that the formation of 5-H-triazole 4 progresses via 5-stibanotriazole 3a, the cycloaddition product of 1 with 2a.
Table 1: Cu-catalyzed reaction of ethynylstibane 1 with benzylazide 2aa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-123-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Cu cat. | Solvent | Yield [%]b | |
|---|---|---|---|---|
| 3a | 4 | |||
| 1 | CuI | THF | 76 | 10 |
| 2 | CuBr | THF | 93 | 3 |
| 3 | CuCl | THF | 61 | 19 |
| 4 | CuOAc | THF | 18 | 30 |
| 5 | Cu2O | THF | 25 | 26 |
| 6 | CuBr2 | THF | 49 | 12 |
| 7 | Cu(OAc)2 | THF | 9 | 83 |
| 8 | CuO | THF | 9 | 8 |
| 9 | CuSO4 | THF | 9 | 30 |
| 10 | – | THF | – | 2 |
| 11 | CuBr | 1,4-dioxane | 61 | 17 |
| 12 | CuBr | CH3CN | 46 | 42 |
| 13 | CuBr | DMSO | 32 | 56 |
| 14 | CuBr | 1,2-DCE | 18 | 28 |
| 15 | CuBr | EtOH | 18 | 14 |
| 16 | CuBr | DMF | 12 | 83 |
| 17 | CuBr | toluene | 6 | 3 |
aReaction conditions: 1 (0.5 mmol), 2a (0.5 mmol), Cu cat. (0.025 mmol). bIsolated yield.
To investigate the scope and limitations of the CuAAC reaction of stibane, ethynylstibane 1 was reacted with a series of organic azides 2 under optimized conditions (CuBr/THF/60 °C). The results are summarized in Scheme 1. Organic azides having functional groups such as o-bromobenzyl, 1-naphthalenemethyl, 2-phenylethyl, (ethoxycarbonyl)methyl, cinnamyl, (phenylthio)methyl and (phenylseleno)methyl afforded the corresponding products 3b–h in good to excellent yields. In the case of the reaction with o-bromobenzyl azide, the carbon–bromine bond of 3b remained intact, and other byproducts were not observed. Azides containing a linear alkyl group, acetal moiety, and a heteroaromatic ring such as pyridine gave the corresponding triazoles 3i–k in moderate yields. The CuAAC reaction of 1 with aryl azides such as 4-methylphenyl and 4-cyanophenyl azide gave a complex mixture, presumably due to the steric hindrance introduced by the aryl groups.
![[1860-5397-12-123-i1]](/bjoc/content/inline/1860-5397-12-123-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Copper-catalyzed [3 + 2] cycloaddition of 1 with organic azides 2. Reaction conditions: 1 (0.5 mmol), 2a (0.5 mmol), Cu cat. (0.025 mmol). Isolated yield are shown.
Scheme 1: Copper-catalyzed [3 + 2] cycloaddition of 1 with organic azides 2. Reaction conditions: 1 (0.5 mmol...
The regiochemistry of 5-stibanotriazole 3a was elucidated by 1H NMR and confirmed by single-crystal X-ray analysis (Figure 1). A nuclear Overhauser effect (NOE) was observed between the benzyl protons and the aromatic protons of the antimony p-tolyl groups. Other triazole 3 showed similar NOE signals between the 1-N-substituent protons and the p-tolyl protons.
![[1860-5397-12-123-1]](/bjoc/content/figures/1860-5397-12-123-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Ortep drawing of 3a with 50% probability. All hydrogen atoms are omitted for clarity. Two independent molecules exist in the asymmetric unit, one of them is shown.
Figure 1: Ortep drawing of 3a with 50% probability. All hydrogen atoms are omitted for clarity. Two independe...
The reaction mechanism of the cyclization is unclear at present. We consider that the catalytic cycle of this reaction would be similar to that for the reaction of 1-iodoalkynes [21,33] and 1-bismuthanoalkynes [24] with organic azides. A possible mechanism of the present Cu-catalyzed cycloaddition is shown in Scheme 2. Initially, π-complex A is generated by the reaction of the Cu(I) catalyst and ethynylstibane 1. Complex A coordinates with an organic azide to give complex B. Cyclization proceeds via a vinylidene-like transition state C to give 5-stibanotriazole 3.
![[1860-5397-12-123-i2]](/bjoc/content/inline/1860-5397-12-123-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
To test the reactivity of 5-stibanotriazole 3a was treated with hydrochloric acid, halogens, and nitrosyl tetrafluoroborate (NOBF4) (Scheme 3). The deantimonation of 3a with HCl gave 1-benzyl-4-phenyl-1,2,3-triazole (4) in 98% yield. Iodination of 3a using I2 afforded 5-iodo-4-phenyl-1,2,3-triazole (5) in 71% yield. However, the reaction of 3a with Br2 gave a complex mixture. The reaction of 3a with NOBF4 afforded pentavalent organoantimony compound 6 in 85% yield. It is noteworthy that 5-bismuthanotriazole was demetallated upon reaction with NOBF4 to give the corresponding 5-nitroso compound [24].
![[1860-5397-12-123-i3]](/bjoc/content/inline/1860-5397-12-123-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of 3a with HCl, I2 and NOBF4.
Scheme 3: Reaction of 3a with HCl, I2 and NOBF4.
Conclusion
In conclusion, the Cu-catalyzed azide–alkyne cycloaddition of (phenylethynyl)di-p-tolylstibane with organic azides afforded novel 1,4,5-trisubstituted 5-organostibano-1H-1,2,3-triazoles, which could be further derivatized using I2 and NOBF4. Studies on the cycloaddition of diversely-functionalized ethynylstibanes and functionalization at the 5-position of 5-stibanotriazole by electrophilic substitution and cross-coupling reaction are in progress.
Experimental
General procedure for the preparation of compounds 3: CuBr (3.6 mg, 0.025 mmol, 5 mol %), (phenylethynyl)di-p-tolylstibane (1, 203 mg, 0.5 mmol), and an organic azide (2, 0.5 mmol) were dissolved in THF (5 mL). The reaction mixture was stirred at 60 °C and monitored by TLC. Upon disappearance of the starting materials, the reaction mixture was diluted with CH2Cl2 (30 mL) and water (20 mL). The phases were separated and the aqueous layer was extracted with CH2Cl2 (20 mL × 2). The combined organic layers were washed 5% aqueous ammonia and water, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by silica gel chromatography (n-hexane:AcOEt) to give 3b, 3i (8:1), 3f (6:1), 3g, 3h (5:1), 3c, 3d, 3e, 3j (4:1), 3k (1:1).
Supporting Information
| Supporting Information File 1: Experimental procedures, full compound characterisation data and X-ray crystallographic data. | ||
| Format: PDF | Size: 3.8 MB | Download |
References
-
Padwa, A. 1,3-Dipolar Cycloaddition Chemistry; Wiley: New York, 1984.
Return to citation in text: [1] -
Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4
Return to citation in text: [1] -
Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057–3064. doi:10.1021/jo011148j
Return to citation in text: [1] -
Wu, P.; Fokin, V. V. Aldrichimica Acta 2007, 40, 7–17.
Return to citation in text: [1] [2] -
Moses, J. E.; Moorhouse, A. D. Chem. Soc. Rev. 2007, 36, 1249–1262. doi:10.1039/b613014n
Return to citation in text: [1] [2] -
Meldal, M.; Tornøe, C. W. Chem. Rev. 2008, 108, 2952–3015. doi:10.1021/cr0783479
Return to citation in text: [1] [2] -
Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302–1315. doi:10.1039/b904091a
Return to citation in text: [1] [2] -
Aragão-Leoneti, V.; Campo, V. L.; Gomes, A. S.; Field, R. A.; Carvalho, I. Tetrahedron 2010, 66, 9475–9492. doi:10.1016/j.tet.2010.10.001
Return to citation in text: [1] [2] -
Majumdar, K. C.; Ray, K. Synthesis 2011, 3767–3783. doi:10.1055/s-0031-1289295
Return to citation in text: [1] [2] -
Berg, R.; Straub, B. F. Beilstein J. Org. Chem. 2013, 9, 2715–2750. doi:10.3762/bjoc.9.308
Return to citation in text: [1] [2] -
Sokolova, N. V.; Nenajdenko, V. G. RSC Adv. 2013, 3, 16212–16242. doi:10.1039/c3ra42482k
Return to citation in text: [1] [2] -
Haldón, E.; Nicasio, M. C.; Pérez, P. J. Org. Biomol. Chem. 2015, 13, 9528–9550. doi:10.1039/C5OB01457C
Return to citation in text: [1] [2] -
Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Chem. Rev. 2013, 113, 4905–4979. doi:10.1021/cr200409f
Return to citation in text: [1] [2] -
Debets, M. F.; van Berkel, S. S.; Dommerholt, J.; Dirks, A. J.; Rutjes, F. P. J. T.; van Delft, F. L. Acc. Chem. Res. 2011, 44, 805–815. doi:10.1021/ar200059z
Return to citation in text: [1] [2] -
Agalave, S. G.; Maujan, S. R.; Pore, V. S. Chem. – Asian J. 2011, 6, 2696–2718. doi:10.1002/asia.201100432
Return to citation in text: [1] [2] -
Mamidyala, S. K.; Finn, M. G. Chem. Soc. Rev. 2010, 39, 1252–1261. doi:10.1039/b901969n
Return to citation in text: [1] [2] -
Jewett, J. C.; Bertozzi, C. R. Chem. Soc. Rev. 2010, 39, 1272–1279. doi:10.1039/b901970g
Return to citation in text: [1] [2] -
Golas, P. L.; Matyjaszewski, K. Chem. Soc. Rev. 2010, 39, 1338–1354. doi:10.1039/B901978M
Return to citation in text: [1] [2] -
Qin, A.; Lam, J. W. Y.; Tang, B. Z. Chem. Soc. Rev. 2010, 39, 2522–2544. doi:10.1039/b909064a
Return to citation in text: [1] [2] -
Bryant, J. J.; Bunz, U. H. F. Chem. – Asian J. 2013, 8, 1354–1367. doi:10.1002/asia.201300260
Return to citation in text: [1] [2] -
Spiteri, C.; Moses, J. E. Angew. Chem., Int. Ed. 2010, 49, 31–33. doi:10.1002/anie.200905322
Return to citation in text: [1] [2] -
Wang, B.; Liu, N.; Chen, W.; Huang, D.; Wang, X.; Hu, Y. Adv. Synth. Catal. 2015, 357, 401–407. doi:10.1002/adsc.201400471
Return to citation in text: [1] -
Li, L.; Hao, G.; Zhu, A.; Fan, X.; Zhang, G.; Zhang, L. Chem. – Eur. J. 2013, 19, 14403–14406. doi:10.1002/chem.201303324
Return to citation in text: [1] -
Worrell, B. T.; Ellery, S. P.; Fokin, V. V. Angew. Chem., Int. Ed. 2013, 52, 13037–13041. doi:10.1002/anie.201306192
Return to citation in text: [1] [2] [3] -
Matano, Y. In Main Group Metals in Organic Synthesis; Yamamoto, H.; Oshima, K., Eds.; Wiley-VCH, Verlag GmbH & Co. KGaA: Weinheim, 2004; Vol. 2, pp 753–811.
Return to citation in text: [1] -
Burton, J. W. In Science of Synthesis; Fleming, I., Ed.; Georg Thieme Verlag: Stuttgart, 2002; Vol. 4, pp 53–75.
Return to citation in text: [1] -
Kakusawa, N.; Yamaguchi, K.; Kurita, J.; Tsuchiya, T. Tetrahedron Lett. 2000, 41, 4143–4146. doi:10.1016/S0040-4039(00)00554-2
Return to citation in text: [1] -
Kakusawa, N.; Yamaguchi, K.; Kurita, J. J. Organomet. Chem. 2005, 690, 2956–2966. doi:10.1016/j.jorganchem.2005.03.021
Return to citation in text: [1] -
Kakusawa, N.; Kurita, J. Chem. Pharm. Bull. 2005, 53, 1369–1371. doi:10.1248/cpb.53.1369
Return to citation in text: [1] -
Kakusawa, N.; Tobiyasu, Y.; Yasuike, S.; Yamaguchi, K.; Seki, H.; Kurita, J. J. Organomet. Chem. 2006, 691, 2953–2968. doi:10.1016/j.jorganchem.2006.02.041
Return to citation in text: [1] -
Kakusawa, N.; Kurita, J. Chem. Pharm. Bull. 2006, 54, 699–702. doi:10.1248/cpb.54.699
Return to citation in text: [1] -
Kakusawa, N.; Kurita, J. Heterocycles 2006, 68, 1335–1348. doi:10.3987/COM-05-10590
Return to citation in text: [1] -
Hein, J. E.; Tripp, J. C.; Krasnova, L. B.; Sharpless, K. B.; Fokin, V. V. Angew. Chem., Int. Ed. 2009, 48, 8018–8021. doi:10.1002/anie.200903558
Return to citation in text: [1]
| 13. | Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Chem. Rev. 2013, 113, 4905–4979. doi:10.1021/cr200409f |
| 14. | Debets, M. F.; van Berkel, S. S.; Dommerholt, J.; Dirks, A. J.; Rutjes, F. P. J. T.; van Delft, F. L. Acc. Chem. Res. 2011, 44, 805–815. doi:10.1021/ar200059z |
| 15. | Agalave, S. G.; Maujan, S. R.; Pore, V. S. Chem. – Asian J. 2011, 6, 2696–2718. doi:10.1002/asia.201100432 |
| 16. | Mamidyala, S. K.; Finn, M. G. Chem. Soc. Rev. 2010, 39, 1252–1261. doi:10.1039/b901969n |
| 17. | Jewett, J. C.; Bertozzi, C. R. Chem. Soc. Rev. 2010, 39, 1272–1279. doi:10.1039/b901970g |
| 24. | Worrell, B. T.; Ellery, S. P.; Fokin, V. V. Angew. Chem., Int. Ed. 2013, 52, 13037–13041. doi:10.1002/anie.201306192 |
| 4. | Wu, P.; Fokin, V. V. Aldrichimica Acta 2007, 40, 7–17. |
| 5. | Moses, J. E.; Moorhouse, A. D. Chem. Soc. Rev. 2007, 36, 1249–1262. doi:10.1039/b613014n |
| 6. | Meldal, M.; Tornøe, C. W. Chem. Rev. 2008, 108, 2952–3015. doi:10.1021/cr0783479 |
| 7. | Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302–1315. doi:10.1039/b904091a |
| 8. | Aragão-Leoneti, V.; Campo, V. L.; Gomes, A. S.; Field, R. A.; Carvalho, I. Tetrahedron 2010, 66, 9475–9492. doi:10.1016/j.tet.2010.10.001 |
| 9. | Majumdar, K. C.; Ray, K. Synthesis 2011, 3767–3783. doi:10.1055/s-0031-1289295 |
| 10. | Berg, R.; Straub, B. F. Beilstein J. Org. Chem. 2013, 9, 2715–2750. doi:10.3762/bjoc.9.308 |
| 11. | Sokolova, N. V.; Nenajdenko, V. G. RSC Adv. 2013, 3, 16212–16242. doi:10.1039/c3ra42482k |
| 12. | Haldón, E.; Nicasio, M. C.; Pérez, P. J. Org. Biomol. Chem. 2015, 13, 9528–9550. doi:10.1039/C5OB01457C |
| 3. | Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057–3064. doi:10.1021/jo011148j |
| 21. | Spiteri, C.; Moses, J. E. Angew. Chem., Int. Ed. 2010, 49, 31–33. doi:10.1002/anie.200905322 |
| 33. | Hein, J. E.; Tripp, J. C.; Krasnova, L. B.; Sharpless, K. B.; Fokin, V. V. Angew. Chem., Int. Ed. 2009, 48, 8018–8021. doi:10.1002/anie.200903558 |
| 2. | Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4 |
| 24. | Worrell, B. T.; Ellery, S. P.; Fokin, V. V. Angew. Chem., Int. Ed. 2013, 52, 13037–13041. doi:10.1002/anie.201306192 |
| 23. | Li, L.; Hao, G.; Zhu, A.; Fan, X.; Zhang, G.; Zhang, L. Chem. – Eur. J. 2013, 19, 14403–14406. doi:10.1002/chem.201303324 |
| 25. | Matano, Y. In Main Group Metals in Organic Synthesis; Yamamoto, H.; Oshima, K., Eds.; Wiley-VCH, Verlag GmbH & Co. KGaA: Weinheim, 2004; Vol. 2, pp 753–811. |
| 26. | Burton, J. W. In Science of Synthesis; Fleming, I., Ed.; Georg Thieme Verlag: Stuttgart, 2002; Vol. 4, pp 53–75. |
| 21. | Spiteri, C.; Moses, J. E. Angew. Chem., Int. Ed. 2010, 49, 31–33. doi:10.1002/anie.200905322 |
| 22. | Wang, B.; Liu, N.; Chen, W.; Huang, D.; Wang, X.; Hu, Y. Adv. Synth. Catal. 2015, 357, 401–407. doi:10.1002/adsc.201400471 |
| 27. | Kakusawa, N.; Yamaguchi, K.; Kurita, J.; Tsuchiya, T. Tetrahedron Lett. 2000, 41, 4143–4146. doi:10.1016/S0040-4039(00)00554-2 |
| 28. | Kakusawa, N.; Yamaguchi, K.; Kurita, J. J. Organomet. Chem. 2005, 690, 2956–2966. doi:10.1016/j.jorganchem.2005.03.021 |
| 29. | Kakusawa, N.; Kurita, J. Chem. Pharm. Bull. 2005, 53, 1369–1371. doi:10.1248/cpb.53.1369 |
| 30. | Kakusawa, N.; Tobiyasu, Y.; Yasuike, S.; Yamaguchi, K.; Seki, H.; Kurita, J. J. Organomet. Chem. 2006, 691, 2953–2968. doi:10.1016/j.jorganchem.2006.02.041 |
| 31. | Kakusawa, N.; Kurita, J. Chem. Pharm. Bull. 2006, 54, 699–702. doi:10.1248/cpb.54.699 |
| 32. | Kakusawa, N.; Kurita, J. Heterocycles 2006, 68, 1335–1348. doi:10.3987/COM-05-10590 |
| 4. | Wu, P.; Fokin, V. V. Aldrichimica Acta 2007, 40, 7–17. |
| 5. | Moses, J. E.; Moorhouse, A. D. Chem. Soc. Rev. 2007, 36, 1249–1262. doi:10.1039/b613014n |
| 6. | Meldal, M.; Tornøe, C. W. Chem. Rev. 2008, 108, 2952–3015. doi:10.1021/cr0783479 |
| 7. | Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302–1315. doi:10.1039/b904091a |
| 8. | Aragão-Leoneti, V.; Campo, V. L.; Gomes, A. S.; Field, R. A.; Carvalho, I. Tetrahedron 2010, 66, 9475–9492. doi:10.1016/j.tet.2010.10.001 |
| 9. | Majumdar, K. C.; Ray, K. Synthesis 2011, 3767–3783. doi:10.1055/s-0031-1289295 |
| 10. | Berg, R.; Straub, B. F. Beilstein J. Org. Chem. 2013, 9, 2715–2750. doi:10.3762/bjoc.9.308 |
| 11. | Sokolova, N. V.; Nenajdenko, V. G. RSC Adv. 2013, 3, 16212–16242. doi:10.1039/c3ra42482k |
| 12. | Haldón, E.; Nicasio, M. C.; Pérez, P. J. Org. Biomol. Chem. 2015, 13, 9528–9550. doi:10.1039/C5OB01457C |
| 13. | Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Chem. Rev. 2013, 113, 4905–4979. doi:10.1021/cr200409f |
| 14. | Debets, M. F.; van Berkel, S. S.; Dommerholt, J.; Dirks, A. J.; Rutjes, F. P. J. T.; van Delft, F. L. Acc. Chem. Res. 2011, 44, 805–815. doi:10.1021/ar200059z |
| 15. | Agalave, S. G.; Maujan, S. R.; Pore, V. S. Chem. – Asian J. 2011, 6, 2696–2718. doi:10.1002/asia.201100432 |
| 16. | Mamidyala, S. K.; Finn, M. G. Chem. Soc. Rev. 2010, 39, 1252–1261. doi:10.1039/b901969n |
| 17. | Jewett, J. C.; Bertozzi, C. R. Chem. Soc. Rev. 2010, 39, 1272–1279. doi:10.1039/b901970g |
| 18. | Golas, P. L.; Matyjaszewski, K. Chem. Soc. Rev. 2010, 39, 1338–1354. doi:10.1039/B901978M |
| 19. | Qin, A.; Lam, J. W. Y.; Tang, B. Z. Chem. Soc. Rev. 2010, 39, 2522–2544. doi:10.1039/b909064a |
| 20. | Bryant, J. J.; Bunz, U. H. F. Chem. – Asian J. 2013, 8, 1354–1367. doi:10.1002/asia.201300260 |
| 18. | Golas, P. L.; Matyjaszewski, K. Chem. Soc. Rev. 2010, 39, 1338–1354. doi:10.1039/B901978M |
| 19. | Qin, A.; Lam, J. W. Y.; Tang, B. Z. Chem. Soc. Rev. 2010, 39, 2522–2544. doi:10.1039/b909064a |
| 20. | Bryant, J. J.; Bunz, U. H. F. Chem. – Asian J. 2013, 8, 1354–1367. doi:10.1002/asia.201300260 |
| 24. | Worrell, B. T.; Ellery, S. P.; Fokin, V. V. Angew. Chem., Int. Ed. 2013, 52, 13037–13041. doi:10.1002/anie.201306192 |
© 2016 Yamada et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)