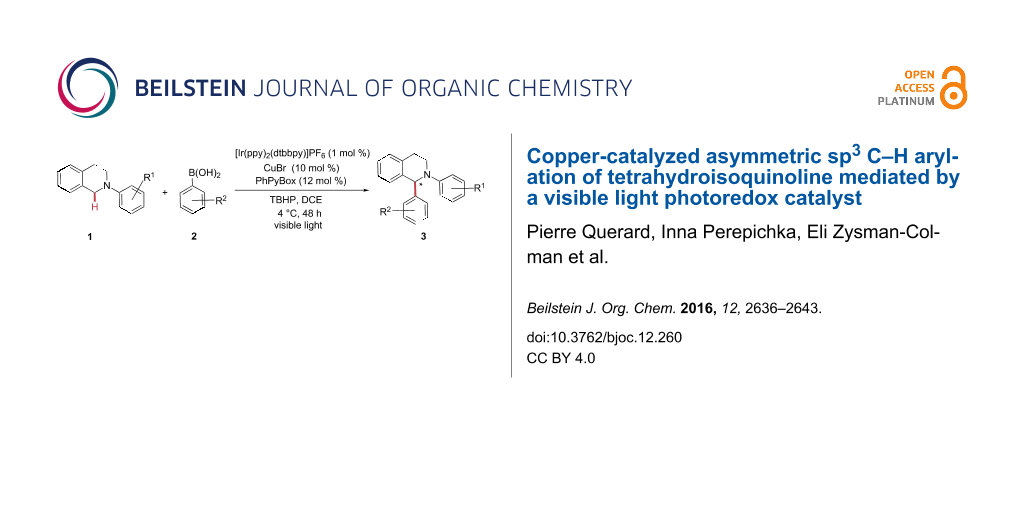
Abstract
This report describes a highly enantioselective oxidative sp3 C–H arylation of N-aryltetrahydroisoquinolines (THIQs) through a dual catalysis platform. The combination of the photoredox catalyst, [Ir(ppy)2(dtbbpy)]PF6, and chiral copper catalysts provide a mild and highly effective sp3 C–H asymmetric arylation of THIQs.

Graphical Abstract
Introduction
Functionalization of sp3 C–H bonds is a unique and powerful transformation in modern organic synthesis, which remains a challenging process despite the advances that have been made in this field [1]. The directing group strategies are widely used and developed to achieve enantioselective metal-catalyzed C–H bond functionalizations in recent years. Unactivated alkyl C–H bond activation (i.e., without any directing group) is of great interest in terms of atom economy, nevertheless enantioselectivity is difficult to control due to often-required harsh reaction conditions. Therefore, the development of simple and facile processes to functionalize sp3 C–H bonds under mild conditions in the absence of directing groups is of great interest [2].
The emerging and expanding field of visible-light-mediated photoredox catalysis presents unique opportunities for the conception of new synthetic routes [3-12]. Upon exposure to visible light, photoredox catalysts can function as both reductant and oxidant, thereby providing extremely important tools for potential transition-metal-catalyzed enantioselective reactions of sp3 C–H bonds, which could be carried out at low temperature and under mild reaction conditions [13,14]. We envisioned that combining photoredox catalysis with typical cross-coupling methods will allow us to design a visible-light-mediated photoredox asymmetric arylation of tetrahydroisoquinolines (THIQs) [15-20].
During the last decade, numerous examples of sp3 C–H bond arylation procedures have been developed [1,21-29]. In 2008, our group developed the first direct sp3 C–H arylation of THIQ with arylboronic acids using a copper catalyst (Scheme 1) [30]. Oxygen gas and tert-butyl hydroperoxide (TBHP) were used as external oxidants, which gave moderate to good isolated yields (up to 75%). In addition, we demonstrated the first enantioselective arylation of THIQ using phenylboronic acid with 44% enantiomeric excess (ee), but very poor yield of the optically active products. Lowering the reaction temperature, in order to increase the corresponding ee, resulted in inhibition of the reaction.
![[1860-5397-12-260-i1]](/bjoc/content/inline/1860-5397-12-260-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Design light-mediated arylation of THIQs.
Scheme 1: Design light-mediated arylation of THIQs.
More recently, Liu et al. have demonstrated the arylation of THIQs with arylboronic esters via asymmetric organocatalysis methodology [25,28]. The use of chiral tartaric acid derivatives, 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and high temperature (70 °C) were found to be the optimal conditions to obtain the desired arylated product with acceptable yield and good enantioselectivity. However, this methodology has shown limitations in terms of substrate scope: only phenylboronic esters with electron-donating substituents yielded the corresponding products.
We herein report the first visible light-mediated asymmetric cross-coupling arylation of sp3 C–H bonds adjacent to nitrogen, combining photoredox catalysis with metal-catalyzed transformations.
Results and Discussion
Optimisation of reaction conditions
In our previous work on arylation of N-aryltetrahydroisoquinoline [30], we demonstrated that lowering the temperature from 90 °C to room temperature in the reaction with copper(I) bromide caused a significant drop in yield. During optimisation of the reaction system, TBHP was found to be the best external oxidant for this reaction over many others [31]. To accelerate the reaction at lower temperature, we reasoned that a light-mediated photoredox system might help, which indeed has improved the reaction yield and enantioselectivity. Different iridium and ruthenium photoredox catalysts were evaluated and [Ir(ppy)2(dtbbpy)]PF6 was found to be the most efficient [32]. With this iridium photoredox catalyst, TBHP, and copper(I) bromide co-catalyst in DME as solvent, we observed a trace amount of the desired product at room temperature. When different copper salts were evaluated, it was found that CuBr was less active (Table 1, entry 1) and copper(II) bromide provided the highest yield for the arylation of THIQ with phenylboronic acid (2, Table 1, entry 2). Other copper salts such as Cu(OTf)2 and Cu(OAc)2 were much less effective (Table 1, entries 3 and 4). A significant increase of yield was observed when the stoichiometry of the system was changed to a slight excess of arylboronic acid. When more than 1.6 equivalents of 2 were involved in the reaction, a drastic acceleration of the reaction was observed, leading to up to 85% yield (Table 1, entries 5 and 6). During the investigation of solvent influence on the formation of 3a, it was found that polar solvents such as DCE gave the best yields, compared to less polar solvents such as toluene and THF (Table 1, entries 7 and 8). On the other hand, highly polar solvents such as MeCN and MeOH were not beneficial for the formation of the desired product 3a (Table 1, entries 9 and 10). Control experiments performed in the absence of photoredox catalyst and/or transition metal copper(II) salt (Table 1, entries 11–13) showed very poor reactivity. Moreover, in the absence of light, an extremely poor yield was obtained (Table 1, entry 14).
Table 1: Optimization of reaction conditionsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-260-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Catalyst | Solvent | 2 (equiv) | Yield of 3a (%) |
|---|---|---|---|---|
| 1 | CuBr | DCE | 1.6 | 19 |
| 2 | CuBr2 | DCE | 1.6 | 29 |
| 3 | Cu(OTf)2 | DCE | 1.6 | 2 |
| 4 | Cu(OAc)2 | DCE | 1.6 | 14 |
| 5 | CuBr2 | DCE | 2 | 72 |
| 6 | CuBr2 | DCE | 3 | 85 |
| 7 | CuBr2 | THF | 3 | 15 |
| 8 | CuBr2 | toluene | 3 | 23 |
| 9 | CuBr2 | MeCN | 3 | 11 |
| 10 | CuBr2 | MeOH | 3 | 13 |
| 11b | CuBr2 | DCE | 3 | 12 |
| 12b | – | DCE | 3 | 0 |
| 13b,c | CuBr2 | DCE | 3 | 0 |
| 14d | CuBr2 | DCE | 3 | 12 |
aReaction conditions: THIQs (0.10 mmol), arylboronic acid (0.30 mmol), TBHP (0.16 mmol), [Ir(ppy)2(dtbbpy)]PF6 (0.001 mmol), CuBr2 (0.02 mmol), DCE (0.5 mL), under argon atmosphere. NMR yields are reported. bReaction carried out without Ir(III) photoredox catalyst. cReaction carried out without TBHP. dReaction performed in absence of light. All reported yields were determined by 1H NMR spectroscopy using dibromomethane as an internal standard.
General scope of reaction
With the optimized reaction conditions in hand, the substrate scope was investigated (Figure 1). N-Phenyltetrahydroisoquinoline (1) combined with phenylboronic acid (2) gave rise to 85% yield of the corresponding arylated product 3a. N-Phenyl-substituted THIQs bearing electron-donating groups (EDG), such a OMe and Me, were tolerated in our reaction system. We were surprised to see that strong electron-donating substituents such as OMe gave lower yields (3b, 3c and 3d), which we attribute to the lowered oxidation potentials of the tertiary amine, favouring side reactions. It is notable that weaker EDG substituents on the aryl moiety (e.g., Me) resulted in higher yields (3e). Electron-withdrawing groups (EWG) such as Br were tolerated and yielded the desired product in 80% yield (3f). Aromatic boronic acids possessing both electron-withdrawing and electron-donating substituents were evaluated under our reaction conditions and all resulted in good yields. While aromatic boronic acids substituted with electron-withdrawing groups (e.g., acyl, F or CF3) were likewise tolerated well (3g–j). Aromatic boronic acids substituted with electron-donating groups resulted in the formation of the corresponding arylated products with higher yields (3k–n).
![[1860-5397-12-260-1]](/bjoc/content/figures/1860-5397-12-260-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Reaction scope. Reaction conditions: THIQs (0.10 mmol), arylboronic acid (0.30 mmol), TBHP (0.2 mmol), [Ir(ppy)2(dtbbpy)]PF6 (0.001 mmol), CuBr2 (0.02 mmol), DCE (0.5 mL), under argon atmosphere.
Figure 1: Reaction scope. Reaction conditions: THIQs (0.10 mmol), arylboronic acid (0.30 mmol), TBHP (0.2 mmo...
Enantioselective arylation reaction
Subsequently, we explored the asymmetric version of this arylation reaction with various chiral ligands (see Scheme 2 and Supporting Information File 1, Table S3, for a detailed screening table). Among them, Box-type ligands have demonstrated a good performance in this reaction, affording low to good enantioselectivities.
![[1860-5397-12-260-i2]](/bjoc/content/inline/1860-5397-12-260-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
We began our study by evaluating the efficiency of the ligands using the standard arylation of THIQ with phenylboronic acid. A modest enantiomeric ratio (er) of the C−H coupling reaction was obtained using L1 ligand (Table 2, entry 1) at low temperature (4 °C). On the other hand, the commercially available mono-arylated PyBox L2 gave very good er under our reaction conditions (Table 2, entry 2). It is noteworthy that the er observed was higher when copper(I) bromide was used as a co–catalyst, compared to copper(II) bromide (Table 2, entry 3), possibly due to the Lewis acidity difference of Cu(I) and Cu(II). However, the yield of the desired optically active product 3a dropped by about half, when CuBr was used as catalyst. Alkyl-substituted PyBox such as L3 was not beneficial to the enantioselectivity (Table 2, entry 4). The efficacy of N,N-Box ligand (L4) was investigated and it appeared that the pyridine motif was extremely important to achieve high enantioselectivity (Table 2, entry 5).
Table 2: Effect of chiral ligand on the enantioselectivity of coupling of N-phenyltetrahydroisoquinoline with phenylboronic acida.
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-260-i5.svg?max-width=637&scale=1.0)
|
||
| Entry | L* | er |
|---|---|---|
| 1 | L1 | 69:31 |
| 2 | L2 | 82:18 |
| 3b | L2 | 68:32 |
| 4 | L3 | 54:46 |
| 5 | L4 | 54:46 |
aReaction conditions: THIQs (0.10 mmol), arylboronic acid (0.30 mmol), TBHP (0.2 mmol), [Ir(ppy)2(dtbbpy)]PF6 (0.001 mmol), CuBr (0.01 mmol), L* (0.012 mmol), DCE (0.5 mL), under argon atmosphere. bCuBr2 was used. All reported enantiomeric ratios were determined using a Chiralcel OD-H column and 96:4 hexane/isopropanol as an eluent (Supporting Information File 1).
To evaluate the scope of the enantiomeric selectivity of the arylation reaction, copper(I) bromide together with (R,R)-PhPyBox L2 at 4 °C was used as the standard conditions. We were pleased to see that our model reaction yielded 3a with good enantiomeric ratio (Table 3, entry 1). In the presence of the other enantiomer of L2, (S,S)-PhPyBox, the reaction afforded good er. When N-(2-methhoxyphenyl)tetrahydroisoquinoline was used, the corresponding enantiomer was obtained with similar enantioselectivity (Table 3, entry 2). N-Aryl-substituted THIQs gave high er, when either EDG or EWG were present (Table 3, entries 3–6). High and moderate enantiomeric ratios were obtained, respectively, when vinyl-substituted arylboronic acids and fluoro-substituted arylboronic acids were subjected to the reaction system (Table 3, entries 7 and 8).
Table 3: Enantioselective arylation reactiona.
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-260-i6.svg?max-width=637&scale=1.0)
|
||||
| Entry | Product | R1 | R2 | er |
|---|---|---|---|---|
| 1 | 3a | H | H | 19:81 |
| 2b | 3b | 2-OMe | H | 84:16 |
| 3 | 3c | 3-OMe | H | 10:90 |
| 4 | 3d | 4-OMe | H | 15:85 |
| 5 | 3e | 4-Me | H | 24:76 |
| 6 | 3f | 4-Br | H | 19:81 |
| 7 | 3m | H | 4-vinyl | 19:81 |
| 8 | 3j | H | 2,4-difluoro | 37:63 |
aReaction conditions: THIQs (0.10 mmol), arylboronic acid (0.30 mmol), TBHP (0.2 mmol), [Ir(ppy)2(dtbbpy)]PF6 (0.001 mmol), CuBr (0.01 mmol), (R,R)-2,6-Bis(4-phenyl-2-oxazolinyl)pyridine (0.012 mmol), DCE (0.5 mL), under argon atmosphere. b(S,S)-2,6-bis(4-phenyl-2-oxazolinyl)pyridine was used instead. All reported yields enantiomeric ratios were determined using a Chiralcel OD-H column and 96:4 hexane/isopropanol as an eluent (Supporting Information File 1).
A tentative reaction mechanism has been proposed in Scheme 3, in order to rationalize this arylation reaction. Upon visible light irradiation, [Ir(ppy)2(dtbbpy)]PF6 I was converted into an excited state II, Ir(III)* [11,33-37]. The THIQ undergoes a single electron transfer (SET), reducing the iridium complex to Ir(II) III and oxidizing the the nitrogen of THIQ IV to its radical cation V, which then undergoes a hydride abstraction to form the iminium salt form VI, of the THIQ. The pre-formed chiral PhCu–PyBox complex [38], coordinates to the iminium cation VI, followed by stereofacial nucleophilic addition of the arylboronic acid to produce the desired enantioenriched arylated product VII. The Ir(III) is regenerated in the presence of the sacrificial external oxidant TBHP.
![[1860-5397-12-260-i3]](/bjoc/content/inline/1860-5397-12-260-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, we have successfully developed a highly efficient light-mediated coupling method for the direct asymmetric arylation of N-arylated tetrahydroisoquinolines (THIQs) with arylboronic acids. Using [Ir(ppy)2(dtbbpy)]PF6 as photoredox catalyst provided a novel facile method to build important arylated compounds in very high yields under very mild conditions. The combination of copper salts and PhPyBox as chiral ligand have demonstrated its efficiency producing good enantioselectivity and tolerated a fairly diverse substrate scope. We envisioned that this visible light-mediated asymmetric arylation reaction could be extended to other sp3 C–H bonds. The development of new light-mediated processes for stereoselective functionalization of unactivated C−H bonds is currently undergoing in our laboratory.
Experimental
General procedure for the sp3 C–H arylation of THIQs with boronic acid derivatives (Figure 1). A V-shaped 10 mL Biotage reaction vial was charged with [Ir(ppy)2(dtbbpy)]PF6 (1 mol %, 1.0 mg), CuBr2 (10 mol %, 2.23 mg), N-phenyltetrahydroisoquinoline (0.1 mmol), and the corresponding phenylboronic acid (0.3 mmol), evacuated and refilled with argon three times. DCE (0.5 mL) was added, followed by subsequent slow addition of TBHP (0.16 mmol). The reaction vessel was sealed, placed under white light bulbs irradiation with vigorous stirring (approx. 1000 rpm) and hold for 24 h. The mixture was diluted with ethyl acetate (2 mL), washed with water (2 mL), filtered through a pad of silica, and rinsed with additional ethyl acetate. The combined organic phase was concentrated and purified by column chromatography or preparative thin-layer chromatography on silica gel to yield the corresponding arylated compound 3. Dibromomethane was used as internal standard for 1H NMR analysis.
Variation for enantioselective sp3 C–H arylation of THIQs with boronic acid derivatives (Table 3). A V-shaped 10 mL Biotage reaction vial was charged with CuBr (10 mol %, 1.43 mg) and PhPybox (12 mol %, 4.43 mg), evacuated and refilled with argon three times, and then 0.1 mL of DCE was added. The reaction was stirred for 30 min. N-Phenyltetrahydroisoquinoline (0.1 mmol), [Ir(ppy)2(dtbbpy)]PF6 (1 mol %, 1.0 mg) and the corresponding phenylboronic acid (0.3 mmol) were added, and then the atmosphere was evacuated and refilled with argon three times. DCE (0.4 mL) was added followed by subsequent slow addition of TBHP (0.16 mmol). The reaction vessel was sealed, placed under white light bulbs irradiation with vigorous stirring (approx. 1000 rpm) and held for 48 h in a cold room (4 °C). The mixture was diluted with ethyl acetate (2 mL), washed with water (2 mL), filtered through a pad of silica, and rinsed with additional ethyl acetate. The combined organic phase was concentrated and purified by column chromatography or preparative thin-layer chromatography on silica gel to yield the corresponding arylated compound 3. Dibromomethane was used as internal standard for 1H NMR analysis.
Supporting Information
| Supporting Information File 1: Experimental and copies of spectra. | ||
| Format: PDF | Size: 2.4 MB | Download |
References
-
He, J.; Li, S.; Deng, Y.; Fu, H.; Laforteza, B. N.; Spangler, J. E.; Homs, A.; Yu, J.-Q. Science 2014, 343, 1216–1220. doi:10.1126/science.1249198
Return to citation in text: [1] [2] -
Li, C.-J. From C-H to C-C Bonds: Cross-Dehydrogenative-Coupling; RSC Green Chemistry, 2015.
Return to citation in text: [1] -
Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r
Return to citation in text: [1] -
Cismesia, M. A.; Yoon, T. P. Chem. Sci. 2015, 6, 5426–5434. doi:10.1039/C5SC02185E
Return to citation in text: [1] -
Nicewicz, D. A.; MacMillan, D. W. C. Science 2008, 322, 77–80. doi:10.1126/science.1161976
Return to citation in text: [1] -
Nagib, D. A.; Scott, M. E.; MacMillan, D. W. C. J. Am. Chem. Soc. 2009, 131, 10875–10877. doi:10.1021/ja9053338
Return to citation in text: [1] -
Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/B913880N
Return to citation in text: [1] -
Kalyani, D.; McMurtrey, K. B.; Neufeldt, S. R.; Sanford, M. S. J. Am. Chem. Soc. 2011, 133, 18566–18569. doi:10.1021/ja208068w
Return to citation in text: [1] -
Ischay, M. A.; Anzovino, M. E.; Du, J.; Yoon, T. P. J. Am. Chem. Soc. 2008, 130, 12886–12887. doi:10.1021/ja805387f
Return to citation in text: [1] -
Yoon, T. P.; Ischay, M. A.; Du, J. Nat. Chem. 2010, 2, 527–532. doi:10.1038/nchem.687
Return to citation in text: [1] -
Du, J.; Skubi, K. L.; Schultz, D. M.; Yoon, T. P. Science 2014, 344, 392–396. doi:10.1126/science.1251511
Return to citation in text: [1] [2] -
Beatty, J. W.; Stephenson, C. R. J. Acc. Chem. Res. 2015, 48, 1474–1484. doi:10.1021/acs.accounts.5b00068
Return to citation in text: [1] -
Perepichka, I.; Kundu, S.; Hearne, Z.; Li, C.-J. Org. Biomol. Chem. 2015, 13, 447–451. doi:10.1039/C4OB02138J
Return to citation in text: [1] -
Buschmann, H.; Scharf, H.-D.; Hoffmann, N.; Esser, P. Angew. Chem., Int. Ed. 1991, 30, 477–515. doi:10.1002/anie.199104771
Return to citation in text: [1] -
Meggers, E. Chem. Commun. 2015, 51, 3290–3301. doi:10.1039/C4CC09268F
Return to citation in text: [1] -
Wang, C.; Harms, K.; Meggers, E. Angew. Chem., Int. Ed. 2016, 55, 13495–13498. doi:10.1002/anie.201607305
Return to citation in text: [1] -
Murphy, J. J.; Bastida, D.; Paria, S.; Fagnoni, M.; Melchiorre, P. Nature 2016, 532, 218–222. doi:10.1038/nature17438
Return to citation in text: [1] -
Kizu, T.; Uraguchi, D.; Ooi, T. J. Org. Chem. 2016, 81, 6953–6958. doi:10.1021/acs.joc.6b00445
Return to citation in text: [1] -
Amador, A. G.; Sherbrook, E. M.; Yoon, T. P. J. Am. Chem. Soc. 2016, 138, 4722–4725. doi:10.1021/jacs.6b01728
Return to citation in text: [1] -
Maturi, M. M.; Bach, T. Angew. Chem., Int. Ed. 2014, 53, 7661–7664. doi:10.1002/anie.201403885
Return to citation in text: [1] -
Barham, J. P.; John, M. P.; Murphy, J. A. Beilstein J. Org. Chem. 2014, 10, 2981–2988. doi:10.3762/bjoc.10.316
Return to citation in text: [1] -
Cuthbertson, J. D.; MacMillan, D. W. C. Nature 2015, 519, 74–77. doi:10.1038/nature14255
Return to citation in text: [1] -
Muramatsu, W.; Nakano, K.; Li, C.-J. Org. Biomol. Chem. 2014, 12, 2189–2192. doi:10.1039/c3ob42354a
Return to citation in text: [1] -
Dhineshkumar, J.; Lamani, M.; Alagiri, K.; Prabhu, K. R. Org. Lett. 2013, 15, 1092–1095. doi:10.1021/ol4001153
Return to citation in text: [1] -
Liu, X.; Sun, S.; Meng, Z.; Lou, H.; Liu, L. Org. Lett. 2015, 17, 2396–2399. doi:10.1021/acs.orglett.5b00909
Return to citation in text: [1] [2] -
Li, Z.; Bohle, D. S.; Li, C.-J. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 8928–8933. doi:10.1073/pnas.0601687103
Return to citation in text: [1] -
Yan, C.; Li, L.; Liu, Y.; Wang, Q. Org. Lett. 2016, 18, 4686–4689. doi:10.1021/acs.orglett.6b02326
Return to citation in text: [1] -
Liu, X.; Meng, Z.; Li, C.; Lou, H.; Liu, L. Angew. Chem., Int. Ed. 2015, 54, 6012–6015. doi:10.1002/anie.201500703
Return to citation in text: [1] [2] -
Bergonzini, G.; Schindler, C. S.; Wallentin, C.-J.; Jacobsen, E. N.; Stephenson, C. R. J. Chem. Sci. 2014, 5, 112–116. doi:10.1039/C3SC52265B
Return to citation in text: [1] -
Baslé, O.; Li, C.-J. Org. Lett. 2008, 10, 3661–3663. doi:10.1021/ol8012588
Return to citation in text: [1] [2] -
For more information, see Supporting Information File 1, Table S1.
Return to citation in text: [1] -
For more information, see Supporting Information File 1, Table S2.
Return to citation in text: [1] -
Condie, A. G.; González-Gómez, J. C.; Stephenson, C. R. J. J. Am. Chem. Soc. 2010, 132, 1464–1465. doi:10.1021/ja909145y
Return to citation in text: [1] -
Rueping, M.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C.; Leonori, D.; Vila, C. Chem. – Eur. J. 2012, 18, 5170–5174. doi:10.1002/chem.201200050
Return to citation in text: [1] -
Boess, E.; Schmitz, C.; Klussmann, M. J. Am. Chem. Soc. 2012, 134, 5317–5325. doi:10.1021/ja211697s
Return to citation in text: [1] -
Lowry, M. S.; Goldsmith, J. I.; Slinker, J. D.; Rohl, R.; Pascal, R. A., Jr.; Malliaras, G. G.; Bernhard, S. Chem. Mater. 2005, 17, 5712–5719. doi:10.1021/cm051312+
Return to citation in text: [1] -
Ladouceur, S.; Fortin, D.; Zysman-Colman, E. Inorg. Chem. 2011, 50, 11514–11526. doi:10.1021/ic2014013
Return to citation in text: [1] -
Panera, M.; Díez, J.; Merino, I.; Rubio, E.; Gamasa, M. P. Inorg. Chem. 2009, 48, 11147–11160. doi:10.1021/ic901527x
Return to citation in text: [1]
| 1. | He, J.; Li, S.; Deng, Y.; Fu, H.; Laforteza, B. N.; Spangler, J. E.; Homs, A.; Yu, J.-Q. Science 2014, 343, 1216–1220. doi:10.1126/science.1249198 |
| 15. | Meggers, E. Chem. Commun. 2015, 51, 3290–3301. doi:10.1039/C4CC09268F |
| 16. | Wang, C.; Harms, K.; Meggers, E. Angew. Chem., Int. Ed. 2016, 55, 13495–13498. doi:10.1002/anie.201607305 |
| 17. | Murphy, J. J.; Bastida, D.; Paria, S.; Fagnoni, M.; Melchiorre, P. Nature 2016, 532, 218–222. doi:10.1038/nature17438 |
| 18. | Kizu, T.; Uraguchi, D.; Ooi, T. J. Org. Chem. 2016, 81, 6953–6958. doi:10.1021/acs.joc.6b00445 |
| 19. | Amador, A. G.; Sherbrook, E. M.; Yoon, T. P. J. Am. Chem. Soc. 2016, 138, 4722–4725. doi:10.1021/jacs.6b01728 |
| 20. | Maturi, M. M.; Bach, T. Angew. Chem., Int. Ed. 2014, 53, 7661–7664. doi:10.1002/anie.201403885 |
| 13. | Perepichka, I.; Kundu, S.; Hearne, Z.; Li, C.-J. Org. Biomol. Chem. 2015, 13, 447–451. doi:10.1039/C4OB02138J |
| 14. | Buschmann, H.; Scharf, H.-D.; Hoffmann, N.; Esser, P. Angew. Chem., Int. Ed. 1991, 30, 477–515. doi:10.1002/anie.199104771 |
| 3. | Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r |
| 4. | Cismesia, M. A.; Yoon, T. P. Chem. Sci. 2015, 6, 5426–5434. doi:10.1039/C5SC02185E |
| 5. | Nicewicz, D. A.; MacMillan, D. W. C. Science 2008, 322, 77–80. doi:10.1126/science.1161976 |
| 6. | Nagib, D. A.; Scott, M. E.; MacMillan, D. W. C. J. Am. Chem. Soc. 2009, 131, 10875–10877. doi:10.1021/ja9053338 |
| 7. | Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/B913880N |
| 8. | Kalyani, D.; McMurtrey, K. B.; Neufeldt, S. R.; Sanford, M. S. J. Am. Chem. Soc. 2011, 133, 18566–18569. doi:10.1021/ja208068w |
| 9. | Ischay, M. A.; Anzovino, M. E.; Du, J.; Yoon, T. P. J. Am. Chem. Soc. 2008, 130, 12886–12887. doi:10.1021/ja805387f |
| 10. | Yoon, T. P.; Ischay, M. A.; Du, J. Nat. Chem. 2010, 2, 527–532. doi:10.1038/nchem.687 |
| 11. | Du, J.; Skubi, K. L.; Schultz, D. M.; Yoon, T. P. Science 2014, 344, 392–396. doi:10.1126/science.1251511 |
| 12. | Beatty, J. W.; Stephenson, C. R. J. Acc. Chem. Res. 2015, 48, 1474–1484. doi:10.1021/acs.accounts.5b00068 |
| 38. | Panera, M.; Díez, J.; Merino, I.; Rubio, E.; Gamasa, M. P. Inorg. Chem. 2009, 48, 11147–11160. doi:10.1021/ic901527x |
| 2. | Li, C.-J. From C-H to C-C Bonds: Cross-Dehydrogenative-Coupling; RSC Green Chemistry, 2015. |
| 25. | Liu, X.; Sun, S.; Meng, Z.; Lou, H.; Liu, L. Org. Lett. 2015, 17, 2396–2399. doi:10.1021/acs.orglett.5b00909 |
| 28. | Liu, X.; Meng, Z.; Li, C.; Lou, H.; Liu, L. Angew. Chem., Int. Ed. 2015, 54, 6012–6015. doi:10.1002/anie.201500703 |
| 11. | Du, J.; Skubi, K. L.; Schultz, D. M.; Yoon, T. P. Science 2014, 344, 392–396. doi:10.1126/science.1251511 |
| 33. | Condie, A. G.; González-Gómez, J. C.; Stephenson, C. R. J. J. Am. Chem. Soc. 2010, 132, 1464–1465. doi:10.1021/ja909145y |
| 34. | Rueping, M.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C.; Leonori, D.; Vila, C. Chem. – Eur. J. 2012, 18, 5170–5174. doi:10.1002/chem.201200050 |
| 35. | Boess, E.; Schmitz, C.; Klussmann, M. J. Am. Chem. Soc. 2012, 134, 5317–5325. doi:10.1021/ja211697s |
| 36. | Lowry, M. S.; Goldsmith, J. I.; Slinker, J. D.; Rohl, R.; Pascal, R. A., Jr.; Malliaras, G. G.; Bernhard, S. Chem. Mater. 2005, 17, 5712–5719. doi:10.1021/cm051312+ |
| 37. | Ladouceur, S.; Fortin, D.; Zysman-Colman, E. Inorg. Chem. 2011, 50, 11514–11526. doi:10.1021/ic2014013 |
| 1. | He, J.; Li, S.; Deng, Y.; Fu, H.; Laforteza, B. N.; Spangler, J. E.; Homs, A.; Yu, J.-Q. Science 2014, 343, 1216–1220. doi:10.1126/science.1249198 |
| 21. | Barham, J. P.; John, M. P.; Murphy, J. A. Beilstein J. Org. Chem. 2014, 10, 2981–2988. doi:10.3762/bjoc.10.316 |
| 22. | Cuthbertson, J. D.; MacMillan, D. W. C. Nature 2015, 519, 74–77. doi:10.1038/nature14255 |
| 23. | Muramatsu, W.; Nakano, K.; Li, C.-J. Org. Biomol. Chem. 2014, 12, 2189–2192. doi:10.1039/c3ob42354a |
| 24. | Dhineshkumar, J.; Lamani, M.; Alagiri, K.; Prabhu, K. R. Org. Lett. 2013, 15, 1092–1095. doi:10.1021/ol4001153 |
| 25. | Liu, X.; Sun, S.; Meng, Z.; Lou, H.; Liu, L. Org. Lett. 2015, 17, 2396–2399. doi:10.1021/acs.orglett.5b00909 |
| 26. | Li, Z.; Bohle, D. S.; Li, C.-J. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 8928–8933. doi:10.1073/pnas.0601687103 |
| 27. | Yan, C.; Li, L.; Liu, Y.; Wang, Q. Org. Lett. 2016, 18, 4686–4689. doi:10.1021/acs.orglett.6b02326 |
| 28. | Liu, X.; Meng, Z.; Li, C.; Lou, H.; Liu, L. Angew. Chem., Int. Ed. 2015, 54, 6012–6015. doi:10.1002/anie.201500703 |
| 29. | Bergonzini, G.; Schindler, C. S.; Wallentin, C.-J.; Jacobsen, E. N.; Stephenson, C. R. J. Chem. Sci. 2014, 5, 112–116. doi:10.1039/C3SC52265B |
© 2016 Querard et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)