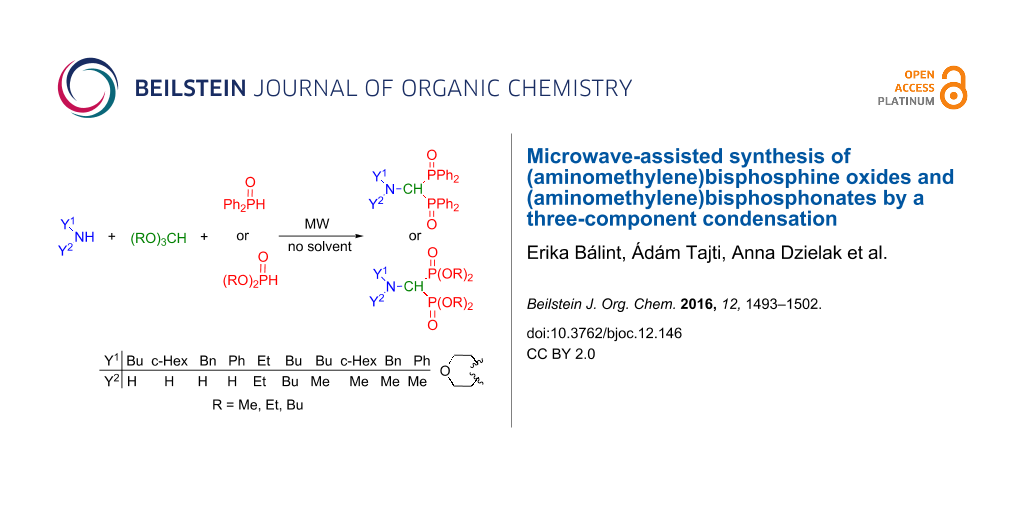
Abstract
A practical method was elaborated for the synthesis of (aminomethylene)bisphosphine oxides comprising the catalyst- and solvent-free microwave-assisted three-component condensation of primary amines, triethyl orthoformate and two equivalents of diphenylphosphine oxide. The method is also suitable for the preparation of (aminomethylene)bisphosphonates using (MeO)2P(O)H/(MeO)3CH or (EtO)2P(O)H/(EtO)3CH reactant pairs and even secondary amines. Several intermediates referring to the reaction mechanism together with a few by-products could also be identified.

Graphical Abstract
Introduction
Substituted (hydroxymethylene)bisphosphonic acid derivatives form an important group of drugs used in the treatment of osteoporosis and related bone diseases [1-3]. In the last decades, at least three generations of dronic acid derivatives appeared [4].
(Aminomethylene)bisphosphonic acid derivatives are analogous species, that also have potential bioactivity in bone diseases, besides they display antibacterial, antiparasitic, anticancer and herbicidal activities [5].
(Aminomethylene)bisphosphonates may be prepared in different ways [5]. One of the most convenient and widespread methods is the three-component condensation involving an amine, an orthoformate and a dialkyl phosphite. Usually, primary or secondary amines were reacted with an equivalent, or a small excess of triethyl orthoformate and 2–7 equivalents of diethyl phosphite [6-21]. In most cases, the corresponding acids were the target molecules that were obtained by hydrolysis of the esters [15-21]. The use of crown ethers with an NH unit, or thienopyrimidine amines as starting materials was also reported [22,23]. The catalyst- and solvent-free methods required long reaction times and/or a high temperature [6-14,21-23]. Ionic liquids and a few catalysts were also tried out [24-27], and the synthesis was also described under microwave (MW) irradiation [28-32]. However, most of the MW-assisted syntheses were performed in kitchen ovens [28-30], hence these results cannot be reproduced.
The mechanism of the three-component condensation has been investigated by the research group of Krutikov and Kafarski [6,7]. A detailed proposal is shown in Scheme 1 [7]. The first step of the condensation is the reaction of the amine with the orthoformate, in which imine-type intermediates I or II may be formed. The next step is the nucleophilic addition of diethyl phosphite to the C=N bond of the imines resulting in phosphonates III or IV, respectively. Then, the elimination of an amine or ethanol and the addition of another unit of diethyl phosphite may lead to (aminomethylene)bisphosphonates (VI). If the amine is in predominance over the phosphite in the reaction, the pathway A is more likely, but if the phosphite is used in excess, the pathway B comes to the fore.
![[1860-5397-12-146-i1]](/bjoc/content/inline/1860-5397-12-146-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Proposed routes for the three-component condensation [7].
Scheme 1: Proposed routes for the three-component condensation [7].
There are other possibilities to synthesize (aminomethylene)bisphosphonates, such as by the reaction of dimethylformamide diethyl acetal with diethyl phosphite (Scheme 2a) [33], by the condensation of formamides and diethyl phosphite using 2,6-di-tert-butyl-4-methylpyridine (DTBMP) as the base, and trifluoromethanesulfonic anhydride (Tf2O) as the catalyst (Scheme 2b) [34], or by the reaction of isonitriles with triethyl phosphite (Scheme 2c) [35,36]. (Aminomethylene)bisphosphonates can also be obtained starting from amides, triethyl phosphite and phosphorus oxychloride (Scheme 3a) [37], or in the reaction of amines with diazophosphonate in the presence of a rhodium catalyst (Scheme 3b) [38].
![[1860-5397-12-146-i2]](/bjoc/content/inline/1860-5397-12-146-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthetic methods for (aminomethylene)bisphosphonates I.
Scheme 2: Synthetic methods for (aminomethylene)bisphosphonates I.
![[1860-5397-12-146-i3]](/bjoc/content/inline/1860-5397-12-146-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthetic methods for (aminomethylene)bisphosphonates II.
Scheme 3: Synthetic methods for (aminomethylene)bisphosphonates II.
(Aminomethylene)bisphosphine oxides are analogous to (aminomethylene)bisphosphonates, but they are much less studied. Only a few publications were found, which focus on their synthesis [33,39-42], however, a three-component condensation has not been described. They can also be prepared starting from dimethylformamide dimethyl acetal, as in the synthesis of (aminomethylene)bisphosphonates, but in the latter case a secondary phosphine oxide is the P-reagent [33,39]. In addition, (aminomethylene)bisphosphine oxides can be synthesized by the reaction of two molecules of (dialkylamino)(diphenylphosphinoyl)chloromethane (Scheme 4a) [40,41], or by the addition of diphenylphosphine oxide to an isonitrile (Scheme 4b) [36,42].
![[1860-5397-12-146-i4]](/bjoc/content/inline/1860-5397-12-146-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthetic methods for (aminomethylene)bisphosphine oxides.
Scheme 4: Synthetic methods for (aminomethylene)bisphosphine oxides.
In this paper, we wish to report the results of our investigations on the synthetic protocol utilizing the three-component condensations of primary or secondary amines, orthoformates and >P(O)H species, such as dialkyl phosphites or diphenylphosphine oxide, and we aimed at the preparation of new derivatives.
Results and Discussion
Synthesis of alkylamino- and (phenylaminomethylene)bisphosphine oxides
In the first series of experiments, the condensation of primary amines, such as butyl-, cyclohexyl- and benzylamine or aniline with triethyl orthoformate, and 2 equivalents of diphenylphosphine oxide at 150 °C for 1 h under MW conditions was studied (Scheme 5). To avoid the formation of by-products, benzylamine was reacted at a lower temperature of 125 °C (Table 1, entry 3). The reactions were carried out without any catalyst and solvent. After column chromatography, the new amino-methylenebisphosphine oxides 1a–d were obtained in yields of 72–82% (Table 1, entries 1–4).
![[1860-5397-12-146-i5]](/bjoc/content/inline/1860-5397-12-146-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of alkylamino- and (phenylaminomethylene)bisphosphine oxides.
Scheme 5: Synthesis of alkylamino- and (phenylaminomethylene)bisphosphine oxides.
Table 1: Synthesis of alkylamino- and (phenylaminomethylene)bisphosphine oxides 1a–d.
| Entry | Y | T (°C) | Yield (%)a |
|---|---|---|---|
| 1 | Bu | 150 | 82 (1a) |
| 2 | c-Hex | 150 | 79 (1b) |
| 3 | Bn | 125b | 72 (1c) |
| 4 | Ph | 150 | 80 (1d) |
aIsolated yield. bAt 150 °C by-products were formed.
The condensation of simple secondary amines (diethyl-, dibutyl-, N-butylmethyl-, N-cyclohexylmethyl-, N-benzylmethylamine, N-methylaniline and morpholine) was also investigated with triethyl orthoformate, and 2 equivalents of diphenylphosphine oxide (Scheme 6, Table 2). The MW-assisted reactions were performed at 150 °C for 1 h under solvent- and catalyst-free conditions, and the (dialkylaminomethylene)bisphosphine oxides 2a–g were obtained in yields of 60–85% after column chromatography (Table 2, entries 1–7). Except for compound 2g, all (aminomethylene)bisphosphine oxides (2a–f) prepared are new compounds. According to the literature method [41], 2g was synthesized by the reaction of two molecules of (diphenylphosphinoyl)morpholinochloromethane in the presence of chloroform at 5 °C for 18 h in a yield of 41% (Scheme 4a). Using the MW-assisted three-component condensation method, this compound (2g) can be synthesized without any catalyst and solvent in a short time (1 h), and in a yield of 85% (Table 2, entry 7).
![[1860-5397-12-146-i6]](/bjoc/content/inline/1860-5397-12-146-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of (dialkylaminomethylene)bisphosphine oxides.
Scheme 6: Synthesis of (dialkylaminomethylene)bisphosphine oxides.
Table 2: Synthesis of (dialkylaminomethylene)bisphosphine oxides 2a–g.
| Entry | Y1 | Y2 | Yield (%)a |
|---|---|---|---|
| 1 | Et | Et | 82 (2a) |
| 2 | Bu | Bu | 73 (2b) |
| 3 | Bu | Me | 69 (2c) |
| 4 | c-Hex | Me | 66 (2d) |
| 5 | Bn | Me | 64 (2e) |
| 6 | Ph | Me | 60 (2f) |
| 7 | -(CH2)2-O-(CH2)2- | 85 (2g) | |
aIsolated yield.
Synthesis of alkylamino- and (phenylaminomethylene)bisphosphonates
In the next stage, our method was extended to the synthesis of alkyl- and (phenylaminomethylene)bisphosphonates by reacting butyl- and cyclohexylamine or aniline, and triethyl orthoformate with diethyl phosphite under MW irradiation in the absence of catalyst and solvent (Table 3). First, the condensation of butylamine, triethyl orthoformate with 2 equivalents of diethyl phosphite was studied at 125 °C. After a 2 h’s reaction time, the conversion was 91%, and beside the expected (aminomethylene)bisphosphonate 3a formed in 81%, the N-ethylated by-product 4a was formed in 19% (Table 3, entry 1). Increasing the temperature to 150 °C, the reaction was completed after 30 min, but the proportion of the main product 3a was somewhat lower (78%), and another by-product 5a also appeared in 7% (Table 3, entry 2). The target compound 3a could be obtained in a yield of 61%. Using 3.5 equivalents of diethyl phosphite at 125 °C for 1 h, the conversion was only 75%, but the expected product 3a was formed exclusively (Table 3, entry 3). After a longer reaction time of 1.5 h, by-product 4a also appeared in 22% (Table 3, entry 4). In the reaction with cyclohexylamine, the same tendency was observed (Table 3, entries 5–8), and the corresponding (cyclohexylaminomethylene)bisphosphonate 3b was obtained in a yield of 68% after column chromatography (Table 3, entry 6). Finally, the three-component condensation of aniline, triethyl orthoformate and diethyl phosphite was studied (Table 3, entries 9–11). Applying 2 equivalents of phosphite, the reaction was not complete, neither at 125 °C, nor at 150 °C (Table 3, entries 9 and 10). Two types of imine intermediates (6a and 6b) could be observed in the reaction mixture beside the expected product 3c. These intermediates refer to the mechanism of the condensation (see compounds I and V in Scheme 1, pathway A). Previously, iminephosphonate 6b was only an assumed intermediate [7], but now we could prove it by 31P NMR and HRMS (Table 4). Increasing the amount of diethyl phosphite to 3 equivalents, the reaction was complete at 125 °C after 1 h, and only 3c was formed with a yield of 82% (Table 3, entry 11). In the cases discussed, no ethylated or formylated by-products (4 and 5, respectively) were formed. (Aminomethylene)bisphosphonates 3b and 3c were synthesized earlier in unoptimized experiments to provide compounds 3b and 3c in yields of 36% [9] and 53% [8], respectively. The former compound 3b was characterized only by 1H NMR [9]. Compound 3c was also synthesized under MW irradiation in a yield of 75% [31]. It can be seen, that the refined MW-assisted method elaborated by us may give the (aminomethylene)bisphosphonates 3b and 3c in yields of 68% and 82%, respectively.
Table 3: The reactions of primary amines with triethyl orthoformate and diethyl phosphite.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-146-i9.svg?max-width=637&scale=1.0)
|
|||||||||
| Entry | Y | DEP (equiv) | T (°C) | t (h) | Conversion (%)a | Product composition (%)a |
Yield of 3
(%)c |
||
|---|---|---|---|---|---|---|---|---|---|
| 3 | by-productsb | ||||||||
| 4 | 5 | ||||||||
| 1 | Bu | 2 | 125 | 2 | 91 | 81 | 19 | 0 | – |
| 2 | Bu | 2 | 150 | 0.5 | 100 | 78 | 15 | 7 | 61 (3a) |
| 3 | Bu | 3.5 | 125 | 1 | 75 | 100 | 0 | 0 | – |
| 4 | Bu | 3.5 | 125 | 1.5 | 90 | 78 | 22 | 0 | – |
| 5 | c-Hex | 2 | 125 | 2 | 76 | 83 | 17 | 0 | – |
| 6 | c-Hex | 2 | 150 | 0.5 | 100 | 88 | 10 | 2 | 68 (3b) |
| 7 | c-Hex | 3.5 | 125 | 1 | 63 | 100 | 0 | 0 | – |
| 8 | c-Hex | 3.5 | 125 | 1.5 | 83 | 86 | 14 | 0 | – |
| 9 | Ph | 2 | 125 | 2 | 68 | 56d | 0 | 0 | 36 (3c) |
| 10 | Ph | 2 | 150 | 1 | 90 | 70d | 0 | 0 | 52 (3c) |
| 11 | Ph | 3 | 125 | 1 | 100 | 100 | 0 | 0 | 82 (3c) |
aOn the basis of GC (entries 1–8) or on the basis of HPLC (entries 9–11). bThe by-products identified:
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-146-i10.svg?max-width=637&scale=1.18182)
cIsolated yield. dThe following intermediates were also formed based on LC–MS:
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-146-i11.svg?max-width=637&scale=1.18182)
Table 4: Spectral characterization of N-ethyl- (4) and N-formyl- (5) (aminomethylene)bisphosphonates and imine-type intermediates 6a and 6b.
| Compounds | δP in CDCl3 | δP [lit.] | [M + H]+found | [M + H]+requires |
|---|---|---|---|---|
| 4a | 19.98 | – | 388.2020 | 388.2012 |
| 4b | 20.63 | – | 414.2162 | 414.2169 |
| 5a |
16.08 and 16.16
(E and Z isomers) |
15.69 and 15.98a
(E and Z isomers) [43] |
388.1659 | 388.1649 |
| 5b |
16.00 and 16.06
(E and Z isomers) |
– | 414.1797 | 414.1805 |
| 6a | – | – | 197.1075 | 197.1073 |
| 6b | 17.67 | – | 242.0936 | 242.0941 |
aIn CCl4.
Next, the condensation of aniline with trimethyl orthoformate and dimethyl phosphite was also performed (Scheme 7). In this case, the reaction was complete after a 1 h heating at 110 °C using 3.5 equivalents of dimethyl phosphite. After column chromatography, the corresponding product 7a was isolated in a yield of 63%. At higher temperatures, decomposition was observed.
![[1860-5397-12-146-i7]](/bjoc/content/inline/1860-5397-12-146-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of tetramethyl (phenylaminomethylene)bisphosphonate.
Scheme 7: Synthesis of tetramethyl (phenylaminomethylene)bisphosphonate.
In the next stage, the MW-assisted reaction of secondary amines was studied with triethyl orthoformate and diethyl phosphite (Scheme 8). The condensations were carried out applying 3.5 equivalents of diethyl phosphite at 125 °C for 1 h in the absence of a catalyst and a solvent. In case of N-methylaniline, 4.5 equivalents of the P-reagent was necessary to attain complete conversion (Table 5, entry 6). The corresponding (dialkylaminomethylene)bisphosphonates (8a–g) were obtained in yields of 65–86% after purification by chromatography (Table 5, entries 1–7).
![[1860-5397-12-146-i8]](/bjoc/content/inline/1860-5397-12-146-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of (dialkylaminomethylene)bisphosphonates.
Scheme 8: Synthesis of (dialkylaminomethylene)bisphosphonates.
Table 5: Synthesis of (dialkylaminomethylene)bisphosphonates 8a–g.
| Entry | Y1 | Y2 | Yield (%)a |
|---|---|---|---|
| 1 | Et | Et | 86 (8a) |
| 2 | Bu | Bu | 68 (8b) |
| 3 | Bu | Me | 79 (8c) |
| 4 | c-Hex | Me | 72 (8d) |
| 5 | Bn | Me | 70 (8e)b |
| 6c | Ph | Me | 65 (8f) |
| 7 | -(CH2)2-O-(CH2)2- | 81 (8g)d | |
aIsolated yield. bIt was synthesized in a yield of 61% [10]. c4.5 equivalents of diethyl phosphonate was used.dIt was synthesized in a yield of 46% [4].
Finally, the three-component condensation of aniline, triethyl orthoformate and dimethyl or dibutyl phosphite was studied (Table 6, Figure 1). Using dimethyl phosphite, the reactions were performed at 110 °C for 1 h, but in case of dibutyl phosphite, the conditions applied were the same as those in the condensations with diethyl phosphite (125–150 °C, 1 h). Using 2 equivalents of dialkyl phosphite, more or less transesterified (aminomethylene)bisphosphonates (9–11 and 3c) were also formed beside the expected (phenylaminomethylene)bisphosphonates 7a or 7b (Table 6,entries 1 and 6, 7). The transesterified by-products (9-11 and 3c) were indentified by GC–MS (Figure 2) or LC–MS, and were proved by HRMS (Table 7). The composition of the reaction mixture for the experiment marked by Table 6, entry 6 was analyzed by 31P NMR (see Figure 3). It was observed that increasing the quantity of dialkyl phosphite, the proportion of the by-products was decreased, and the condensations became more selective for the desired product (7a or 7b) (Table 6, Figure 1). In the reaction with dimethyl phosphite, the best result was achieved using 20 equivalents of the P-reagent, but in case of dibutyl phosphite, a 15-fold excess was sufficient (Table 6, entries 5 and 10).
Table 6: Condensation of aniline, triethyl orthoformate and dimethyl or dibutyl phosphite.
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-146-i12.svg?max-width=637&scale=1.0)
|
||||||||
| Entry | R | Dialkyl phosphite (equiv) | T (°C) | Product composition (%)a | ||||
|---|---|---|---|---|---|---|---|---|
| 7 | 9 | 10 | 11 | 3c | ||||
| 1 | Me | 2 | 110 | 14 | 36 | 36 | 11 | 3 |
| 2 | Me | 6 | 110 | 54 | 37 | 9 | 0 | 0 |
| 3 | Me | 10 | 110 | 77 | 18 | 5 | 0 | 0 |
| 4 | Me | 15 | 110 | 87 | 13 | 0 | 0 | 0 |
| 5 | Me | 20b | 110 | 91 | 9 | 0 | 0 | 0 |
| 6 | Bu | 2 | 150 | 19 | 23 | 29 | 26 | 3 |
| 7 | Bu | 2 | 125 | 54 | 33 | 10 | 3 | 0 |
| 8 | Bu | 6 | 125 | 82 | 16 | 2 | 0 | 0 |
| 9 | Bu | 10 | 125 | 93 | 7 | 0 | 0 | 0 |
| 10 | Bu | 15b | 125 | 95 | 5 | 0 | 0 | 0 |
aOn the basis of GC (entries 1–5) or on the basis of HPLC (entries 6–9). bThe product composition has not changed using a larger excess of dialkyl phosphite.
![[1860-5397-12-146-1]](/bjoc/content/figures/1860-5397-12-146-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Effect of the quantity of dimethyl phosphite (DMP) on the product composition (from Table 6, entries 1–5.)
Figure 1: Effect of the quantity of dimethyl phosphite (DMP) on the product composition (from Table 6, entries 1–5.)
![[1860-5397-12-146-2]](/bjoc/content/figures/1860-5397-12-146-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: GC–MS chromatogram for the condensation of aniline, triethyl orthoformate and 2 equivalents of dimethyl phosphite (from the exp. marked by Table 6, entry 1).
Figure 2: GC–MS chromatogram for the condensation of aniline, triethyl orthoformate and 2 equivalents of dime...
Table 7: Mass spectral characterization of (aminomethylene)bisphosphonates.
| R = Me (b) | R = Bu (c) | |||
|---|---|---|---|---|
| [M + H]+found | [M + H]+requires | [M + H]+found | [M + H]+requires | |
| 7 | 324.0760 | 324.0760 | 492.2648 | 492.2638 |
| 9 | 338.0921 | 338.0917 | 464.2329 | 464.2325 |
| 10 | 352.1072 | 352.1073 | 436.2011 | 436.2012 |
| 11 | 366.1231 | 366.1230 | 408.1711 | 408.1699 |
| 3c | 380.1382 | 380.1386 | 380.1382 | 380.1386 |
![[1860-5397-12-146-3]](/bjoc/content/figures/1860-5397-12-146-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: 31P NMR spectrum for the condensation of aniline, triethyl orthoformate and 2 equivalents of dibutyl phosphite (from the exp. marked by Table 6, entry 6).
Figure 3: 31P NMR spectrum for the condensation of aniline, triethyl orthoformate and 2 equivalents of dibuty...
Conclusion
In summary, we have developed a facile, solvent- and catalyst-free MW-assisted method for the synthesis of (aminomethylene)bisphosphine oxides (AMBPOs) and (aminomethylene)bisphosphonates by the condensation of a primary or secondary amine, an orthoformate, and diphenylphosphine oxide or a dialkyl phosphite. This method is a novel approach for the preparation of AMBPOs and an optimized process for the synthesis of (aminomethylene)bisphosphonates. Twenty-two derivatives were isolated and characterized, except two, all of them are new compounds. Furthermore, a few intermediates supporting the mechanism of the condensation, and several by-products were also identified.
Supporting Information
Experimental procedures, characterization data, details of the NMR structural determination of all products and copies of 31P, 1H, and 13C NMR spectra for all compounds synthesized are presented in Supporting Information File 1.
| Supporting Information File 1: Experimental, NMR spectra. | ||
| Format: PDF | Size: 3.6 MB | Download |
References
-
Breuer, E. The Development of Bisphosphonates as Drugs. In Analogue-based Drug Discovery; Fischer, J.; Ganelli, C. R., Eds.; Wiley-VCH: Weinheim, 2006; pp 371–384.
Return to citation in text: [1] -
Russell, R. G. G. Pediatrics 2007, 119 (Suppl. 2), S150–S162. doi:10.1542/peds.2006-2023H
Return to citation in text: [1] -
Russell, R. G. G. Bone 2011, 49, 2–19. doi:10.1016/j.bone.2011.04.022
Return to citation in text: [1] -
Hudson, H. R.; Wardle, N. J.; Bligh, S. W. A.; Greiner, I.; Grün, A.; Keglevich, G. Mini-Rev. Med. Chem. 2012, 12, 313–325. doi:10.2174/138955712799829285
Return to citation in text: [1] [2] -
Romanenko, V. D.; Kukhar, V. P. ARKIVOC 2012, No. iv, 127–166. doi:10.3998/ark.5550190.0013.411
Return to citation in text: [1] [2] -
Krutikov, V. I.; Erkin, A. V.; Pautov, P. A.; Zolotukhina, M. M. Russ. J. Gen. Chem. 2003, 73, 187–191. doi:10.1023/A:1024775501781
Return to citation in text: [1] [2] [3] -
Dąbrowska, E.; Burzyńska, A.; Mucha, A.; Matczak-Jon, E.; Sawka-Dobrowolska, W.; Berlicki, Ł.; Kafarski, P. J. Organomet. Chem. 2009, 694, 3806–3813. doi:10.1016/j.jorganchem.2009.07.025
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Tauro, M.; Laghezza, A.; Loiodice, F.; Agamennone, M.; Campestre, C.; Tortorella, P. Bioorg. Med. Chem. 2013, 21, 6456–6465. doi:10.1016/j.bmc.2013.08.054
Return to citation in text: [1] [2] [3] -
Takeuchi, M.; Sakamoto, S.; Yoshida, M.; Abe, T.; Isomura, Y. Chem. Pharm. Bull. 1993, 41, 688–693. doi:10.1248/cpb.41.688
Return to citation in text: [1] [2] [3] [4] -
Ekimoto, H. Metal Complex Compound, Cancer Therapeutic Composition Comprising the Metal Complex Compound as Active Ingredient, and Intermediate for Production of the Metal Complex Compound. Eur. Pat. EP2177525 A1, April 21, 2010.
Return to citation in text: [1] [2] [3] -
Rose, Y. S.; Ciblat, S.; Kang, T.; Far, A. R.; Dietrich, E.; Lafontaine, Y.; Reddy, R. Phosphonated Rifamycins and Uses Thereof or the Prevention and Treatment of Bone and Joint Infections. U.S. Patent US20110263534 A1, Oct 27, 2011.
Return to citation in text: [1] [2] -
Zhang, Q. M.; Serpe, M. J. Macromolecules 2014, 47, 8018–8025. doi:10.1021/ma501997x
Return to citation in text: [1] [2] -
Kantoci, D.; Denike, J. K.; Wechter, W. J. Synth. Commun. 1996, 26, 2037–2043. doi:10.1080/00397919608003560
Return to citation in text: [1] [2] -
Kubíček, V.; Rudovský, J.; Kotek, J.; Hermann, P.; Vander Elst, L.; Muller, R. N.; Kolar, Z. I.; Wolterbeek, H. T.; Peters, J. A.; Lukeš, I. J. Am. Chem. Soc. 2005, 127, 16477–16485. doi:10.1021/ja054905u
Return to citation in text: [1] [2] -
Forlani, G.; Occhipinti, A.; Berlicki, Ł.; Dziedzioła, G.; Wieczorek, A.; Kafarski, P. J. Agric. Food Chem. 2008, 56, 3193–3199. doi:10.1021/jf800029t
Return to citation in text: [1] [2] -
Martin, M. B.; Grimley, J. S.; Lewis, J. C.; Heath, H. T., III; Bailey, B. N.; Kendrick, H.; Yardley, V.; Caldera, A.; Lira, R.; Urbina, J. A.; Moreno, S. N. J.; Docampo, R.; Croft, S. L.; Oldfield, E. J. Med. Chem. 2001, 44, 909–916. doi:10.1021/jm0002578
Return to citation in text: [1] [2] -
Widler, L.; Jaeggi, K. A.; Glatt, M.; Müller, K.; Bachmann, R.; Bisping, M.; Born, A.-R.; Cortesi, R.; Guiglia, G.; Jeker, H.; Klein, R.; Ramseier, U.; Schmid, J.; Schreiber, G.; Seltenmeyer, Y.; Green, J. R. J. Med. Chem. 2002, 45, 3721–3738. doi:10.1021/jm020819i
Return to citation in text: [1] [2] -
Forlani, G.; Berlicki, Ł.; Duò, M.; Dziędzioła, G.; Giberti, S.; Bertazzini, M.; Kafarski, P. J. Agric. Food Chem. 2013, 61, 6792–6798. doi:10.1021/jf401234s
Return to citation in text: [1] [2] -
Kotsikorou, E.; Song, Y.; Chan, J. M. W.; Faelens, S.; Tovian, Z.; Broderick, E.; Bakalara, N.; Docampo, R.; Oldfield, E. J. Med. Chem. 2005, 48, 6128–6139. doi:10.1021/jm058220g
Return to citation in text: [1] [2] -
Parniak, M.; Mellors, J. W.; Oldfield, E.; Tovian, Z.; Chan, J. M. W. Composition and Methods for Use of Antiviral Drugs in the Treatment of Retroviral Diseases Resistant to Nucleoside Reverse Transcriptase Inhibitors. U.S. Patent Appl. US20050113331 A1, May 26, 2005.
Return to citation in text: [1] [2] -
Chmielewska, E.; Mazur, Z.; Kempińska, K.; Wietrzyk, J.; Piątek, A.; Kuryszko, J. J.; Kiełbowicz, Z.; Kafarski, P. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 2164–2172. doi:10.1080/10426507.2015.1085046
Return to citation in text: [1] [2] [3] -
Fallouh, F.; Bernier, D.; Virieux, D.; Cristau, H. J.; Pirat, J. L. Phosphorus, Sulfur Silicon Relat. Elem. 2006, 181, 219–225. doi:10.1080/104265090969766
Return to citation in text: [1] [2] -
Leung, C.-Y.; Langille, A. M.; Mancuso, J.; Tsantrizos, Y. S. Bioorg. Med. Chem. 2013, 21, 2229–2240. doi:10.1016/j.bmc.2013.02.006
Return to citation in text: [1] [2] -
Reddy, M. V.; Kalla, R. M. N.; Dong, L. S.; Jeong, Y. T. Catal. Commun. 2015, 61, 102–106. doi:10.1016/j.catcom.2014.12.021
Return to citation in text: [1] -
Kunda, U. M. R.; Balam, S. K.; Nemallapudi, B. R.; Chereddy, S. S.; Nayak, S. K.; Cirandur, S. R. Chem. Pharm. Bull. 2012, 60, 104–109. doi:10.1248/cpb.60.104
Return to citation in text: [1] -
Reddy, M. V. N.; Kim, J.; Jeong, Y. T. J. Fluorine Chem. 2012, 135, 155–158. doi:10.1016/j.jfluchem.2011.10.005
Return to citation in text: [1] -
Prasad, S. S.; Jayaprakash, S. H.; Syamasundar, C.; Sreelakshmi, P.; Bhuvaneswar, C.; Bhaskar, B. V.; Rajendra, W.; Nayak, S. K.; Reddy, C. S. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 2040–2050. doi:10.1080/10426507.2015.1054928
Return to citation in text: [1] -
Minaeva, L. I.; Patrikeeva, L. S.; Kabachnik, M. M.; Beletskaya, I. P.; Orlinson, B. S.; Novakov, I. A. Heteroat. Chem. 2011, 22, 55–58. doi:10.1002/hc.20656
Return to citation in text: [1] [2] -
Minaeva, L. I.; Kabachnik, M. M.; Ponomarev, G. V.; Morozova, J. V.; Beletskaya, I. P. Synthesis 2010, 2451–2455. doi:10.1055/s-0029-1218781
Return to citation in text: [1] [2] -
Reddy, G. C. S.; Reddy, M. V. N.; Reddy, N. B.; Reddy, C. S. Phosphorus, Sulfur Silicon Relat. Elem. 2010, 186, 74–80. doi:10.1080/10426507.2010.482542
Return to citation in text: [1] [2] -
Kaboudin, B.; Alipour, S. Tetrahedron Lett. 2009, 50, 4243–4245. doi:10.1016/j.tetlet.2009.05.016
Return to citation in text: [1] [2] -
Lacbay, C. M.; Mancuso, J.; Lin, Y.-S.; Bennett, N.; Götte, M.; Tsantrizos, Y. S. J. Med. Chem. 2014, 57, 7435–7449. doi:10.1021/jm501010f
Return to citation in text: [1] -
Prishchenko, A. A.; Livantsov, M. V.; Novikova, O. P.; Livantsova, L. I.; Erschov, I. S.; Petrosyan, V. S. Heteroat. Chem. 2015, 26, 101–105. doi:10.1002/hc.21220
Return to citation in text: [1] [2] [3] -
Wang, A.-E.; Chang, Z.; Sun, W.-T.; Huang, P.-Q. Org. Lett. 2015, 17, 732–735. doi:10.1021/acs.orglett.5b00004
Return to citation in text: [1] -
Goldeman, W.; Kluczyński, A.; Soroka, M. Tetrahedron Lett. 2012, 53, 5290–5292. doi:10.1016/j.tetlet.2012.07.085
Return to citation in text: [1] -
Pudovik, A. N.; Nikitina, V. I.; Zimin, M. G.; Vostretsova, N. L. J. Gen. Chem. USSR 1975, 45, 1450–1455.
Return to citation in text: [1] [2] -
Olive, G.; Jacques, A. Phosphorus, Sulfur Silicon Relat. Elem. 2003, 178, 33–46. doi:10.1080/10426500307821
Return to citation in text: [1] -
Lecerclé, D.; Gabillet, S.; Gomis, J.-M.; Taran, F. Tetrahedron Lett. 2008, 49, 2083–2087. doi:10.1016/j.tetlet.2008.01.127
Return to citation in text: [1] -
Gross, H.; Costisella, B. J. Prakt. Chem. 1969, 311, 925–929. doi:10.1002/prac.19693110610
Return to citation in text: [1] [2] -
Morgalyuk, V. P.; Strelkova, T. V.; Nifant'ev, E. E. Bull. Chem. Soc. Jpn. 2012, 85, 93–100. doi:10.1246/bcsj.20110157
Return to citation in text: [1] [2] -
Morgalyuk, V. P.; Strelkova, T. V.; Nifant'ev, E. E. Russ. Chem. Bull. 2012, 61, 380–385. doi:10.1007/s11172-012-0053-2
Return to citation in text: [1] [2] [3] -
Hirai, T.; Han, L.-B. J. Am. Chem. Soc. 2006, 128, 7422–7423. doi:10.1021/ja060984d
Return to citation in text: [1] [2] -
Costisella, B.; Gross, H. J. Prakt. Chem. 1979, 321, 361–369. doi:10.1002/prac.19793210303
Return to citation in text: [1]
| 9. | Takeuchi, M.; Sakamoto, S.; Yoshida, M.; Abe, T.; Isomura, Y. Chem. Pharm. Bull. 1993, 41, 688–693. doi:10.1248/cpb.41.688 |
| 31. | Kaboudin, B.; Alipour, S. Tetrahedron Lett. 2009, 50, 4243–4245. doi:10.1016/j.tetlet.2009.05.016 |
| 43. | Costisella, B.; Gross, H. J. Prakt. Chem. 1979, 321, 361–369. doi:10.1002/prac.19793210303 |
| 1. | Breuer, E. The Development of Bisphosphonates as Drugs. In Analogue-based Drug Discovery; Fischer, J.; Ganelli, C. R., Eds.; Wiley-VCH: Weinheim, 2006; pp 371–384. |
| 2. | Russell, R. G. G. Pediatrics 2007, 119 (Suppl. 2), S150–S162. doi:10.1542/peds.2006-2023H |
| 3. | Russell, R. G. G. Bone 2011, 49, 2–19. doi:10.1016/j.bone.2011.04.022 |
| 6. | Krutikov, V. I.; Erkin, A. V.; Pautov, P. A.; Zolotukhina, M. M. Russ. J. Gen. Chem. 2003, 73, 187–191. doi:10.1023/A:1024775501781 |
| 7. | Dąbrowska, E.; Burzyńska, A.; Mucha, A.; Matczak-Jon, E.; Sawka-Dobrowolska, W.; Berlicki, Ł.; Kafarski, P. J. Organomet. Chem. 2009, 694, 3806–3813. doi:10.1016/j.jorganchem.2009.07.025 |
| 8. | Tauro, M.; Laghezza, A.; Loiodice, F.; Agamennone, M.; Campestre, C.; Tortorella, P. Bioorg. Med. Chem. 2013, 21, 6456–6465. doi:10.1016/j.bmc.2013.08.054 |
| 9. | Takeuchi, M.; Sakamoto, S.; Yoshida, M.; Abe, T.; Isomura, Y. Chem. Pharm. Bull. 1993, 41, 688–693. doi:10.1248/cpb.41.688 |
| 10. | Ekimoto, H. Metal Complex Compound, Cancer Therapeutic Composition Comprising the Metal Complex Compound as Active Ingredient, and Intermediate for Production of the Metal Complex Compound. Eur. Pat. EP2177525 A1, April 21, 2010. |
| 11. | Rose, Y. S.; Ciblat, S.; Kang, T.; Far, A. R.; Dietrich, E.; Lafontaine, Y.; Reddy, R. Phosphonated Rifamycins and Uses Thereof or the Prevention and Treatment of Bone and Joint Infections. U.S. Patent US20110263534 A1, Oct 27, 2011. |
| 12. | Zhang, Q. M.; Serpe, M. J. Macromolecules 2014, 47, 8018–8025. doi:10.1021/ma501997x |
| 13. | Kantoci, D.; Denike, J. K.; Wechter, W. J. Synth. Commun. 1996, 26, 2037–2043. doi:10.1080/00397919608003560 |
| 14. | Kubíček, V.; Rudovský, J.; Kotek, J.; Hermann, P.; Vander Elst, L.; Muller, R. N.; Kolar, Z. I.; Wolterbeek, H. T.; Peters, J. A.; Lukeš, I. J. Am. Chem. Soc. 2005, 127, 16477–16485. doi:10.1021/ja054905u |
| 15. | Forlani, G.; Occhipinti, A.; Berlicki, Ł.; Dziedzioła, G.; Wieczorek, A.; Kafarski, P. J. Agric. Food Chem. 2008, 56, 3193–3199. doi:10.1021/jf800029t |
| 16. | Martin, M. B.; Grimley, J. S.; Lewis, J. C.; Heath, H. T., III; Bailey, B. N.; Kendrick, H.; Yardley, V.; Caldera, A.; Lira, R.; Urbina, J. A.; Moreno, S. N. J.; Docampo, R.; Croft, S. L.; Oldfield, E. J. Med. Chem. 2001, 44, 909–916. doi:10.1021/jm0002578 |
| 17. | Widler, L.; Jaeggi, K. A.; Glatt, M.; Müller, K.; Bachmann, R.; Bisping, M.; Born, A.-R.; Cortesi, R.; Guiglia, G.; Jeker, H.; Klein, R.; Ramseier, U.; Schmid, J.; Schreiber, G.; Seltenmeyer, Y.; Green, J. R. J. Med. Chem. 2002, 45, 3721–3738. doi:10.1021/jm020819i |
| 18. | Forlani, G.; Berlicki, Ł.; Duò, M.; Dziędzioła, G.; Giberti, S.; Bertazzini, M.; Kafarski, P. J. Agric. Food Chem. 2013, 61, 6792–6798. doi:10.1021/jf401234s |
| 19. | Kotsikorou, E.; Song, Y.; Chan, J. M. W.; Faelens, S.; Tovian, Z.; Broderick, E.; Bakalara, N.; Docampo, R.; Oldfield, E. J. Med. Chem. 2005, 48, 6128–6139. doi:10.1021/jm058220g |
| 20. | Parniak, M.; Mellors, J. W.; Oldfield, E.; Tovian, Z.; Chan, J. M. W. Composition and Methods for Use of Antiviral Drugs in the Treatment of Retroviral Diseases Resistant to Nucleoside Reverse Transcriptase Inhibitors. U.S. Patent Appl. US20050113331 A1, May 26, 2005. |
| 21. | Chmielewska, E.; Mazur, Z.; Kempińska, K.; Wietrzyk, J.; Piątek, A.; Kuryszko, J. J.; Kiełbowicz, Z.; Kafarski, P. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 2164–2172. doi:10.1080/10426507.2015.1085046 |
| 33. | Prishchenko, A. A.; Livantsov, M. V.; Novikova, O. P.; Livantsova, L. I.; Erschov, I. S.; Petrosyan, V. S. Heteroat. Chem. 2015, 26, 101–105. doi:10.1002/hc.21220 |
| 5. | Romanenko, V. D.; Kukhar, V. P. ARKIVOC 2012, No. iv, 127–166. doi:10.3998/ark.5550190.0013.411 |
| 34. | Wang, A.-E.; Chang, Z.; Sun, W.-T.; Huang, P.-Q. Org. Lett. 2015, 17, 732–735. doi:10.1021/acs.orglett.5b00004 |
| 5. | Romanenko, V. D.; Kukhar, V. P. ARKIVOC 2012, No. iv, 127–166. doi:10.3998/ark.5550190.0013.411 |
| 7. | Dąbrowska, E.; Burzyńska, A.; Mucha, A.; Matczak-Jon, E.; Sawka-Dobrowolska, W.; Berlicki, Ł.; Kafarski, P. J. Organomet. Chem. 2009, 694, 3806–3813. doi:10.1016/j.jorganchem.2009.07.025 |
| 4. | Hudson, H. R.; Wardle, N. J.; Bligh, S. W. A.; Greiner, I.; Grün, A.; Keglevich, G. Mini-Rev. Med. Chem. 2012, 12, 313–325. doi:10.2174/138955712799829285 |
| 7. | Dąbrowska, E.; Burzyńska, A.; Mucha, A.; Matczak-Jon, E.; Sawka-Dobrowolska, W.; Berlicki, Ł.; Kafarski, P. J. Organomet. Chem. 2009, 694, 3806–3813. doi:10.1016/j.jorganchem.2009.07.025 |
| 24. | Reddy, M. V.; Kalla, R. M. N.; Dong, L. S.; Jeong, Y. T. Catal. Commun. 2015, 61, 102–106. doi:10.1016/j.catcom.2014.12.021 |
| 25. | Kunda, U. M. R.; Balam, S. K.; Nemallapudi, B. R.; Chereddy, S. S.; Nayak, S. K.; Cirandur, S. R. Chem. Pharm. Bull. 2012, 60, 104–109. doi:10.1248/cpb.60.104 |
| 26. | Reddy, M. V. N.; Kim, J.; Jeong, Y. T. J. Fluorine Chem. 2012, 135, 155–158. doi:10.1016/j.jfluchem.2011.10.005 |
| 27. | Prasad, S. S.; Jayaprakash, S. H.; Syamasundar, C.; Sreelakshmi, P.; Bhuvaneswar, C.; Bhaskar, B. V.; Rajendra, W.; Nayak, S. K.; Reddy, C. S. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 2040–2050. doi:10.1080/10426507.2015.1054928 |
| 28. | Minaeva, L. I.; Patrikeeva, L. S.; Kabachnik, M. M.; Beletskaya, I. P.; Orlinson, B. S.; Novakov, I. A. Heteroat. Chem. 2011, 22, 55–58. doi:10.1002/hc.20656 |
| 29. | Minaeva, L. I.; Kabachnik, M. M.; Ponomarev, G. V.; Morozova, J. V.; Beletskaya, I. P. Synthesis 2010, 2451–2455. doi:10.1055/s-0029-1218781 |
| 30. | Reddy, G. C. S.; Reddy, M. V. N.; Reddy, N. B.; Reddy, C. S. Phosphorus, Sulfur Silicon Relat. Elem. 2010, 186, 74–80. doi:10.1080/10426507.2010.482542 |
| 6. | Krutikov, V. I.; Erkin, A. V.; Pautov, P. A.; Zolotukhina, M. M. Russ. J. Gen. Chem. 2003, 73, 187–191. doi:10.1023/A:1024775501781 |
| 7. | Dąbrowska, E.; Burzyńska, A.; Mucha, A.; Matczak-Jon, E.; Sawka-Dobrowolska, W.; Berlicki, Ł.; Kafarski, P. J. Organomet. Chem. 2009, 694, 3806–3813. doi:10.1016/j.jorganchem.2009.07.025 |
| 8. | Tauro, M.; Laghezza, A.; Loiodice, F.; Agamennone, M.; Campestre, C.; Tortorella, P. Bioorg. Med. Chem. 2013, 21, 6456–6465. doi:10.1016/j.bmc.2013.08.054 |
| 9. | Takeuchi, M.; Sakamoto, S.; Yoshida, M.; Abe, T.; Isomura, Y. Chem. Pharm. Bull. 1993, 41, 688–693. doi:10.1248/cpb.41.688 |
| 10. | Ekimoto, H. Metal Complex Compound, Cancer Therapeutic Composition Comprising the Metal Complex Compound as Active Ingredient, and Intermediate for Production of the Metal Complex Compound. Eur. Pat. EP2177525 A1, April 21, 2010. |
| 11. | Rose, Y. S.; Ciblat, S.; Kang, T.; Far, A. R.; Dietrich, E.; Lafontaine, Y.; Reddy, R. Phosphonated Rifamycins and Uses Thereof or the Prevention and Treatment of Bone and Joint Infections. U.S. Patent US20110263534 A1, Oct 27, 2011. |
| 12. | Zhang, Q. M.; Serpe, M. J. Macromolecules 2014, 47, 8018–8025. doi:10.1021/ma501997x |
| 13. | Kantoci, D.; Denike, J. K.; Wechter, W. J. Synth. Commun. 1996, 26, 2037–2043. doi:10.1080/00397919608003560 |
| 14. | Kubíček, V.; Rudovský, J.; Kotek, J.; Hermann, P.; Vander Elst, L.; Muller, R. N.; Kolar, Z. I.; Wolterbeek, H. T.; Peters, J. A.; Lukeš, I. J. Am. Chem. Soc. 2005, 127, 16477–16485. doi:10.1021/ja054905u |
| 21. | Chmielewska, E.; Mazur, Z.; Kempińska, K.; Wietrzyk, J.; Piątek, A.; Kuryszko, J. J.; Kiełbowicz, Z.; Kafarski, P. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 2164–2172. doi:10.1080/10426507.2015.1085046 |
| 22. | Fallouh, F.; Bernier, D.; Virieux, D.; Cristau, H. J.; Pirat, J. L. Phosphorus, Sulfur Silicon Relat. Elem. 2006, 181, 219–225. doi:10.1080/104265090969766 |
| 23. | Leung, C.-Y.; Langille, A. M.; Mancuso, J.; Tsantrizos, Y. S. Bioorg. Med. Chem. 2013, 21, 2229–2240. doi:10.1016/j.bmc.2013.02.006 |
| 6. | Krutikov, V. I.; Erkin, A. V.; Pautov, P. A.; Zolotukhina, M. M. Russ. J. Gen. Chem. 2003, 73, 187–191. doi:10.1023/A:1024775501781 |
| 7. | Dąbrowska, E.; Burzyńska, A.; Mucha, A.; Matczak-Jon, E.; Sawka-Dobrowolska, W.; Berlicki, Ł.; Kafarski, P. J. Organomet. Chem. 2009, 694, 3806–3813. doi:10.1016/j.jorganchem.2009.07.025 |
| 22. | Fallouh, F.; Bernier, D.; Virieux, D.; Cristau, H. J.; Pirat, J. L. Phosphorus, Sulfur Silicon Relat. Elem. 2006, 181, 219–225. doi:10.1080/104265090969766 |
| 23. | Leung, C.-Y.; Langille, A. M.; Mancuso, J.; Tsantrizos, Y. S. Bioorg. Med. Chem. 2013, 21, 2229–2240. doi:10.1016/j.bmc.2013.02.006 |
| 10. | Ekimoto, H. Metal Complex Compound, Cancer Therapeutic Composition Comprising the Metal Complex Compound as Active Ingredient, and Intermediate for Production of the Metal Complex Compound. Eur. Pat. EP2177525 A1, April 21, 2010. |
| 15. | Forlani, G.; Occhipinti, A.; Berlicki, Ł.; Dziedzioła, G.; Wieczorek, A.; Kafarski, P. J. Agric. Food Chem. 2008, 56, 3193–3199. doi:10.1021/jf800029t |
| 16. | Martin, M. B.; Grimley, J. S.; Lewis, J. C.; Heath, H. T., III; Bailey, B. N.; Kendrick, H.; Yardley, V.; Caldera, A.; Lira, R.; Urbina, J. A.; Moreno, S. N. J.; Docampo, R.; Croft, S. L.; Oldfield, E. J. Med. Chem. 2001, 44, 909–916. doi:10.1021/jm0002578 |
| 17. | Widler, L.; Jaeggi, K. A.; Glatt, M.; Müller, K.; Bachmann, R.; Bisping, M.; Born, A.-R.; Cortesi, R.; Guiglia, G.; Jeker, H.; Klein, R.; Ramseier, U.; Schmid, J.; Schreiber, G.; Seltenmeyer, Y.; Green, J. R. J. Med. Chem. 2002, 45, 3721–3738. doi:10.1021/jm020819i |
| 18. | Forlani, G.; Berlicki, Ł.; Duò, M.; Dziędzioła, G.; Giberti, S.; Bertazzini, M.; Kafarski, P. J. Agric. Food Chem. 2013, 61, 6792–6798. doi:10.1021/jf401234s |
| 19. | Kotsikorou, E.; Song, Y.; Chan, J. M. W.; Faelens, S.; Tovian, Z.; Broderick, E.; Bakalara, N.; Docampo, R.; Oldfield, E. J. Med. Chem. 2005, 48, 6128–6139. doi:10.1021/jm058220g |
| 20. | Parniak, M.; Mellors, J. W.; Oldfield, E.; Tovian, Z.; Chan, J. M. W. Composition and Methods for Use of Antiviral Drugs in the Treatment of Retroviral Diseases Resistant to Nucleoside Reverse Transcriptase Inhibitors. U.S. Patent Appl. US20050113331 A1, May 26, 2005. |
| 21. | Chmielewska, E.; Mazur, Z.; Kempińska, K.; Wietrzyk, J.; Piątek, A.; Kuryszko, J. J.; Kiełbowicz, Z.; Kafarski, P. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 2164–2172. doi:10.1080/10426507.2015.1085046 |
| 28. | Minaeva, L. I.; Patrikeeva, L. S.; Kabachnik, M. M.; Beletskaya, I. P.; Orlinson, B. S.; Novakov, I. A. Heteroat. Chem. 2011, 22, 55–58. doi:10.1002/hc.20656 |
| 29. | Minaeva, L. I.; Kabachnik, M. M.; Ponomarev, G. V.; Morozova, J. V.; Beletskaya, I. P. Synthesis 2010, 2451–2455. doi:10.1055/s-0029-1218781 |
| 30. | Reddy, G. C. S.; Reddy, M. V. N.; Reddy, N. B.; Reddy, C. S. Phosphorus, Sulfur Silicon Relat. Elem. 2010, 186, 74–80. doi:10.1080/10426507.2010.482542 |
| 31. | Kaboudin, B.; Alipour, S. Tetrahedron Lett. 2009, 50, 4243–4245. doi:10.1016/j.tetlet.2009.05.016 |
| 32. | Lacbay, C. M.; Mancuso, J.; Lin, Y.-S.; Bennett, N.; Götte, M.; Tsantrizos, Y. S. J. Med. Chem. 2014, 57, 7435–7449. doi:10.1021/jm501010f |
| 4. | Hudson, H. R.; Wardle, N. J.; Bligh, S. W. A.; Greiner, I.; Grün, A.; Keglevich, G. Mini-Rev. Med. Chem. 2012, 12, 313–325. doi:10.2174/138955712799829285 |
| 38. | Lecerclé, D.; Gabillet, S.; Gomis, J.-M.; Taran, F. Tetrahedron Lett. 2008, 49, 2083–2087. doi:10.1016/j.tetlet.2008.01.127 |
| 35. | Goldeman, W.; Kluczyński, A.; Soroka, M. Tetrahedron Lett. 2012, 53, 5290–5292. doi:10.1016/j.tetlet.2012.07.085 |
| 36. | Pudovik, A. N.; Nikitina, V. I.; Zimin, M. G.; Vostretsova, N. L. J. Gen. Chem. USSR 1975, 45, 1450–1455. |
| 37. | Olive, G.; Jacques, A. Phosphorus, Sulfur Silicon Relat. Elem. 2003, 178, 33–46. doi:10.1080/10426500307821 |
| 9. | Takeuchi, M.; Sakamoto, S.; Yoshida, M.; Abe, T.; Isomura, Y. Chem. Pharm. Bull. 1993, 41, 688–693. doi:10.1248/cpb.41.688 |
| 8. | Tauro, M.; Laghezza, A.; Loiodice, F.; Agamennone, M.; Campestre, C.; Tortorella, P. Bioorg. Med. Chem. 2013, 21, 6456–6465. doi:10.1016/j.bmc.2013.08.054 |
| 41. | Morgalyuk, V. P.; Strelkova, T. V.; Nifant'ev, E. E. Russ. Chem. Bull. 2012, 61, 380–385. doi:10.1007/s11172-012-0053-2 |
| 7. | Dąbrowska, E.; Burzyńska, A.; Mucha, A.; Matczak-Jon, E.; Sawka-Dobrowolska, W.; Berlicki, Ł.; Kafarski, P. J. Organomet. Chem. 2009, 694, 3806–3813. doi:10.1016/j.jorganchem.2009.07.025 |
| 40. | Morgalyuk, V. P.; Strelkova, T. V.; Nifant'ev, E. E. Bull. Chem. Soc. Jpn. 2012, 85, 93–100. doi:10.1246/bcsj.20110157 |
| 41. | Morgalyuk, V. P.; Strelkova, T. V.; Nifant'ev, E. E. Russ. Chem. Bull. 2012, 61, 380–385. doi:10.1007/s11172-012-0053-2 |
| 36. | Pudovik, A. N.; Nikitina, V. I.; Zimin, M. G.; Vostretsova, N. L. J. Gen. Chem. USSR 1975, 45, 1450–1455. |
| 42. | Hirai, T.; Han, L.-B. J. Am. Chem. Soc. 2006, 128, 7422–7423. doi:10.1021/ja060984d |
| 33. | Prishchenko, A. A.; Livantsov, M. V.; Novikova, O. P.; Livantsova, L. I.; Erschov, I. S.; Petrosyan, V. S. Heteroat. Chem. 2015, 26, 101–105. doi:10.1002/hc.21220 |
| 39. | Gross, H.; Costisella, B. J. Prakt. Chem. 1969, 311, 925–929. doi:10.1002/prac.19693110610 |
| 40. | Morgalyuk, V. P.; Strelkova, T. V.; Nifant'ev, E. E. Bull. Chem. Soc. Jpn. 2012, 85, 93–100. doi:10.1246/bcsj.20110157 |
| 41. | Morgalyuk, V. P.; Strelkova, T. V.; Nifant'ev, E. E. Russ. Chem. Bull. 2012, 61, 380–385. doi:10.1007/s11172-012-0053-2 |
| 42. | Hirai, T.; Han, L.-B. J. Am. Chem. Soc. 2006, 128, 7422–7423. doi:10.1021/ja060984d |
| 33. | Prishchenko, A. A.; Livantsov, M. V.; Novikova, O. P.; Livantsova, L. I.; Erschov, I. S.; Petrosyan, V. S. Heteroat. Chem. 2015, 26, 101–105. doi:10.1002/hc.21220 |
| 39. | Gross, H.; Costisella, B. J. Prakt. Chem. 1969, 311, 925–929. doi:10.1002/prac.19693110610 |
© 2016 Bálint et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)