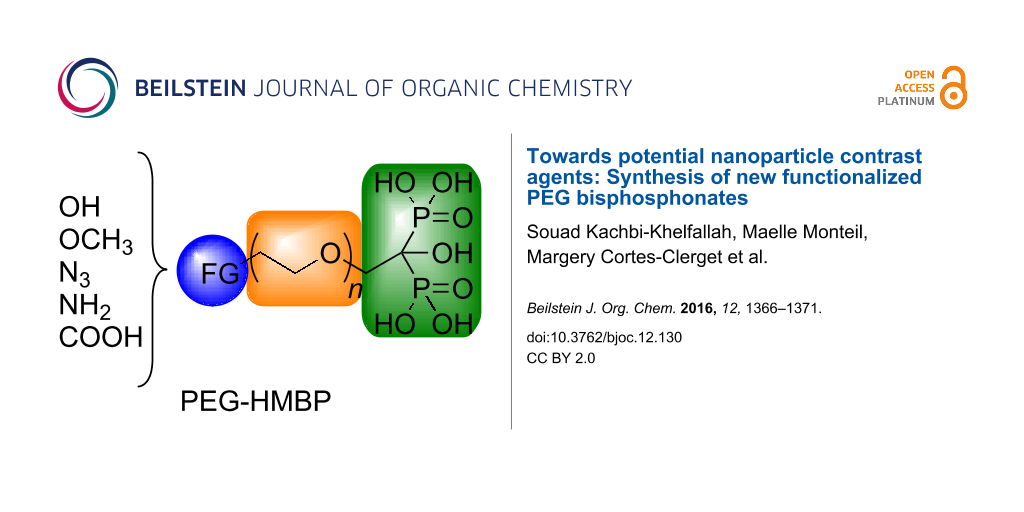
Abstract
The use of nanotechnologies for biomedical applications took a real development during these last years. To allow an effective targeting for biomedical imaging applications, the adsorption of plasmatic proteins on the surface of nanoparticles must be prevented to reduce the hepatic capture and increase the plasmatic time life. In biologic media, metal oxide nanoparticles are not stable and must be coated by biocompatible organic ligands. The use of phosphonate ligands to modify the nanoparticle surface drew a lot of attention in the last years for the design of highly functional hybrid materials. Here, we report a methodology to synthesize bisphosphonates having functionalized PEG side chains with different lengths. The key step is a procedure developed in our laboratory to introduce the bisphosphonate from acyl chloride and tris(trimethylsilyl)phosphite in one step.

Graphical Abstract
Introduction
Numerous researchers are interested in the development of superparamagnetic iron oxide nanoparticles (SPIONPs) because of their biocompatibility which allows there in vivo use both for diagnosis in magnetic resonance imaging and in therapy [1,2]. Most often, it is necessary to modify the surface of SPIONPs to increase the metabolic stability.
To overcome this main drawback, the NP surface could be derivatized by various functional groups. These ligands have to possess certain chemical and biological properties as the flexibility, the hydrophilicity and an absence of in vivo toxicity. In addition, the nanoparticulate systems so obtained must be stable in the various biological compartments and they must be stealthy to avoid the elimination by macrophages.
For this purpose, appropriate coatings have already been reported [3,4]. Some of which consist in the NP surface modification using hydrophilic polymers (dextran, PEG) or bifunctional molecules substituted by amines, thiols, carboxylates, sulfonates, phosphonates or bisphosphonates [5-7]. Particularly, a strong interaction between the NPs and the phosphonic moiety was observed and more interestingly the best results were obtained with bisphosphonate products [8,9]. For the past years, our group has focused its interest in the synthesis of various functionalized hydroxymethylene bisphosphonates (HMBPs) [10] and their applications in health science, especially in antitumor therapy [11-13]. Herein, we described the synthesis of novel bifunctional PEG-HMBP compounds in order to employ them as anchoring agents for SPIONPs (Figure 1).
![[1860-5397-12-130-1]](/bjoc/content/figures/1860-5397-12-130-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
For this family of compounds, the HMBP moiety has to be built starting from a modified PEG chain. The HMBP introduction could be achieved by several reported methodologies starting from an acid chloride (Scheme 1).
![[1860-5397-12-130-i1]](/bjoc/content/inline/1860-5397-12-130-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Direct methods for the 1-hydroxyalkylidenebisphosphonic acid synthesis.
Scheme 1: Direct methods for the 1-hydroxyalkylidenebisphosphonic acid synthesis.
The first method, used in the industry, allows accessing the desired products in one step under rather harsh conditions [14]. 1-Hydroxyalkylidenebisphosphonic acids have also been obtained in good yields. However, this widely used method seems not to be compatible with breakable and delicate functionalized substrates. In contrast, our lab has developed a new synthetic strategy starting from an acid chloride and tris(trimethylsilyl) phosphite, followed by a methanolysis step [15].
This one-pot procedure allows the synthesis of various aliphatic and aromatic bisphosphonic acids under mild conditions. Moreover, reactions were very fast and pure products were obtained after evaporation of the volatile fraction. The scope of this reaction was successfully widened in aliphatic and aromatic anhydride [15-21]. The introduction of the HMBP moiety in presence of the PEG tether seems to be critical due to its high sensitivity under harsh conditions. Wherefore, our methodology, which exhibits a high tolerance to various functionalized groups, appears to be an adequate way to introduce the HMBP chain in presence of the PEG moiety.
To obtain the PEG-HMBP 1 compound family, the synthetic strategy consists in mono-protecting and/or mono-functionalizing commercially available PEGs followed by the lab-made HMBP methodology introduced previously (Scheme 2).
![[1860-5397-12-130-i2]](/bjoc/content/inline/1860-5397-12-130-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthetic strategy of PEG-HMBPs 1.
Scheme 2: Synthetic strategy of PEG-HMBPs 1.
Starting materials, the free alcohol PEG and monomethyl ether PEGs (compounds 3a,b) with various chain lengths (n = 4, 7 and 12) were commercially available. Firstly, the free alcohol PEG was selectively monoprotected with a benzyl group (Scheme 3).
![[1860-5397-12-130-i3]](/bjoc/content/inline/1860-5397-12-130-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of PEG-HMBPs 1 and 1’.
Scheme 3: Synthesis of PEG-HMBPs 1 and 1’.
Only one alcohol function was indeed deprotonated with one equivalent of sodium hydride at −78 °C in THF after the solution was stirred for 12 hours at room temperature. The alcoholate intermediate reacted smoothly with benzyl bromide at room temperature to afford the monobenzylated PEG 2 in 77% yield (Table 1, entry 1). Afterwards, the alcohols 2 and 3a,b have to be oxidized to the corresponding carboxylic acids 6 and 7a,b. First of all, the direct oxidation reported in the literature in one step has been performed. Thus, tested oxidants were the Jones reagent [22], potassium permanganate [23], with catalytic o-iodoxybenzoic acid (IBX) in oxone [24] and catalytic 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) with bis(acetoxy)iodobenzene (BAIB) [25]. The first two conditions led to a PEG chain cleavage and the recovery of benzoic acid from alcohol 2. Besides, the mixture IBX/oxone gave the expected product inseparable of IBX byproducts. Only oxidation using TEMPO and BAIB furnished the pure corresponding carboxylic acid. Nevertheless, the low obtained yields encouraged us to test a strategy in two steps via an aldehyde. Fortunately, the following two-step procedure was more effective. The alcohol derivatives 2 and 3a,b were treated with dimethyl sulfoxide, oxalyl chloride and triethylamine in dichloromethane at −55 °C. Under these classical Swern conditions, the corresponding products 4 and 5a,b were isolated in excellent yields from 79% to 89% (Table 1, entries 2–4). The aldehydes 4 and 5a,b were next smoothly oxidized in the presence of a catalytic amount of PCC and a the co-oxidative agent H5IO6 in acetonitrile at 0 °C. The PEG 6 and 7a,b were obtained in good yields (Table 1, entries 5–7).
Table 1: Synthesis of PEG-HMBPs 1,1’and 10.
| Entry | Compound | R | n | Yield (%) | 31P δ (ppm) |
|---|---|---|---|---|---|
| 1 | 2 | Bn | 4 | 77 | – |
| 2 | 4 | Bn | 4 | 89 | – |
| 3 | 5a | Me | 7 | 79 | – |
| 4 | 5b | Me | 12 | 80 | – |
| 5 | 6 | Bn | 4 | 72 | – |
| 6 | 7a | Me | 7 | 82 | – |
| 7 | 7b | Me | 12 | 72 | – |
| 8 | 8 | Bn | 4 | quant. | – |
| 9 | 9a | Me | 7 | quant. | – |
| 10 | 9b | Me | 12 | quant. | – |
| 11 | 1 | Bn | 4 | 78 | 16.8 |
| 12 | 1’a | Me | 7 | 47 | 17.2 |
| 13 | 1’b | Me | 12 | 43 | 17.2 |
| 14 | 10 | H | 4 | 72 | 16.2 |
aIsolated yield. bproton decoupling 31P NMR experiment.
Finally, the optimized two-step procedure enabled us to isolate the expected carboxylic acids 6 and 7a,b which are key intermediates in the synthesis of PEG-HMBPs.
The carboxylic acids 6 and 7a,b reacted quantitatively with oxalyl chloride to give acyl chlorides 8 and 9a,b at room temperature after 24 hours (Table 1, entries 8–10). The completion of the reaction was monitored by infrared spectroscopy with the disappearance of the hydroxy absorption band and the shifting of the carbonyl vibration band to about 1800 cm−1. The addition of two equivalents of tris(trimethylsilyl) phosphite to the acyl chloride derivatives 8 and 9a,b yielded the corresponding silylated PEG-HMBP.
The formation of silylated bisphosphonate was monitored by 31P NMR. After evaporation of volatile compounds under vacuum the silylated PEG-HMBP was hydrolyzed with methanol at room temperature for 24 hours. After methanol evaporation, the crude PEG-HMBP containing phosphorous acid was purified by successive washes with dry diethyl ether. The pure targeted PEG-HMBPs 1 and 1’a,b were then obtained in moderate yields (Table 1, entries 11–13). The treatment of 1 with dihydrogen and palladium on charcoal in water allowed cleavage of the benzyl moiety and led to the HO-PEG-HMBP 10 in 72% yield (Table 1, entry 14). The ligand 10 permitted to obtain new gadolinium phosphate nanocrystals with luminescent properties [26].
Therefore, we considered the syntheses of other compounds which possess azido or amino functional groups. The azido products will allow us to perform click chemistry to introduce various functionalities. Moreover, the amino derivatives will be easily obtained by reducing the azido group. As previously mentioned, the first step was a selective mono-activation of PEG using para-toluenesulfonyl chloride in the presence of sodium hydroxide in a water/THF mixture (Scheme 4).
![[1860-5397-12-130-i4]](/bjoc/content/inline/1860-5397-12-130-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Syntheses of HMBP-PEG-N3 16 and HMBP-PEG-NH3+ 17.
Scheme 4: Syntheses of HMBP-PEG-N3 16 and HMBP-PEG-NH3+ 17.
The tosylated products 11a,b were obtained after three hours at 0 °C with a 95% yields. Next, the monoactivated compounds 11a,b were substituted by sodium azide in DMF at 60 °C within five hours. The azido compounds 12a,b were obtained in 83% and 81% yield, respectively. The alcohols 12a,b were subsequently firstly deprotonated with NaH in DMF and the generated alcoholates were stirred 16 hours with ethyl bromoacetate giving the expected esters 13a,b in moderate yields.
The saponification reactions of the esters 13a,b were carried out with sodium hydroxide in methanol. The completion of the reactions was controlled by TLC. After neutralization with a cationic exchange Dowex® 50WX2 resin, the corresponding carboxylic acids 14a,b were isolated in quantitative yields. The HMBP moiety was subsequently incorporated by the lab-made methodology previously described. The carboxylic acids 14a,b were converted into the expected HMBP-PEG-N3 16a,b via the corresponding acyl chloride 15a,b. The reactions were monitored by 31P NMR, compounds 16a and 16b were obtained after 12 and 18 hours, respectively.
Thus, HMBP-PEG-N3 16a,b were obtained after purification in 72% and 74% yield and characterized by a singlet in 31P [27] NMR at about 17 ppm. Finally, the reduction of the azido compounds 16a,b in the presence of palladium on charcoal and dihydrogen led to the targeted amino-PEG-HMBPs 17a and 17b, respectively in moderate 62% and 68% yields.
In order to access available PEG-HMBPs functionalized with a primary amine or a carboxylic acid group usable in peptidic coupling with various molecules for example, the HMBP-PEG-COOH 23 was synthesized (Scheme 5). This compound was obtained in six steps starting from a free alcohol four-unit PEG. It reacted with ethyl bromoacetate after mono-deprotonation using sodium hydride in THF at −78 °C for 12 hours at room temperature giving PEG 18. HMBP-PEG 22 was next synthesized in four steps with satisfying yields in a similar strategy previously described for compounds 1 and 1’a,b. The last step was the saponification of the ethyl ester group. Different usual conditions were tested, leading to partial degradation of the HMBP. The use of a diluted aqueous solution of potassium hydroxide (0.1 M) followed by a protonation with a Dowex® 50WX2 H+ resin allowed us to obtain the expected HMBP-PEG-COOH 23 in 85% yield.
![[1860-5397-12-130-i5]](/bjoc/content/inline/1860-5397-12-130-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, novel bifunctional PEG-HMBPs ligands for the coating of iron oxide nanoparticles have been synthesized. The procedure is efficient to introduce different functional groups such as azide, carboxylic acid, amine permitting further coupling reactions with a drug, protein or antibody. The use of PEG polymers with chains of different lengths has also given satisfying results. This modulation would allow improving the nanoparticles stealth in vivo. Further studies in this area and their applications will be reported in due course.
Supporting Information
| Supporting Information File 1: Experimental and analytical data of all new compounds as well as copies of their 1H, 31P and 13C NMR spectra. | ||
| Format: PDF | Size: 3.1 MB | Download |
References
-
Laurent, S.; Saei, A. A.; Behzadi, S.; Panahifar, A.; Mahmoudi, M. Expert Opin. Drug Delivery 2014, 11, 1449–1470. doi:10.1517/17425247.2014.924501
Return to citation in text: [1] -
Yen, S. K.; Padmanabhan, P.; Selvan, S. T. Theranostics 2013, 3, 986–1003. doi:10.7150/thno.4827
Return to citation in text: [1] -
Ma, N.; Ma, C.; Li, C.; Wang, T.; Tang, Y.; Wang, H.; Moul, X.; Chen, Z.; Hel, N. J. Nanosci. Nanotechnol. 2013, 13, 6485–6498. doi:10.1166/jnn.2013.7525
Return to citation in text: [1] -
Lu, Z.; Yin, Y. Chem. Soc. Rev. 2012, 41, 6874–6887. doi:10.1039/c2cs35197h
Return to citation in text: [1] -
Hak, S.; Helgesen, E.; Hektoen, H. H.; Huuse, E. M.; Jarzyna, P. A.; Mulder, W. J. M.; Haraldseth, O.; de Lange Davies, C. ACS Nano 2012, 6, 5648–5658. doi:10.1021/nn301630n
Return to citation in text: [1] -
Queffélec, C.; Petit, M.; Janvier, P.; Knight, D. A.; Bujoli, B. Chem. Rev. 2012, 112, 3777–3807. doi:10.1021/cr2004212
Return to citation in text: [1] -
Karimi, A.; Denizot, B.; Hindré, F.; Filmon, R.; Greneche, J.-M.; Laurent, S.; Daou, T. J.; Begin-Colin, S.; Le Jeune, J.-J. J. Nanopart. Res. 2010, 12, 1239–1248. doi:10.1007/s11051-009-9815-7
Return to citation in text: [1] -
Sandiford, L.; Phinikaridou, A.; Protti, A.; Meszaros, L. K.; Cui, X.; Yan, Y.; Frodsham, G.; Williamson, P. A.; Gaddum, N.; Botnar, R. M.; Blower, P. J.; Green, M. A.; de Rosales, R. T. M. ACS Nano 2012, 7, 500–512. doi:10.1021/nn3046055
Return to citation in text: [1] -
de Rosales, R. T. M.; Tavaré, R.; Paul, R. L.; Jauregui-Osoro, M.; Protti, A.; Glaria, A.; Varma, G.; Szanda, I.; Blower, P. J. Angew. Chem., Int. Ed. 2011, 50, 5509–5513. doi:10.1002/anie.201007894
Return to citation in text: [1] -
Lalatonne, Y.; Paris, C.; Serfaty, J. M.; Weinmann, P.; Lecouvey, M.; Motte, L. Chem. Commun. 2008, 2553–2555. doi:10.1039/b801911h
Return to citation in text: [1] -
Benyettou, F.; Lalatonne, Y.; Chebbi, I.; Di Benedetto, M.; Serfaty, J.-M.; Lecouvey, M.; Motte, L. Phys. Chem. Chem. Phys. 2011, 13, 10020–10027. doi:10.1039/c0cp02034f
Return to citation in text: [1] -
Benyettou, F.; Lalatonne, Y.; Sainte-Catherine, O.; Monteil, M.; Motte, L. Int. J. Pharm. 2009, 379, 324–327. doi:10.1016/j.ijpharm.2009.04.010
Return to citation in text: [1] -
Bolley, J.; Guenin, E.; Lievre, N.; Lecouvey, M.; Soussan, M.; Lalatonne, Y.; Motte, L. Langmuir 2013, 29, 14639–14647. doi:10.1021/la403245h
Return to citation in text: [1] -
Kieczykowski, G. R.; Jobson, R. B.; Melillo, D. G.; Reinhold, D. F.; Grenda, V. J.; Shinkai, I. J. Org. Chem. 1995, 60, 8310–8312. doi:10.1021/jo00130a036
Return to citation in text: [1] -
Lecouvey, M.; Mallard, I.; Bailly, T.; Burgada, R.; Leroux, Y. Tetrahedron Lett. 2001, 42, 8475–8478. doi:10.1016/S0040-4039(01)01844-5
Return to citation in text: [1] [2] -
Guenin, E.; Degache, E.; Liquier, J.; Lecouvey, M. Eur. J. Org. Chem. 2004, 2983–2987. doi:10.1002/ejoc.200400053
Return to citation in text: [1] -
Guenin, E.; Ledoux, D.; Oudar, O.; Lecouvey, M.; Kraemer, M. Anticancer Res. 2005, 25, 1139–1145.
Return to citation in text: [1] -
Migianu-Griffoni, E.; Mbemba, C.; Burgada, R.; Lecerclé, D.; Taran, F.; Lecouvey, M. Tetrahedron 2009, 65, 1517–1523. doi:10.1016/j.tet.2008.11.076
Return to citation in text: [1] -
Monteil, M.; Guenin, E.; Migianu, E.; Lutomski, D.; Lecouvey, M. Tetrahedron 2005, 61, 7528–7537. doi:10.1016/j.tet.2005.05.053
Return to citation in text: [1] -
Monteil, M.; Migianu-Griffoni, E.; Sainte-Catherine, O.; Di Benedetto, M.; Lecouvey, M. Eur. J. Med. Chem. 2014, 77, 56–64. doi:10.1016/j.ejmech.2014.02.054
Return to citation in text: [1] -
Kachbi Khelfallah, S.; Monteil, M.; Deschamp, J.; Gager, O.; Migianu-Griffoni, E.; Lecouvey, M. Org. Biomol. Chem. 2015, 13, 11382–11392. doi:10.1039/C5OB01967B
Return to citation in text: [1] -
Fishman, A.; Acton, A.; Lee-Ruff, E. Synth. Commun. 2004, 34, 2309–2312. doi:10.1081/SCC-120038518
Return to citation in text: [1] -
Hojo, K.; Susuki, Y.; Sasaki, M.; Maeda, M.; Smith, T. J.; Kawasaki, K. Chem. Pharm. Bull. 2002, 50, 1001–1003. doi:10.1248/cpb.50.1001
Return to citation in text: [1] -
Thottumkara, A. P.; Bowsher, M. S.; Vinod, T. K. Org. Lett. 2005, 7, 2933–2936. doi:10.1021/ol050875o
Return to citation in text: [1] -
Greenwald, R. B.; Gilbert, C. W.; Pendri, A.; Conover, C. D.; Xia, J.; Martinez, A. J. Med. Chem. 1996, 39, 424–431. doi:10.1021/jm950475e
Return to citation in text: [1] -
Abécassis, B.; Lerouge, F.; Bouquet, F.; Kachbi, S.; Monteil, M.; Davidson, P. J. Phys. Chem. B 2012, 116, 7590–7595. doi:10.1021/jp303161a
Return to citation in text: [1] -
Cubberley, M. S.; Iverson, B. L. J. Am. Chem. Soc. 2001, 123, 7560–7563. doi:10.1021/ja015817m
Return to citation in text: [1]
| 1. | Laurent, S.; Saei, A. A.; Behzadi, S.; Panahifar, A.; Mahmoudi, M. Expert Opin. Drug Delivery 2014, 11, 1449–1470. doi:10.1517/17425247.2014.924501 |
| 2. | Yen, S. K.; Padmanabhan, P.; Selvan, S. T. Theranostics 2013, 3, 986–1003. doi:10.7150/thno.4827 |
| 10. | Lalatonne, Y.; Paris, C.; Serfaty, J. M.; Weinmann, P.; Lecouvey, M.; Motte, L. Chem. Commun. 2008, 2553–2555. doi:10.1039/b801911h |
| 27. | Cubberley, M. S.; Iverson, B. L. J. Am. Chem. Soc. 2001, 123, 7560–7563. doi:10.1021/ja015817m |
| 8. | Sandiford, L.; Phinikaridou, A.; Protti, A.; Meszaros, L. K.; Cui, X.; Yan, Y.; Frodsham, G.; Williamson, P. A.; Gaddum, N.; Botnar, R. M.; Blower, P. J.; Green, M. A.; de Rosales, R. T. M. ACS Nano 2012, 7, 500–512. doi:10.1021/nn3046055 |
| 9. | de Rosales, R. T. M.; Tavaré, R.; Paul, R. L.; Jauregui-Osoro, M.; Protti, A.; Glaria, A.; Varma, G.; Szanda, I.; Blower, P. J. Angew. Chem., Int. Ed. 2011, 50, 5509–5513. doi:10.1002/anie.201007894 |
| 5. | Hak, S.; Helgesen, E.; Hektoen, H. H.; Huuse, E. M.; Jarzyna, P. A.; Mulder, W. J. M.; Haraldseth, O.; de Lange Davies, C. ACS Nano 2012, 6, 5648–5658. doi:10.1021/nn301630n |
| 6. | Queffélec, C.; Petit, M.; Janvier, P.; Knight, D. A.; Bujoli, B. Chem. Rev. 2012, 112, 3777–3807. doi:10.1021/cr2004212 |
| 7. | Karimi, A.; Denizot, B.; Hindré, F.; Filmon, R.; Greneche, J.-M.; Laurent, S.; Daou, T. J.; Begin-Colin, S.; Le Jeune, J.-J. J. Nanopart. Res. 2010, 12, 1239–1248. doi:10.1007/s11051-009-9815-7 |
| 25. | Greenwald, R. B.; Gilbert, C. W.; Pendri, A.; Conover, C. D.; Xia, J.; Martinez, A. J. Med. Chem. 1996, 39, 424–431. doi:10.1021/jm950475e |
| 3. | Ma, N.; Ma, C.; Li, C.; Wang, T.; Tang, Y.; Wang, H.; Moul, X.; Chen, Z.; Hel, N. J. Nanosci. Nanotechnol. 2013, 13, 6485–6498. doi:10.1166/jnn.2013.7525 |
| 4. | Lu, Z.; Yin, Y. Chem. Soc. Rev. 2012, 41, 6874–6887. doi:10.1039/c2cs35197h |
| 26. | Abécassis, B.; Lerouge, F.; Bouquet, F.; Kachbi, S.; Monteil, M.; Davidson, P. J. Phys. Chem. B 2012, 116, 7590–7595. doi:10.1021/jp303161a |
| 15. | Lecouvey, M.; Mallard, I.; Bailly, T.; Burgada, R.; Leroux, Y. Tetrahedron Lett. 2001, 42, 8475–8478. doi:10.1016/S0040-4039(01)01844-5 |
| 16. | Guenin, E.; Degache, E.; Liquier, J.; Lecouvey, M. Eur. J. Org. Chem. 2004, 2983–2987. doi:10.1002/ejoc.200400053 |
| 17. | Guenin, E.; Ledoux, D.; Oudar, O.; Lecouvey, M.; Kraemer, M. Anticancer Res. 2005, 25, 1139–1145. |
| 18. | Migianu-Griffoni, E.; Mbemba, C.; Burgada, R.; Lecerclé, D.; Taran, F.; Lecouvey, M. Tetrahedron 2009, 65, 1517–1523. doi:10.1016/j.tet.2008.11.076 |
| 19. | Monteil, M.; Guenin, E.; Migianu, E.; Lutomski, D.; Lecouvey, M. Tetrahedron 2005, 61, 7528–7537. doi:10.1016/j.tet.2005.05.053 |
| 20. | Monteil, M.; Migianu-Griffoni, E.; Sainte-Catherine, O.; Di Benedetto, M.; Lecouvey, M. Eur. J. Med. Chem. 2014, 77, 56–64. doi:10.1016/j.ejmech.2014.02.054 |
| 21. | Kachbi Khelfallah, S.; Monteil, M.; Deschamp, J.; Gager, O.; Migianu-Griffoni, E.; Lecouvey, M. Org. Biomol. Chem. 2015, 13, 11382–11392. doi:10.1039/C5OB01967B |
| 23. | Hojo, K.; Susuki, Y.; Sasaki, M.; Maeda, M.; Smith, T. J.; Kawasaki, K. Chem. Pharm. Bull. 2002, 50, 1001–1003. doi:10.1248/cpb.50.1001 |
| 15. | Lecouvey, M.; Mallard, I.; Bailly, T.; Burgada, R.; Leroux, Y. Tetrahedron Lett. 2001, 42, 8475–8478. doi:10.1016/S0040-4039(01)01844-5 |
| 24. | Thottumkara, A. P.; Bowsher, M. S.; Vinod, T. K. Org. Lett. 2005, 7, 2933–2936. doi:10.1021/ol050875o |
| 14. | Kieczykowski, G. R.; Jobson, R. B.; Melillo, D. G.; Reinhold, D. F.; Grenda, V. J.; Shinkai, I. J. Org. Chem. 1995, 60, 8310–8312. doi:10.1021/jo00130a036 |
| 11. | Benyettou, F.; Lalatonne, Y.; Chebbi, I.; Di Benedetto, M.; Serfaty, J.-M.; Lecouvey, M.; Motte, L. Phys. Chem. Chem. Phys. 2011, 13, 10020–10027. doi:10.1039/c0cp02034f |
| 12. | Benyettou, F.; Lalatonne, Y.; Sainte-Catherine, O.; Monteil, M.; Motte, L. Int. J. Pharm. 2009, 379, 324–327. doi:10.1016/j.ijpharm.2009.04.010 |
| 13. | Bolley, J.; Guenin, E.; Lievre, N.; Lecouvey, M.; Soussan, M.; Lalatonne, Y.; Motte, L. Langmuir 2013, 29, 14639–14647. doi:10.1021/la403245h |
| 22. | Fishman, A.; Acton, A.; Lee-Ruff, E. Synth. Commun. 2004, 34, 2309–2312. doi:10.1081/SCC-120038518 |
© 2016 Kachbi-Khelfallah et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)