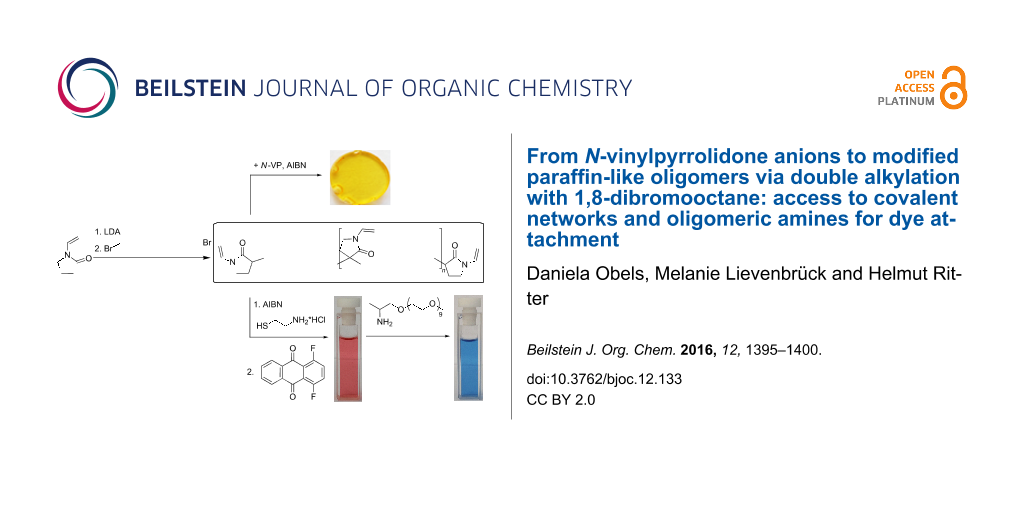
Abstract
The double alkylation of N-vinylpyrrolidone (N-VP) with 1,8-dibromooctane yields paraffin-like oligomeric chains bearing polymerizable vinyl moieties. These oligomers were radically crosslinked in bulk with N-VP as co-monomer yielding swellable polymer disks. The vinylic side groups of the N-VP oligomers allow thiol–ene click reactions with 2-aminoethanethiol hydrochloride to obtain reactive amino-functionalized oligomers. Further modification of the free amino groups with 1,4-difluoro-9,10-anthraquinone (DFA) yields red-colored oligomeric anthraquinone dyes. The final reaction of DFA-substituted N-VP oligomers with Jeffamine® M 600 leads to blue-colored and branched oligomers with poly(ethylene glycol) side chains.

Graphical Abstract
Introduction
Poly(N-vinylpyrrolidone) (PVP) is established in daily life due to its high water solubility and physiological compatibility [1-7]. It is used as additive, e.g., in cosmetics [8], pharmaceutical preparations [9-12] and food industry (additive E 1201) [13]. Monoalkylation reactions of N-VP in α-position via deprotonation by lithium diisopropylamide [14] were carried out to open up access to further potential applications of the corresponding polymers [14-16]. Thereby, the electron-rich double bond of N-VP is stable towards nucleophilic attack of the resulting carbanion intermediates. Some alkylated N-VP-based monomers can be used as agents in click chemistry [17] to obtain thermo- [18] or/and pH [19] responsive polymers. Besides classical alkylation reactions, Reinecke et al. performed ring-opening reactions to insert functional groups and aromatic side chains [20,21]. Furthermore, they investigated the formation of homogeneous [22] and amphiphilic [23] N-VP networks. However, in contrast to the restricted monoalkylation of N-vinylcaprolactam [24], the five-membered ring of N-VP can be double alkylated in α-position. In this connection, 1-bromo-2-(2-bromoethoxy)ethane was used as alkylation reagent to allow the preparation of a spiro-type monomer [25] due to intramolecular reaction. Until now, dibromo compounds were mainly used to synthesize symmetric cross linker [22,26,27]. To the best of our knowledge, the synthesis of modified paraffin-like oligomers via double alkylation of N-VP with aliphatic dibromides was not yet described in literature. In general, the term paraffin means parum affinis or poor reactivity and comprises acyclic alkanes. They are obtained as byproducts in petroleum industry [28,29]. Technical derivatives are chloro- and chlorosulfonated paraffins which are used, e.g., as surfactants [30-33].
In the current paper we wish to present the synthesis of paraffin-like oligomers via double alkylation of N-VP with 1,8-dibromooctane. Additionally, this work focuses on the use of the free double bonds for radical crosslinking as well as thiol-ene modification for subsequent dye attachment.
Results and Discussion
N-Vinylpyrrolidone (1) can be mono or di-deprotonated in α-position to the carbonyl group by the use of the strong and sterically demanding base lithium diisopropylamide [14,34,35]. As mentioned above, the resulting carbanions do not attack the electron-rich N-vinyl double bond. Accordingly, by using 1,8-dibromooctane as alkylation reagent, extending the reaction time and working with a high concentrated solution a mixture of paraffin-like oligomers 2a–c differing in their end groups was obtained. The oligomers were separated by use of column chromatography (Scheme 1).
![[1860-5397-12-133-i1]](/bjoc/content/inline/1860-5397-12-133-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Alkylation of N-VP with 1,8-dibromooctane yielding paraffin-like oligomers 2a–c.
Scheme 1: Alkylation of N-VP with 1,8-dibromooctane yielding paraffin-like oligomers 2a–c.
The chemical structures of the derivatives 2a–c were verified by 1H NMR spectroscopy and ESI mass spectrometry. Accordingly, a maximal number of three repeating units could be detected and the corresponding data are given in Supporting Information File 1.
The vinylic groups of 2a can easily be radically copolymerized in bulk with a molar excess of N-VP. By using either 1.0 mol % (Figure 1) or 2.5 mol % of 2a in relation to N-VP, two different cross-linked polymers 3a,b were obtained as disks showing a certain swelling behavior in distilled water (Figure 1).
![[1860-5397-12-133-1]](/bjoc/content/figures/1860-5397-12-133-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chemical structure of water swellable network 3a and 3b. Photographs of water-swollen polymer disks consisting of 1 mol % (3a) and 2.5 mol % (3b) of 2a as cross linker.
Figure 1: Chemical structure of water swellable network 3a and 3b. Photographs of water-swollen polymer disks...
The water uptake (W) [23,36] of the cross-linked polymers 3a and 3b in distilled water was followed gravimetrically. Equation 1 was used for the calculation,
![[1860-5397-12-133-i3]](/bjoc/content/inline/1860-5397-12-133-i3.svg?max-width=590&scale=1.18182)
where W = water uptake, x = weight of dry polymer disk, and y = weight of swollen polymer disk.
As expected, the water uptake decreased with increasing the amount of used cross linker 2a (Table 1) [37-39].
Table 1: Water uptake of cross-linked polymers 3a and 3b.
| Polymer (disk) | Content of 2a [mol %] | Water uptake [%]a |
|---|---|---|
| 3a | 1 | 189 |
| 3b | 2.5 | 104 |
aMean value of 3 measurements.
Additionally, the free double bonds of 2a were subjected to further modification through a thiol–ene [40-42] click reaction with 2-aminoethanethiol hydrochloride yielding oligomer 4 (Scheme 2). Subsequently, the reactivity of the primary amino groups in 4 was proven by the attachment of 1,4-difluoro-9,10-anthraquinone (DFA). Due to the strongly graduated reactivity of DFA, mono-functionalization is feasible, accompanied by a visible color change from yellow to red. This feature allows a reaction control with the naked eye and also with, e.g., UV–vis spectroscopy. The following reaction of DFA-substituted polymer 5 with Jeffamine® M 600 leads to blue-colored branched oligomer 6 with poly(ethylene glycol) side chains. The conducted reaction sequence is shown in Scheme 2.
![[1860-5397-12-133-i2]](/bjoc/content/inline/1860-5397-12-133-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the branched oligomeric dye 6: a) radical thiol–ene click reaction of 2a with 2-aminoethanethiol hydrochloride yielding 4. b) Subsequent reaction of 4 with DFA leading to red-colored oligomeric dye 5. c) Branching of 5 with Jeffamine® M 600 yielding blue-colored oligomer dye 6.
Scheme 2: Synthesis of the branched oligomeric dye 6: a) radical thiol–ene click reaction of 2a with 2-aminoe...
The quantitative conversion of the double bonds of oligomer 2a was verified by 1H NMR spectroscopy. As described above, the corresponding product 4 bears reactive amino groups for further modifications. The successful reaction of 4 with DFA can be monitored by 1H NMR spectroscopy through the appearance of an aromatic NH signal at about 10 ppm and additionally on the change of the chemical shifts of the protons next to the fluoro substituents in attached DFA. The synthesis was carried out under mild conditions with a slight excess of DFA to achieve high conversions. Therefore, the 1H NMR and UV–vis spectra, respectively still showed signals of ca. 20 mol % of DFA [43] and the corresponding data are given in Supporting Information File 1.
The second fluoro substituent of the attached anthraquinone moiety in 5 was replaced at elevated temperature with O-(2-aminopropyl)-O'-(2-methoxyethyl)propylene glycol (Jeffamine® M 600) yielding the branched blue-colored oligomeric dye 6. The deep blue color is a result of the presence of two amino groups in 1,4-position of the anthraquinone dye [44]. Thus, this reaction can also be followed with the naked eye. Furthermore, the conversion of oligomer 6 was verified with UV–vis spectroscopy (Figure 2).
![[1860-5397-12-133-2]](/bjoc/content/figures/1860-5397-12-133-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: UV–vis spectra of oligomers 5 (c = 0.16 g/mL, red), 6 (c = 0.63 g/mL, blue) and DFA (c = 0.016 g/mL, grey).
Figure 2: UV–vis spectra of oligomers 5 (c = 0.16 g/mL, red), 6 (c = 0.63 g/mL, blue) and DFA (c = 0.016 g/mL...
Accordingly, the different UV–vis spectra (Figure 2) of DFA, oligomer 5 and the disubstituted anthraquinone dye in 6, respectively clearly verify the successful modification reactions. The complete conversion of 5 with Jeffamine® M 600 is proven by the shift of the absorption maxima from 320 nm and 505 nm to higher wavelengths at 595 nm and 640 nm. According to literature, the introduction of amino groups in the 1- and 1,4-position of anthraquinones leads to a charge transfer of electrons from the amino group to the carbonyl functionality which results in additional π–π* absorption bands in the spectra of these compounds [45].
Conclusion
The synthesis of paraffin-like oligomers 2a–c via double alkylation of N-VP with 1,8-dibromooctane can easily be conducted through a one-pot synthesis. Swellable networks can be obtained by radical copolymerization of 2a with N-VP. Furthermore, the conversion of the double bonds through thiol–ene click reaction with 2-aminoethanethiol hydrochloride leads to paraffinic oligomers 4 bearing primary amino groups. The reactivity of the latter functionalities was exemplarily demonstrated by the reaction with the dye 1,4-difluoro-9,10-anthraquinone resulting in the red-colored derivative 5. Further reaction of 5 with Jeffamine® M 600 yielded the blue-colored branched oligomer 6. Both reactions can easily be monitored with UV–vis spectroscopy as well as with the naked eye. This work contributes to the increasing area of α-alkylation reactions of N-vinyl lactams and the subsequent application of the so obtained compounds to the synthesis of functionalized oligomers.
Supporting Information
| Supporting Information File 1: Experimental. | ||
| Format: PDF | Size: 974.5 KB | Download |
References
-
Hecht, G.; Scholtan, W. Z. Gesamte Exp. Med. 1959, 130, 577–603. doi:10.1007/BF02049339
Return to citation in text: [1] -
Scholtan, W. Makromol. Chem. 1953, 11, 131–230. doi:10.1002/macp.1953.020110114
Return to citation in text: [1] -
Schmidt, K. Klin. Wochenschr. 1979, 57, 1169–1175. doi:10.1007/BF01491757
Return to citation in text: [1] -
Patat, F.; Estupinan, L. Makromol. Chem. 1961, 49, 182–199. doi:10.1002/macp.1961.020490113
Return to citation in text: [1] -
Vijayasekaran, S.; Chirila, T. V.; Hong, Y.; Tahija, S. G.; Dalton, P. D.; Constable, I. J.; McAllister, I. L. J. Biomater. Sci., Polym. Ed. 1996, 7, 685–696. doi:10.1163/156856296X00453
Return to citation in text: [1] -
Göthlich, A.; Koltzenburg, S.; Schornick, G. Chem. Unserer Zeit 2005, 39, 262–273. doi:10.1002/ciuz.200400346
Return to citation in text: [1] -
Fischer, F.; Bauer, S. Chem. Unserer Zeit 2009, 43, 376–383. doi:10.1002/ciuz.200900492
Return to citation in text: [1] -
Haaf, F.; Sanner, A.; Straub, F. Polym. J. 1985, 17, 143–152. doi:10.1295/polymj.17.143
Return to citation in text: [1] -
Franzmann, G.; Ringsdorf, H. Makromol. Chem. 1976, 177, 2547–2552. doi:10.1002/macp.1976.021770823
Return to citation in text: [1] -
Kaneda, Y.; Tsutsumi, Y.; Yoshioka, Y.; Kamada, H.; Yamamoto, Y.; Kodaira, H.; Tsunoda, S.-i.; Okamoto, T.; Mukai, Y.; Shibata, H.; Nagakawa, S.; Mayumi, T. Biomaterials 2004, 25, 3259–3266. doi:10.1016/j.biomaterials.2003.10.003
Return to citation in text: [1] -
Gómez-Tardajos, M.; Pino-Pinto, J. P.; Díaz-Soto, C.; Flores, M. E.; Gallardo, A.; Elvira, C.; Reinecke, H.; Nishide, H.; Moreno-Villoslada, I. Dyes Pigm. 2013, 99, 759–770. doi:10.1016/j.dyepig.2013.06.028
Return to citation in text: [1] -
Widanapathirana, L.; Tale, S.; Reineke, T. M. Mol. Pharmaceutics 2015, 12, 2537–2543. doi:10.1021/acs.molpharmaceut.5b00202
Return to citation in text: [1] -
Scientific Committee on Food European, Commission. Off. J. Eur. Communities: Legis. 2002, 292, 1–28. Note: 2002/82/EC of 15 October 2002.
Return to citation in text: [1] -
Bencini, M.; Ranucci, E.; Ferruti, P.; Oldani, C.; Licandro, E.; Maiorana, S. Macromolecules 2005, 38, 8211–8219. doi:10.1021/ma050422z
Return to citation in text: [1] [2] [3] -
He, W.; Gonsalves, K. E.; Pickett, J. H.; Halberstadt, C. Biomacromolecules 2003, 4, 75–79. doi:10.1021/bm0256505
Return to citation in text: [1] -
del Prado, A.; Navarro, R.; Gallardo, A.; Elvira, C.; Reinecke, H. RSC Adv. 2014, 4, 35950–35958. doi:10.1039/C4RA05354K
Return to citation in text: [1] -
Trellenkamp, T.; Ritter, H. Macromolecules 2010, 43, 5538–5543. doi:10.1021/ma100812q
Return to citation in text: [1] -
Trellenkamp, T.; Ritter, H. Macromol. Rapid Commun. 2009, 30, 1736–1740. doi:10.1002/marc.200900250
Return to citation in text: [1] -
del Prado, A.; Navarro, R.; Levkin, P.; Gallardo, A.; Elvira, C.; Reinecke, H. J. Polym. Sci., Part A: Polym. Chem. 2016, 54, 1098–1108. doi:10.1002/pola.27949
Return to citation in text: [1] -
Pérez Perrino, M.; Navarro, R.; Gómez Tardajos, M.; Gallardo, A.; Reinecke, H. Eur. Polym. J. 2010, 46, 1557–1562. doi:10.1016/j.eurpolymj.2010.04.006
Return to citation in text: [1] -
Pérez Perrino, M.; Navarro, R.; Gómez Tardajos, M.; Gallardo, A.; Reinecke, H. Macromol. Chem. Phys. 2009, 210, 1973–1978. doi:10.1002/macp.200900306
Return to citation in text: [1] -
Tardajos, M. G.; Nash, M.; Rochev, Y.; Reinecke, H.; Elvira, C.; Gallardo, A. Macromol. Chem. Phys. 2012, 213, 529–538. doi:10.1002/macp.201100564
Return to citation in text: [1] [2] -
Tardajos, M. G.; Aranaz, I.; Pérez, M.; López, D.; Reinecke, H.; Elvira, C.; Gallardo, A. Macromolecules 2013, 46, 5018–5025. doi:10.1021/ma400483f
Return to citation in text: [1] [2] -
Schönenberg, L.; Ritter, H. Macromol. Chem. Phys. 2015, 216, 69–76. doi:10.1002/macp.201400376
Return to citation in text: [1] -
Obels, D.; Siegfeld, P.; Ritter, H. Polym. Int. 2016, 65, 109–114. doi:10.1002/pi.5037
Return to citation in text: [1] -
White, L. A.; Jönson, S.; Hoyle, C. E.; Mathias, L. J. Polymer 1999, 40, 6597–6605. doi:10.1016/S0032-3861(99)00189-5
Return to citation in text: [1] -
Engström, J. U. A.; Helgee, B. J. Comb. Chem. 2006, 8, 355–360. doi:10.1021/cc050148k
Return to citation in text: [1] -
Eldridge, R. B. Ind. Eng. Chem. Res. 1993, 32, 2208–2212. doi:10.1021/ie00022a002
Return to citation in text: [1] -
Elder, R. J. Am. Coll. Toxicol. 1984, 3, 43–99. doi:10.3109/10915818409010516
Return to citation in text: [1] -
Bayen, S.; Obbard, J. P.; Thomas, G. O. Environ. Int. 2006, 32, 915–929. doi:10.1016/j.envint.2006.05.009
Return to citation in text: [1] -
de Boer, J.; Hutzinger, O.; T. E.-S., Ali. Chlorinated paraffins; The handbook of environmental chemistry; Springer: Berlin, Heidelberg, 2010.
Return to citation in text: [1] -
Panzel, H.; Ballschmiter, K. Fresenius' Z. Anal. Chem. 1974, 271, 182–191. doi:10.1007/BF00430731
Return to citation in text: [1] -
Alkan, C. Thermochim. Acta 2006, 451, 126–130. doi:10.1016/j.tca.2006.09.010
Return to citation in text: [1] -
Iwema Bakker, W. I.; Lee Wong, P.; Snieckus, V.; Warrington, J. M.; Barriault, L. e-EROS Encyclopedia of Reagents for Organic Synthesis; Wiley, 2001. doi:10.1002/047084289X
Return to citation in text: [1] -
Reetz, M. T.; Maier, W. F. Liebigs Ann. Chem. 1980, 1471–1473. doi:10.1002/jlac.198019801002
Return to citation in text: [1] -
Sánchez-Chaves, M.; Martínez, G.; López Madruga, E.; Fernández-Monreal, C. J. Polym. Sci., Part A: Polym. Chem. 2002, 40, 1192–1199. doi:10.1002/pola.10196
Return to citation in text: [1] -
Ganji, F.; Vasheghani-Farahani, S.; Vasheghani-Farahani, E. Iran. Polym. J. 2010, 19, 375–398.
Return to citation in text: [1] -
Colombo, P. Adv. Drug Delivery Rev. 1993, 11, 37–57. doi:10.1016/0169-409X(93)90026-Z
Return to citation in text: [1] -
Mathur, A. M.; Moorjani, S. K.; Scranton, A. B. J. Macromol. Sci., Rev. Macromol. Chem. Phys. 1996, 36, 405–430. doi:10.1080/15321799608015226
Return to citation in text: [1] -
Hoyle, C. E.; Bowman, C. N. Angew. Chem., Int. Ed. 2010, 49, 1540–1573. doi:10.1002/anie.200903924
Return to citation in text: [1] -
Dondoni, A. Angew. Chem. 2008, 120, 9133–9135. doi:10.1002/ange.200802516
Return to citation in text: [1] -
Parakka, J.; McCrea, K. R.; Ward, R. S. PDMS-PVP block copolymers. U. S. Patent US8,153,728 B2, April 10, 2012.
Return to citation in text: [1] -
Krapcho, A. P.; Getahun, Z. Synth. Commun. 1985, 15, 907–910. doi:10.1080/00397918508063889
Return to citation in text: [1] -
Jacquemin, D.; Preat, J.; Charlot, M.; Wathelet, V.; André, J.-M.; Perpète, E. A. J. Chem. Phys. 2004, 121, 1736–1743. doi:10.1063/1.1764497
Return to citation in text: [1] -
Diaz, A. N. J. Photochem. Photobiol., A: Chem. 1990, 53, 141–167. doi:10.1016/1010-6030(90)87120-Z
Return to citation in text: [1]
| 23. | Tardajos, M. G.; Aranaz, I.; Pérez, M.; López, D.; Reinecke, H.; Elvira, C.; Gallardo, A. Macromolecules 2013, 46, 5018–5025. doi:10.1021/ma400483f |
| 36. | Sánchez-Chaves, M.; Martínez, G.; López Madruga, E.; Fernández-Monreal, C. J. Polym. Sci., Part A: Polym. Chem. 2002, 40, 1192–1199. doi:10.1002/pola.10196 |
| 30. | Bayen, S.; Obbard, J. P.; Thomas, G. O. Environ. Int. 2006, 32, 915–929. doi:10.1016/j.envint.2006.05.009 |
| 31. | de Boer, J.; Hutzinger, O.; T. E.-S., Ali. Chlorinated paraffins; The handbook of environmental chemistry; Springer: Berlin, Heidelberg, 2010. |
| 32. | Panzel, H.; Ballschmiter, K. Fresenius' Z. Anal. Chem. 1974, 271, 182–191. doi:10.1007/BF00430731 |
| 33. | Alkan, C. Thermochim. Acta 2006, 451, 126–130. doi:10.1016/j.tca.2006.09.010 |
| 14. | Bencini, M.; Ranucci, E.; Ferruti, P.; Oldani, C.; Licandro, E.; Maiorana, S. Macromolecules 2005, 38, 8211–8219. doi:10.1021/ma050422z |
| 34. | Iwema Bakker, W. I.; Lee Wong, P.; Snieckus, V.; Warrington, J. M.; Barriault, L. e-EROS Encyclopedia of Reagents for Organic Synthesis; Wiley, 2001. doi:10.1002/047084289X |
| 35. | Reetz, M. T.; Maier, W. F. Liebigs Ann. Chem. 1980, 1471–1473. doi:10.1002/jlac.198019801002 |
| 1. | Hecht, G.; Scholtan, W. Z. Gesamte Exp. Med. 1959, 130, 577–603. doi:10.1007/BF02049339 |
| 2. | Scholtan, W. Makromol. Chem. 1953, 11, 131–230. doi:10.1002/macp.1953.020110114 |
| 3. | Schmidt, K. Klin. Wochenschr. 1979, 57, 1169–1175. doi:10.1007/BF01491757 |
| 4. | Patat, F.; Estupinan, L. Makromol. Chem. 1961, 49, 182–199. doi:10.1002/macp.1961.020490113 |
| 5. | Vijayasekaran, S.; Chirila, T. V.; Hong, Y.; Tahija, S. G.; Dalton, P. D.; Constable, I. J.; McAllister, I. L. J. Biomater. Sci., Polym. Ed. 1996, 7, 685–696. doi:10.1163/156856296X00453 |
| 6. | Göthlich, A.; Koltzenburg, S.; Schornick, G. Chem. Unserer Zeit 2005, 39, 262–273. doi:10.1002/ciuz.200400346 |
| 7. | Fischer, F.; Bauer, S. Chem. Unserer Zeit 2009, 43, 376–383. doi:10.1002/ciuz.200900492 |
| 14. | Bencini, M.; Ranucci, E.; Ferruti, P.; Oldani, C.; Licandro, E.; Maiorana, S. Macromolecules 2005, 38, 8211–8219. doi:10.1021/ma050422z |
| 22. | Tardajos, M. G.; Nash, M.; Rochev, Y.; Reinecke, H.; Elvira, C.; Gallardo, A. Macromol. Chem. Phys. 2012, 213, 529–538. doi:10.1002/macp.201100564 |
| 26. | White, L. A.; Jönson, S.; Hoyle, C. E.; Mathias, L. J. Polymer 1999, 40, 6597–6605. doi:10.1016/S0032-3861(99)00189-5 |
| 27. | Engström, J. U. A.; Helgee, B. J. Comb. Chem. 2006, 8, 355–360. doi:10.1021/cc050148k |
| 13. | Scientific Committee on Food European, Commission. Off. J. Eur. Communities: Legis. 2002, 292, 1–28. Note: 2002/82/EC of 15 October 2002. |
| 28. | Eldridge, R. B. Ind. Eng. Chem. Res. 1993, 32, 2208–2212. doi:10.1021/ie00022a002 |
| 29. | Elder, R. J. Am. Coll. Toxicol. 1984, 3, 43–99. doi:10.3109/10915818409010516 |
| 9. | Franzmann, G.; Ringsdorf, H. Makromol. Chem. 1976, 177, 2547–2552. doi:10.1002/macp.1976.021770823 |
| 10. | Kaneda, Y.; Tsutsumi, Y.; Yoshioka, Y.; Kamada, H.; Yamamoto, Y.; Kodaira, H.; Tsunoda, S.-i.; Okamoto, T.; Mukai, Y.; Shibata, H.; Nagakawa, S.; Mayumi, T. Biomaterials 2004, 25, 3259–3266. doi:10.1016/j.biomaterials.2003.10.003 |
| 11. | Gómez-Tardajos, M.; Pino-Pinto, J. P.; Díaz-Soto, C.; Flores, M. E.; Gallardo, A.; Elvira, C.; Reinecke, H.; Nishide, H.; Moreno-Villoslada, I. Dyes Pigm. 2013, 99, 759–770. doi:10.1016/j.dyepig.2013.06.028 |
| 12. | Widanapathirana, L.; Tale, S.; Reineke, T. M. Mol. Pharmaceutics 2015, 12, 2537–2543. doi:10.1021/acs.molpharmaceut.5b00202 |
| 24. | Schönenberg, L.; Ritter, H. Macromol. Chem. Phys. 2015, 216, 69–76. doi:10.1002/macp.201400376 |
| 45. | Diaz, A. N. J. Photochem. Photobiol., A: Chem. 1990, 53, 141–167. doi:10.1016/1010-6030(90)87120-Z |
| 8. | Haaf, F.; Sanner, A.; Straub, F. Polym. J. 1985, 17, 143–152. doi:10.1295/polymj.17.143 |
| 25. | Obels, D.; Siegfeld, P.; Ritter, H. Polym. Int. 2016, 65, 109–114. doi:10.1002/pi.5037 |
| 19. | del Prado, A.; Navarro, R.; Levkin, P.; Gallardo, A.; Elvira, C.; Reinecke, H. J. Polym. Sci., Part A: Polym. Chem. 2016, 54, 1098–1108. doi:10.1002/pola.27949 |
| 22. | Tardajos, M. G.; Nash, M.; Rochev, Y.; Reinecke, H.; Elvira, C.; Gallardo, A. Macromol. Chem. Phys. 2012, 213, 529–538. doi:10.1002/macp.201100564 |
| 43. | Krapcho, A. P.; Getahun, Z. Synth. Commun. 1985, 15, 907–910. doi:10.1080/00397918508063889 |
| 18. | Trellenkamp, T.; Ritter, H. Macromol. Rapid Commun. 2009, 30, 1736–1740. doi:10.1002/marc.200900250 |
| 23. | Tardajos, M. G.; Aranaz, I.; Pérez, M.; López, D.; Reinecke, H.; Elvira, C.; Gallardo, A. Macromolecules 2013, 46, 5018–5025. doi:10.1021/ma400483f |
| 44. | Jacquemin, D.; Preat, J.; Charlot, M.; Wathelet, V.; André, J.-M.; Perpète, E. A. J. Chem. Phys. 2004, 121, 1736–1743. doi:10.1063/1.1764497 |
| 17. | Trellenkamp, T.; Ritter, H. Macromolecules 2010, 43, 5538–5543. doi:10.1021/ma100812q |
| 37. | Ganji, F.; Vasheghani-Farahani, S.; Vasheghani-Farahani, E. Iran. Polym. J. 2010, 19, 375–398. |
| 38. | Colombo, P. Adv. Drug Delivery Rev. 1993, 11, 37–57. doi:10.1016/0169-409X(93)90026-Z |
| 39. | Mathur, A. M.; Moorjani, S. K.; Scranton, A. B. J. Macromol. Sci., Rev. Macromol. Chem. Phys. 1996, 36, 405–430. doi:10.1080/15321799608015226 |
| 14. | Bencini, M.; Ranucci, E.; Ferruti, P.; Oldani, C.; Licandro, E.; Maiorana, S. Macromolecules 2005, 38, 8211–8219. doi:10.1021/ma050422z |
| 15. | He, W.; Gonsalves, K. E.; Pickett, J. H.; Halberstadt, C. Biomacromolecules 2003, 4, 75–79. doi:10.1021/bm0256505 |
| 16. | del Prado, A.; Navarro, R.; Gallardo, A.; Elvira, C.; Reinecke, H. RSC Adv. 2014, 4, 35950–35958. doi:10.1039/C4RA05354K |
| 20. | Pérez Perrino, M.; Navarro, R.; Gómez Tardajos, M.; Gallardo, A.; Reinecke, H. Eur. Polym. J. 2010, 46, 1557–1562. doi:10.1016/j.eurpolymj.2010.04.006 |
| 21. | Pérez Perrino, M.; Navarro, R.; Gómez Tardajos, M.; Gallardo, A.; Reinecke, H. Macromol. Chem. Phys. 2009, 210, 1973–1978. doi:10.1002/macp.200900306 |
| 40. | Hoyle, C. E.; Bowman, C. N. Angew. Chem., Int. Ed. 2010, 49, 1540–1573. doi:10.1002/anie.200903924 |
| 41. | Dondoni, A. Angew. Chem. 2008, 120, 9133–9135. doi:10.1002/ange.200802516 |
| 42. | Parakka, J.; McCrea, K. R.; Ward, R. S. PDMS-PVP block copolymers. U. S. Patent US8,153,728 B2, April 10, 2012. |
© 2016 Obels et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)