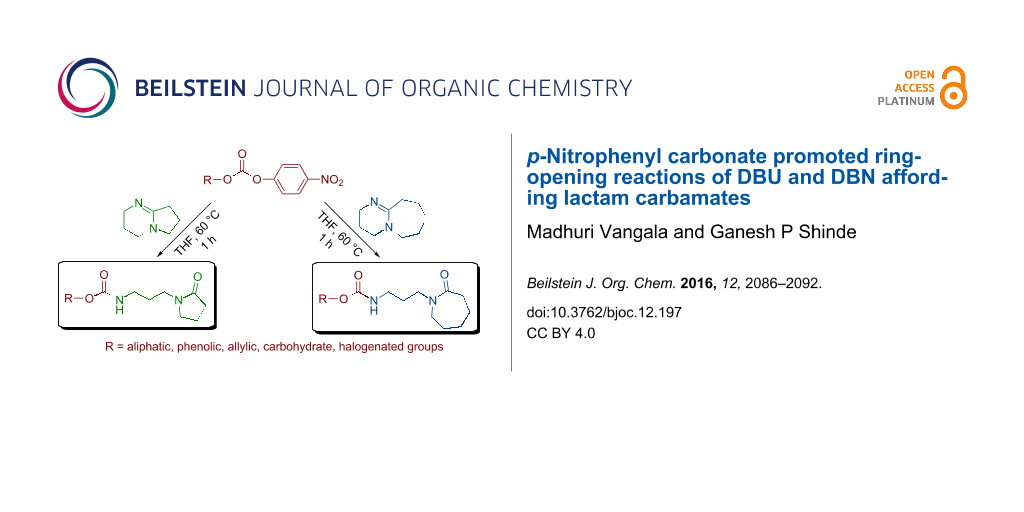
Abstract
The amidine bases DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) and DBN (1,5-diazabicyclo[4.3.0]non-5-ene) display nucleophilic behaviour towards highly electrophilic p-nitrophenyl carbonate derivatives with ring opening of the bicyclic ring to form corresponding substituted ε-caprolactam and γ-lactam derived carbamates. This simple method presents a unified strategy to synthesize structurally diverse ε-caprolactam and γ-lactam compounds with a large substrate scope.

Graphical Abstract
Introduction
Among various organic bases, amidines such as DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) and DBN (1,5-diazabicyclo[4.3.0]non-5-ene) having an imino group attached to the α-carbon of the amine are synthetically useful and strong neutral bases. Besides, DBU and DBN catalyze various organic reactions such as dehydrohalogenations [1], carbonylations [2], amidations [3] and Baylis–Hillman reactions [4]. These bicyclic amidines have been thought to be non-nucleophilic bases, but meanwhile numerous examples unveiled their ability to act as C and N nucleophiles [5-8]. In 1981, McCoy and Mal first isolated an adduct of DBU with dimethyl 1-chloro-3-methyl cyclopropane-1,2-dicarboxylate during the dehydrohalogenation of halocyclopropanes [9]. In 1993, Bertrand and co-workers, showed that DBU and DBN act as nucleophiles towards halo derivatives of main group elements where the DBU and DBN bicyclic rings remained unaffected [10,11]. Later in 1994, Lammers et al. observed the nucleophilicity of amidine bases with 4-halo-3,5-dimethyl-1-nitro-1H-pyrazole and their subsequent ring opening leading to the lactam products [12]. Subsequently, Ma and Dolphin isolated chlorin-e6 lactams from the reaction of methyl pheophorbide with DBU and DBN promoted by trialkyl triflates [13]. Additionally, the conjugate addition reaction of DBU to diarylpyrone [14] and Baylis–Hillman acetates [15] also gave caprolactam products. A closer look at these results suggested that the nucleophilic behavior of DBU highly depends on specific substrates. Vaidyanathan and co-workers reported the DBU-catalyzed addition of amines to acyl imidazoles [16], however, using a stoichiometric amount of DBU, Rajagopal et al. observed nucleophile behavior of DBU towards imidazolides providing ε-caprolactam-derived carbamates and amides [17]. Here, in this report we present the results obtained by the reaction of DBU and DBN with highly electrophilic p-nitrophenyl carbonates leading to ε-caprolactam and γ-lactam carbamates.
p-Nitrophenyl carbonates are highly reactive compounds that are usually treated with alcohols or amines to give either a new carbonate or a carbamate-linked compound depending on the nucleophile. In one of our earlier reports, polycarbamate nucleic acids were synthesized from p-nitrophenyl carbonates with amines of nucleic acid derivatives [18]. Very recently, Hotha et al. utilized 1-ethynylcyclohexyl p-nitrophenyl carbonate to synthesize alkynyl glycosyl carbonate donors from hemiacetals [19]. Also, glycocarbamates [20] obtained from glycosyl p-nitrophenyl carbonates [21-24], were explored in studies of carbohydrate–protein interactions [25], ligation and surfactant properties [26,27]. Although p-nitrophenyl carbonates were extensively utilized in these reactions, the nucleophilicity of amidine bases towards these carbonates was not encountered so far. In continuation of our interest in carbohydrates [28,29] and the synthesis of carbamate-linked compounds using p-nitrophenyl carbonates, we herein report our results from nucleophilic ring opening reactions of DBU and DBN using p-nitrophenyl carbonates.
Results and Discussion
In the course of this study, we observed that in the absence of a nucleophile the p-nitrophenyl carbonate of 1-ethynylcyclohexanol (Table 1, 1a) in THF, exclusively afforded the ε-caprolactam product 1b when 2 equiv of DBU were used. After 1 h reaction at room temperature almost all starting material was consumed (90%, TLC) and heating the reaction mixture to 60 °C led to complete consumption of the starting material within 1 h to give product 1b. The transformation involves a nucleophilic attack of the imine nitrogen onto the carbonyl carbon followed by the elimination of a p-nitrophenoxide ion. Subsequently, the imine carbon of DBU is attacked by water molecules present in the solvent, leading to the ring opening and formation of the corresponding caprolactam carbamates (Scheme 1).
![[1860-5397-12-197-i1]](/bjoc/content/inline/1860-5397-12-197-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reaction of DBU with p-nitrophenyl carbonate.
Scheme 1: Reaction of DBU with p-nitrophenyl carbonate.
The structure of 1b was confirmed by 1H and 13C spectroscopy, which showed the characteristic carbamate NH triplet at 5.80 ppm in the 1H NMR spectrum and the expected peaks at 176.6 and 155.0 ppm in the 13C NMR spectrum for the caprolactam and carbamate carbonyl carbons, respectively. Additionally, HRMS and IR absorptions of the carbonyl groups at 1713, 1624 and carbamate NH at 3301 cm−1 also confirmed the ring opening of DBU. Excited with the outcome of the reaction, we set out to explore the structural diversity using different p-nitrophenyl carbonates which were prepared by treating an alcohol with p-nitrophenyl chloroformate in CH2Cl2 using pyridine as a base. Thus, homopropargyl alcohol, decanol, cholesterol and N-Boc-trans-4-hydroxy-L-proline methyl ester gave the corresponding carbonates (Table 1, 2a–5a) in good to excellent yields. Subsequently, the purified carbonates dissolved in THF were treated with 2 equiv of DBU. Heating the reaction mixture to 60 °C for 1 h gave the caprolactam carbamate products 2b–5b along with p-nitrophenol as byproduct. In case of the ε-caprolactam of N-Boc-trans-4-hydroxy-L-proline methyl ester 5b, rotamers due to flipping of the N-Boc group were obtained. Owing to the importance of sugar caprolactams in polymerizations, 2,3-di-O-benzyl-4-O-p-methoxybenzyl-α-methyl-D-glucopyranoside and 2,3,4-tri-O-benzoyl-α-methyl-D-glucopyranoside [30,31] were converted into the p-nitrophenyl carbonates 6a and 7a in good yields. The corresponding carbonate of per-O-benzoyl glucopyranose 8a [30,31] was obtained in only moderate yield and as a mixture of α and β anomers which was used without further purification. The carbonates were subsequently reacted with DBU under the same conditions as described above, giving 6b and 7b in 82% and 87% yield, and 8b as a mixture of α and β anomers in 48% yield. Encouraged by these results, we turned to evaluate the nucleophilicity of DBN towards p-nitrophenyl carbonate derivatives (Scheme 2).
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-197-i3.svg?max-width=637&scale=1.0)
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-197-i4.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-197-i5.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-197-i6.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-12-197-i7.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-12-197-i8.svg?max-width=637&scale=1.0)
![[Graphic 7]](/bjoc/content/inline/1860-5397-12-197-i9.svg?max-width=637&scale=1.0)
![[Graphic 8]](/bjoc/content/inline/1860-5397-12-197-i10.svg?max-width=637&scale=1.0)
![[Graphic 9]](/bjoc/content/inline/1860-5397-12-197-i11.svg?max-width=637&scale=1.0)
![[Graphic 10]](/bjoc/content/inline/1860-5397-12-197-i12.svg?max-width=637&scale=1.0)
![[Graphic 11]](/bjoc/content/inline/1860-5397-12-197-i13.svg?max-width=637&scale=1.0)
![[Graphic 12]](/bjoc/content/inline/1860-5397-12-197-i14.svg?max-width=637&scale=1.0)
![[Graphic 13]](/bjoc/content/inline/1860-5397-12-197-i15.svg?max-width=637&scale=1.0)
![[Graphic 14]](/bjoc/content/inline/1860-5397-12-197-i16.svg?max-width=637&scale=1.0)
![[Graphic 15]](/bjoc/content/inline/1860-5397-12-197-i17.svg?max-width=637&scale=1.0)
![[Graphic 16]](/bjoc/content/inline/1860-5397-12-197-i18.svg?max-width=637&scale=1.0)
![[1860-5397-12-197-i2]](/bjoc/content/inline/1860-5397-12-197-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction of DBN with p-nitrophenyl carbonates.
Scheme 2: Reaction of DBN with p-nitrophenyl carbonates.
Next, the reaction of the p-nitrophenyl carbonate of homopropargyl alcohol 2a in THF with 2 equiv of DBN was examined. Similar to the results observed with DBU, more than 90% of the reaction was complete at room temperature in 1 h. However, heating the reaction mixture to 60 °C for 1 h resulted in completion of the reaction giving the γ-lactam carbamate 2ab in 56% yield (Table 2). The 1H NMR spectrum of the product 2ab showed a broad singlet at 5.70 ppm assigned to the carbamate NH. The corresponding peaks at 175.6, 156.3 ppm in the 13C NMR spectrum as well as the HRMS and IR data confirmed the ring opening of DBN. To evaluate the substrate feasibility, one phenol, an allylic alcohol and three sugar alcohols were subjected to the reaction. The 3,4-dimethylphenyl p-nitrophenyl carbonate (9a) and geranyl carbonate 10a gave the corresponding γ-lactams 9b and 10b in 62% and 46% yields, respectively. Similarly, the p-nitrophenyl carbonate of D-psicofuranose [28], n-pentenyl 2,3,4-tri-O-benzyl-α-D-mannopyranoside [32-34] and 2,3-di-O-benzyl-α-methyl-D-arabinofuranoside [35] (Table 2, 11a–13a) gave the γ-lactam-derived carbamates 11b–13b in 53%, 63% and 67% yield, respectively.
Table 2: Synthesis of γ-lactam-derived carbamates 2ab, 9b–16b.
| No | carbonate | product | yield |
|---|---|---|---|
| 1 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-12-197-i19.svg?max-width=637&scale=1.0)
2a |
![[Graphic 18]](/bjoc/content/inline/1860-5397-12-197-i20.svg?max-width=637&scale=1.0)
2ab |
56% |
| 2 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-12-197-i21.svg?max-width=637&scale=1.0)
9a |
![[Graphic 20]](/bjoc/content/inline/1860-5397-12-197-i22.svg?max-width=637&scale=1.0)
9b |
62% |
| 3 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-12-197-i23.svg?max-width=637&scale=1.0)
10a |
![[Graphic 22]](/bjoc/content/inline/1860-5397-12-197-i24.svg?max-width=637&scale=1.0)
10b |
46% |
| 4 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-12-197-i25.svg?max-width=637&scale=1.0)
11a |
![[Graphic 24]](/bjoc/content/inline/1860-5397-12-197-i26.svg?max-width=637&scale=1.0)
11b |
53% |
| 5 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-12-197-i27.svg?max-width=637&scale=1.0)
12a |
![[Graphic 26]](/bjoc/content/inline/1860-5397-12-197-i28.svg?max-width=637&scale=1.0)
12b |
63% |
| 6 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-12-197-i29.svg?max-width=637&scale=1.0)
13a |
![[Graphic 28]](/bjoc/content/inline/1860-5397-12-197-i30.svg?max-width=637&scale=1.0)
13b |
67% |
| 7 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-12-197-i31.svg?max-width=637&scale=1.0)
14a |
![[Graphic 30]](/bjoc/content/inline/1860-5397-12-197-i32.svg?max-width=637&scale=1.0)
14b ![[Graphic 31]](/bjoc/content/inline/1860-5397-12-197-i33.svg?max-width=637&scale=1.0)
14c |
64% |
| 8 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-12-197-i34.svg?max-width=637&scale=1.0)
15a |
![[Graphic 33]](/bjoc/content/inline/1860-5397-12-197-i35.svg?max-width=637&scale=1.0)
15b |
41%a
30%b |
| 9 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-12-197-i36.svg?max-width=637&scale=1.0)
16a |
![[Graphic 35]](/bjoc/content/inline/1860-5397-12-197-i37.svg?max-width=637&scale=1.0)
16b |
|
| 10 |
![[Graphic 36]](/bjoc/content/inline/1860-5397-12-197-i38.svg?max-width=637&scale=1.0)
17a |
mixture of products | |
areaction at 60 °C and bat rt.
As DBU and DBN are known to promote dehydrohalogenation reactions, we turned our attention to halogenated alcohols. Thus, the p-nitrophenyl carbonate of 10-bromo-1-decanol 14a was reacted with DBN at 60 °C for 1 h affording a single polar spot on TLC. To our surprise, the 1H NMR spectrum showed the existence of two compounds with the carbamate NH showing a multiplet rather than a triplet and an additional signal for the p-nitrophenyl group. In the 13C NMR spectrum, peaks at 175.7 and 157.0 ppm for the cyclic amide and carbamate carbon confirmed the ring opening of DBN. However, the appearance of new peaks in the 13C NMR at 164.3, 141.2 and 126.0 ppm and the upfield shift of C2 of p-nitrophenyl substituent from 122 ppm to 114.4 ppm, suggested that DBN displaced a bromide with the p-nitrophenoxide ion to give compound 14c. Thus, DBN played a dual role in the reaction with 14a – namely as nucleophile and as base giving products 14b and 14c in a 1:1 ratio. The peaks at 164.3 and 114.4 ppm can therefore be assigned to the ipso carbon and ortho carbon of the p-nitrophenyl substituent in 14c. This observation was particularly interesting as N-alkylation of DBN [36] was not favored and instead underwent substitution. To test if the reaction favors both – nucleophilic addition as well as substitution – at lower temperature, the reaction was performed at room temperature for 1.5 h. Although both products were observed the substitution product was minor product, as seen by the integration of p-nitrophenol peaks in the 1H NMR. On the contrary, the reaction of the 2-(2-chloroethoxy)ethanol carbonate 15a with DBN at room temperature and at 60 °C gave only the ring-opened product of DBN 15b in 30% and 41% yield, due to the poor leaving ability of chloride relative to bromide. Further, addition of DBN to the acidic-proton containing substrates such as the p-nitrophenyl carbonate of 9-fluorenemethanol 16a and N-Cbz-L-serine methyl ester 17a, resulted in the dibenzofulvene product 16b in the former, and a mixture of products in the latter, with DBN acting as a base. Even though the γ-lactam products were the only major compounds noticed, the yields were substantially lower than the caprolactam products, presumably due to the ease of ring opening of DBU.
To check if the nucleophilicity of DBU/DBN was specific to the highly electron deficient p-nitrophenyl carbonate, a set of three different carbonates of 1-ethynylcyclohexanol were synthesized using phenyl, benzyl and ethyl chloroformate, respectively. The reaction of the phenyl carbonate (Table 3, A) with DBU at 60 °C for 20 h gave ε-caprolactam (1b) with 12% yield. In contrast, in the reaction of benzyl and ethyl carbonates (B, C) with DBU no trace of lactam (1b) was formed and the substrates remained unaffected due to the poor leaving nature of the alkoxides in comparison to phenolates. This suggests that a highly electrophilic center is the prerequisite for the nucleophilic behavior of DBU and DBN to come into play.
![[Graphic 37]](/bjoc/content/inline/1860-5397-12-197-i39.svg?max-width=637&scale=1.0)
Finally, nearly quantitative large scale transformations were achieved, when 3.5 g of substrates 3a and 7a were reacted with DBU at 60 °C for 1 h giving lactams 3b and 7b in 90% and 94% yield, respectively.
Conclusion
In conclusion, we have shown an operationally simple synthesis of carbamate-derived ε-caprolactam and γ-lactam compounds utilizing the nucleophilicity of DBU/DBN and highly electrophile p-nitrophenyl carbonate derivatives. The reactions proceeded even at room temperature and displayed the nucleophilic addition and substitution with the p-nitrophenyl carbonate derivative of 10-bromodecanol. These caprolactam derivatives may find application in polymer chemistry.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures, compound characterization and copies of 1H and 13C NMR spectra of all new compounds. | ||
| Format: PDF | Size: 5.7 MB | Download |
Acknowledgements
MV is grateful to Prof. K. N. Ganesh, IISER Pune for research support and infrastructural facilities and Dr. Srinivas Hotha for helpful discussions and lab facility. MV thanks DST SERB, India for Fast Track research grant (SB/FT/CS-159/2012). GPS thanks UGC, New Delhi for Junior Research Fellowship.
References
-
Oediger, H.; Möller, F.; Eiter, K. Synthesis 1972, 591–598. doi:10.1055/s-1972-21943
Return to citation in text: [1] -
Shieh, W.-C.; Dell, S.; Repič, O. J. Org. Chem. 2002, 67, 2188–2191. doi:10.1021/jo011036s
Return to citation in text: [1] -
Price, K. E.; Larrivée-Aboussafy, C.; Lillie, B. M.; McLaughlin, R. W.; Mustakis, J.; Hettenbach, K. W.; Hawkins, J. M.; Vaidyanathan, R. Org. Lett. 2009, 11, 2003–2006. doi:10.1021/ol900435t
Return to citation in text: [1] -
Aggarwal, V. K.; Mereu, A. Chem. Commun. 1999, 2311–2312. doi:10.1039/A907754E
Return to citation in text: [1] -
Baidya, M.; Mayr, H. Chem. Commun. 2008, 1792–1794. doi:10.1039/B801811A
Return to citation in text: [1] -
Taylor, J. E.; Bull, S. D.; Williams, J. M. J. Chem. Soc. Rev. 2012, 41, 2109–2121. doi:10.1039/C2CS15288F
Return to citation in text: [1] -
Sutherland, J. K. Chem. Commun. 1997, 325. doi:10.1039/A607723D
Return to citation in text: [1] -
Poronik, Y. M.; Gryko, D. T. Chem. Commun. 2014, 50, 5688–5690. doi:10.1039/C4CC01106F
Return to citation in text: [1] -
McCoy, L. L.; Mal, D. J. Org. Chem. 1981, 46, 1016–1018. doi:10.1021/jo00318a035
Return to citation in text: [1] -
Reed, R.; Réau, R.; Dahan, F.; Bertrand, G. Angew. Chem., Int. Ed. Engl. 1993, 32, 399–401. doi:10.1002/anie.199303991
Return to citation in text: [1] -
Chambers, R. D.; Roche, A. J.; Batsanov, A. S.; Howard, J. A. K. J. Chem. Soc., Chem. Commun. 1994, 2055–2056. doi:10.1039/C39940002055
Return to citation in text: [1] -
Lammers, H.; Cohen-Fernandes, P.; Habraken, C. L. Tetrahedron 1994, 50, 865–870. doi:10.1016/S0040-4020(01)80801-2
Return to citation in text: [1] -
Ma, L.; Dolphin, D. Tetrahedron 1996, 52, 849–860. doi:10.1016/0040-4020(95)00944-2
Return to citation in text: [1] -
Johnson, M. G.; Foglesong, R. J. Tetrahedron Lett. 1997, 38, 7003–7006. doi:10.1016/S0040-4039(97)01675-4
Return to citation in text: [1] -
Im, Y. J.; Gong, J. H.; Kim, H. J.; Kim, J. N. Bull. Korean Chem. Soc. 2001, 22, 1053–1055.
Return to citation in text: [1] -
Larrivée-Aboussafy, C.; Jones, B. P.; Price, K. E.; Hardink, M. A.; McLaughlin, R. W.; Lillie, B. M.; Hawkins, J. M.; Vaidyanathan, R. Org. Lett. 2010, 12, 324–327. doi:10.1021/ol9026599
Return to citation in text: [1] -
Nirmala, R.; Ponpandian, T.; Venkatraman, B. R.; Rajagopal, S. Tetrahedron Lett. 2013, 54, 5181–5184. doi:10.1016/j.tetlet.2013.07.056
Return to citation in text: [1] -
Madhuri, V.; Kumar, V. A. Org. Biomol. Chem. 2010, 8, 3734–3741. doi:10.1039/C003405N
Return to citation in text: [1] -
Mishra, B.; Neralkar, M.; Hotha, S. Angew. Chem., Int. Ed. 2016, 55, 7786–7791. doi:10.1002/anie.201511695
Return to citation in text: [1] -
Shaikh, A. Y.; Sureshkumar, G.; Pati, D.; Gupta, S. S.; Hotha, S. Org. Biomol. Chem. 2011, 9, 5951–5959. doi:10.1039/C1OB05056G
Return to citation in text: [1] -
Cai, T. B.; Lu, D.; Tang, X.; Zhang, Y.; Landerholm, M.; Wang, P. G. J. Org. Chem. 2005, 70, 3518–3524. doi:10.1021/jo050010o
Return to citation in text: [1] -
André, S.; Specker, D.; Bovin, N. V.; Lensch, M.; Kaltner, H.; Gabius, H.-J.; Wittmann, V. Bioconjugate Chem. 2009, 20, 1716–1728. doi:10.1021/bc900152w
Return to citation in text: [1] -
Madec-Lougerstay, R.; Florent, J.-C.; Monneret, C. J. Chem. Soc., Perkin Trans. 1 1999, 1369–1376. doi:10.1039/A808915I
Return to citation in text: [1] -
Cherif, S.; Leach, M. R.; Williams, D. B.; Monneret, C. Bioorg. Med. Chem. Lett. 2002, 12, 1237–1240. doi:10.1016/S0960-894X(02)00151-8
Return to citation in text: [1] -
Schwefel, D.; Maierhofer, C.; Beck, J. G.; Seeberger, S.; Diederichs, K.; Möller, H. M.; Welte, W.; Wittmann, V. J. Am. Chem. Soc. 2010, 132, 8704–8719. doi:10.1021/ja101646k
Return to citation in text: [1] -
Prata, C.; Mora, N.; Lacombe, J.-M.; Maurizis, J.-C.; Pucci, B. Carbohydr. Res. 1999, 321, 4–14. doi:10.1016/S0008-6215(99)00161-5
Return to citation in text: [1] -
Fernández, C.; Nieto, O.; Fontenla, J. A.; Rivas, E.; de Ceballos, M. L.; Fernández-Mayoralas, A. Org. Biomol. Chem. 2003, 1, 767–771. doi:10.1039/B212066F
Return to citation in text: [1] -
Vangala, M.; Shinde, G. P. Beilstein J. Org. Chem. 2015, 11, 2289–2296. doi:10.3762/bjoc.11.249
Return to citation in text: [1] [2] -
Bhuma, N.; Vangala, M.; Nair, R. J.; Sabharwal, S. G.; Dhavale, D. D. Carbohydr. Res. 2015, 402, 215–224. doi:10.1016/j.carres.2014.10.023
Return to citation in text: [1] -
Zhu, X.; Schmidt, R. R. Angew. Chem., Int. Ed. 2009, 48, 1900–1934. doi:10.1002/anie.200802036
Return to citation in text: [1] [2] -
Vidadala, S. R.; Hotha, S. Chem. Commun. 2009, 2505–2507. doi:10.1039/B822526E
Return to citation in text: [1] [2] -
Fraser-Reid, B.; Udodong, U. E.; Wu, Z.; Ottosson, H.; Merritt, J. R.; Rao, C. S.; Roberts, C.; Madsen, R. Synlett 1992, 12, 927–942. doi:10.1055/s-1992-21543
Return to citation in text: [1] -
Fraser-Reid, B.; Lopez, J. C.; Radhakrishnan, K. V.; Mach, M.; Schlueter, U.; Gomez, A. M.; Uriel, C. J. Am. Chem. Soc. 2002, 124, 3198–3199. doi:10.1021/ja012383m
Return to citation in text: [1] -
Vidadala, S. R.; Thadke, S. A.; Hotha, S. J. Org. Chem. 2009, 74, 9233–9236. doi:10.1021/jo901837z
Return to citation in text: [1] -
Callam, C. S.; Lowary, T. L. J. Chem. Educ. 2001, 78, 73–74. doi:10.1021/ed078p73
Return to citation in text: [1] -
Whitney, R. A.; Penciu, A.; Parent, J. S.; Resendes, R.; Hopkins, W. Macromolecules 2005, 38, 4625–4629. doi:10.1021/ma047850+
Return to citation in text: [1]
| 1. | Oediger, H.; Möller, F.; Eiter, K. Synthesis 1972, 591–598. doi:10.1055/s-1972-21943 |
| 5. | Baidya, M.; Mayr, H. Chem. Commun. 2008, 1792–1794. doi:10.1039/B801811A |
| 6. | Taylor, J. E.; Bull, S. D.; Williams, J. M. J. Chem. Soc. Rev. 2012, 41, 2109–2121. doi:10.1039/C2CS15288F |
| 7. | Sutherland, J. K. Chem. Commun. 1997, 325. doi:10.1039/A607723D |
| 8. | Poronik, Y. M.; Gryko, D. T. Chem. Commun. 2014, 50, 5688–5690. doi:10.1039/C4CC01106F |
| 19. | Mishra, B.; Neralkar, M.; Hotha, S. Angew. Chem., Int. Ed. 2016, 55, 7786–7791. doi:10.1002/anie.201511695 |
| 20. | Shaikh, A. Y.; Sureshkumar, G.; Pati, D.; Gupta, S. S.; Hotha, S. Org. Biomol. Chem. 2011, 9, 5951–5959. doi:10.1039/C1OB05056G |
| 3. | Price, K. E.; Larrivée-Aboussafy, C.; Lillie, B. M.; McLaughlin, R. W.; Mustakis, J.; Hettenbach, K. W.; Hawkins, J. M.; Vaidyanathan, R. Org. Lett. 2009, 11, 2003–2006. doi:10.1021/ol900435t |
| 17. | Nirmala, R.; Ponpandian, T.; Venkatraman, B. R.; Rajagopal, S. Tetrahedron Lett. 2013, 54, 5181–5184. doi:10.1016/j.tetlet.2013.07.056 |
| 2. | Shieh, W.-C.; Dell, S.; Repič, O. J. Org. Chem. 2002, 67, 2188–2191. doi:10.1021/jo011036s |
| 18. | Madhuri, V.; Kumar, V. A. Org. Biomol. Chem. 2010, 8, 3734–3741. doi:10.1039/C003405N |
| 13. | Ma, L.; Dolphin, D. Tetrahedron 1996, 52, 849–860. doi:10.1016/0040-4020(95)00944-2 |
| 15. | Im, Y. J.; Gong, J. H.; Kim, H. J.; Kim, J. N. Bull. Korean Chem. Soc. 2001, 22, 1053–1055. |
| 12. | Lammers, H.; Cohen-Fernandes, P.; Habraken, C. L. Tetrahedron 1994, 50, 865–870. doi:10.1016/S0040-4020(01)80801-2 |
| 16. | Larrivée-Aboussafy, C.; Jones, B. P.; Price, K. E.; Hardink, M. A.; McLaughlin, R. W.; Lillie, B. M.; Hawkins, J. M.; Vaidyanathan, R. Org. Lett. 2010, 12, 324–327. doi:10.1021/ol9026599 |
| 10. | Reed, R.; Réau, R.; Dahan, F.; Bertrand, G. Angew. Chem., Int. Ed. Engl. 1993, 32, 399–401. doi:10.1002/anie.199303991 |
| 11. | Chambers, R. D.; Roche, A. J.; Batsanov, A. S.; Howard, J. A. K. J. Chem. Soc., Chem. Commun. 1994, 2055–2056. doi:10.1039/C39940002055 |
| 9. | McCoy, L. L.; Mal, D. J. Org. Chem. 1981, 46, 1016–1018. doi:10.1021/jo00318a035 |
| 14. | Johnson, M. G.; Foglesong, R. J. Tetrahedron Lett. 1997, 38, 7003–7006. doi:10.1016/S0040-4039(97)01675-4 |
| 26. | Prata, C.; Mora, N.; Lacombe, J.-M.; Maurizis, J.-C.; Pucci, B. Carbohydr. Res. 1999, 321, 4–14. doi:10.1016/S0008-6215(99)00161-5 |
| 27. | Fernández, C.; Nieto, O.; Fontenla, J. A.; Rivas, E.; de Ceballos, M. L.; Fernández-Mayoralas, A. Org. Biomol. Chem. 2003, 1, 767–771. doi:10.1039/B212066F |
| 21. | Cai, T. B.; Lu, D.; Tang, X.; Zhang, Y.; Landerholm, M.; Wang, P. G. J. Org. Chem. 2005, 70, 3518–3524. doi:10.1021/jo050010o |
| 22. | André, S.; Specker, D.; Bovin, N. V.; Lensch, M.; Kaltner, H.; Gabius, H.-J.; Wittmann, V. Bioconjugate Chem. 2009, 20, 1716–1728. doi:10.1021/bc900152w |
| 23. | Madec-Lougerstay, R.; Florent, J.-C.; Monneret, C. J. Chem. Soc., Perkin Trans. 1 1999, 1369–1376. doi:10.1039/A808915I |
| 24. | Cherif, S.; Leach, M. R.; Williams, D. B.; Monneret, C. Bioorg. Med. Chem. Lett. 2002, 12, 1237–1240. doi:10.1016/S0960-894X(02)00151-8 |
| 25. | Schwefel, D.; Maierhofer, C.; Beck, J. G.; Seeberger, S.; Diederichs, K.; Möller, H. M.; Welte, W.; Wittmann, V. J. Am. Chem. Soc. 2010, 132, 8704–8719. doi:10.1021/ja101646k |
| 36. | Whitney, R. A.; Penciu, A.; Parent, J. S.; Resendes, R.; Hopkins, W. Macromolecules 2005, 38, 4625–4629. doi:10.1021/ma047850+ |
| 32. | Fraser-Reid, B.; Udodong, U. E.; Wu, Z.; Ottosson, H.; Merritt, J. R.; Rao, C. S.; Roberts, C.; Madsen, R. Synlett 1992, 12, 927–942. doi:10.1055/s-1992-21543 |
| 33. | Fraser-Reid, B.; Lopez, J. C.; Radhakrishnan, K. V.; Mach, M.; Schlueter, U.; Gomez, A. M.; Uriel, C. J. Am. Chem. Soc. 2002, 124, 3198–3199. doi:10.1021/ja012383m |
| 34. | Vidadala, S. R.; Thadke, S. A.; Hotha, S. J. Org. Chem. 2009, 74, 9233–9236. doi:10.1021/jo901837z |
| 35. | Callam, C. S.; Lowary, T. L. J. Chem. Educ. 2001, 78, 73–74. doi:10.1021/ed078p73 |
| 30. | Zhu, X.; Schmidt, R. R. Angew. Chem., Int. Ed. 2009, 48, 1900–1934. doi:10.1002/anie.200802036 |
| 31. | Vidadala, S. R.; Hotha, S. Chem. Commun. 2009, 2505–2507. doi:10.1039/B822526E |
| 28. | Vangala, M.; Shinde, G. P. Beilstein J. Org. Chem. 2015, 11, 2289–2296. doi:10.3762/bjoc.11.249 |
| 28. | Vangala, M.; Shinde, G. P. Beilstein J. Org. Chem. 2015, 11, 2289–2296. doi:10.3762/bjoc.11.249 |
| 29. | Bhuma, N.; Vangala, M.; Nair, R. J.; Sabharwal, S. G.; Dhavale, D. D. Carbohydr. Res. 2015, 402, 215–224. doi:10.1016/j.carres.2014.10.023 |
| 30. | Zhu, X.; Schmidt, R. R. Angew. Chem., Int. Ed. 2009, 48, 1900–1934. doi:10.1002/anie.200802036 |
| 31. | Vidadala, S. R.; Hotha, S. Chem. Commun. 2009, 2505–2507. doi:10.1039/B822526E |
© 2016 Vangala and Shinde; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)