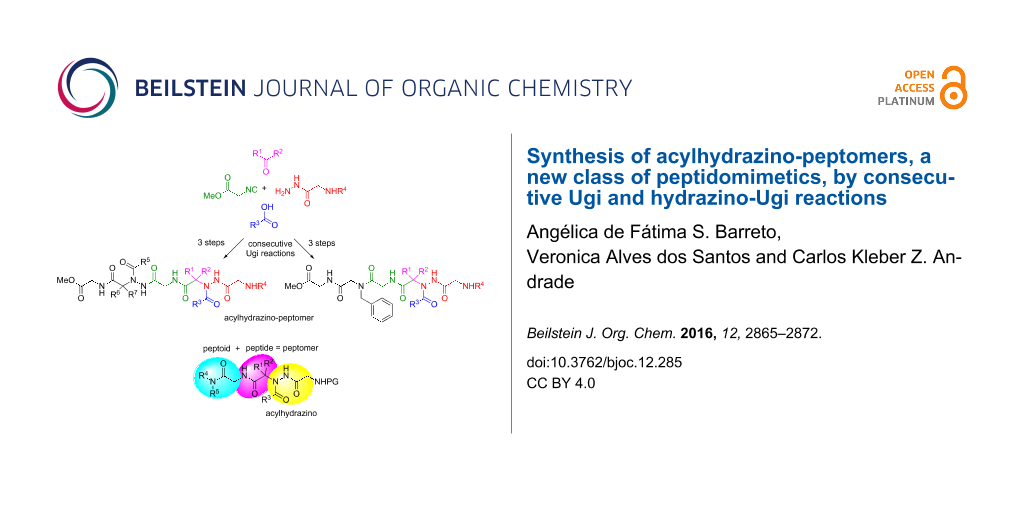
Abstract
Herein we describe a versatile approach for the synthesis of acylhydrazino-peptomers, a new class of peptidomimetics. The key idea in this approach is based on a simple route using a one-pot hydrazino-Ugi four-component reaction followed by a hydrazinolysis or hydrolysis reaction and subsequent hydrazino-Ugi reaction or classical Ugi reaction for the construction of acyclic acylhydrazino-peptomers. The consecutive multicomponent reactions produced a variety of acylhydrazino-peptomers in moderate to excellent yields (47–90%). These compounds are multifunctional intermediates that can be further functionalized to obtain new peptidomimetics with potential biological activity.

Graphical Abstract
Introduction
In the last decades, increasing efforts have been extensively carried out to improve the pharmacological properties of natural peptides by structural modification of the amino acids [1-8]. These modifications allowed the obtention of molecules that mimic the properties of peptides (peptidomimetics) but usually exhibit greater proteolytic stability, increased cellular permeabilities and avoid stereochemical constraints. Figure 1 represents some of the most important classes of peptidomimetics so far obtained and highlights the differences among them. Of these, peptoids (oligomers of N-substituted glycine residues) [9-12] are the most common and may have interesting biological activities. For instance, peptoid 1 is a target for cancer therapeutics for being an antagonist of the vascular endothelial growth factor receptor 2 [13]; peptoid 2 is a ligand of the protooncogene Crk [14]; and peptoids 3 and 4 showed a high affinity for the α1-adrenergic and μ-specific opiate receptors [15], respectively (Figure 2).
![[1860-5397-12-285-1]](/bjoc/content/figures/1860-5397-12-285-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structural features of (a) peptide, (b) peptoid, (c) peptomer, (d) azapeptide, (e) azapeptoid, (f) hydrazinopeptide and (g) hydrazinopeptoid.
Figure 1: Structural features of (a) peptide, (b) peptoid, (c) peptomer, (d) azapeptide, (e) azapeptoid, (f) ...
![[1860-5397-12-285-2]](/bjoc/content/figures/1860-5397-12-285-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Some biologically active peptoids.
Figure 2: Some biologically active peptoids.
Unlike peptides, in peptoids the side chain of the Cα is bound to the nitrogen atom. Due to the consequent lack of the polar N–H bonds, their lipophilicity is increased, which may result in improved membrane permeability [16,17]. Furthermore, peptoids have also found utility in supra- and macromolecular engineering [18] and polymer chemistry [19]. Nevertheless, the lack of constraints in peptoids may result in a lower affinity for macromolecular targets, which limits their utility [20]. This finding has motivated the search for efficient strategies to introduce structural constraints into peptoid structures, which has contributed to increase the plethora of currently available peptidomimetics.
Azapeptides [21-23] are peptide analogs in which the Cα atom is substituted by a nitrogen atom in one or more amino acids (Figure 1) and have been known since 1970 [24]. The introduction of a semicarbazide moiety has an enormous effect on the physical properties of a peptide as well as on its structural characteristics. The semicarbazide constraints tend to facilitate interactions with protein receptors due to turn geometry within the azapeptide. As a result, their stability over enzymes and chemical degradation may be enhanced compared to a natural peptide. Indeed, these compounds have shown to be a useful class of peptidomimetics with interesting biological activities [21-23], including antiviral [25,26] and cysteine protease inhibition [27-30].
Hydrazinopeptides [31-36] (peptide analogs in which one of the CONH links is replaced by a hydrazido fragment CONHNH, Figure 1) represent another class of peptidomimetics with promising conformational and biological activities, such as protease inhibition [37] and antimicrobial activity [38]. There are some natural peptides that contain such an α-hydrazino acid moiety, e.g., the vitamin B6 antagonist linatine and the antibiotic negamycin (Figure 3). In the early 1970s, the first attempts to peptide modifications by hydrazino acids generated bioactive pseudopeptides [39]. Analogously to azapeptides, these compounds possess a conformational constraint (hydrazino turn) due to the presence of intramolecular H-bonding interactions (Figure 3), which is similar in nature to a natural peptide β-turn. As already pointed out, this can improve the proteolytic stability of the natural peptide while preserving its biological activity.
![[1860-5397-12-285-3]](/bjoc/content/figures/1860-5397-12-285-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Biologically active hydrazinopeptides and representation of the hydrazino turn.
Figure 3: Biologically active hydrazinopeptides and representation of the hydrazino turn.
Other changes in peptoid structures have also been carried out to obtain the less common but also biologically active azapeptoids [40], hydrazino-azapeptoids [41,42], retro hydrazino-azapeptoids [43] and peptoid-azapeptoid hybrids [39]. Recently, Seo et al. [44] reported the synthesis of a library of peptide-peptoid hybrid (termed peptomers by Ostergaard and Holm [45]) prodrugs that can be selectively activated by prostate cancer cells.
Peptidomimetics can be conveniently synthesized using the so called "submonomer approach" either in solution [41] or in solid-phase [46,47]. However, some disadvantages have been reported for long or difficult sequences [48]. It is therefore important to have alternative methods for the fast and easy construction of such important compounds. In this sense, the Ugi four-component reaction (U-4CR) has proven to be a robust and versatile method for the synthesis of peptoids and peptide-peptoid hybrids (peptomers) [49-53]. This reaction has also been combined with other protocols for the synthesis of bioactive peptides [54,55] and hydrazinopeptide motifs [33,34]. In continuing our research on the synthesis of peptoids [53,56,57], herein we describe the synthesis of a new class of peptidomimetics, which we have called acylhydrazino-peptomers (Figure 4), by analogy with the existing classes of peptidomimetics shown in Figure 1, using consecutive Ugi and hydrazino-Ugi reactions, respectively. These compounds comprise both a peptoid and a peptide moiety (hence a peptomer) along with an acylhydrazino portion. The use of a consecutive Ugi reaction to access peptidomimetics has proven very useful [56-62]. It is important to point out that these molecules cannot be obtained directly via the “submonomer approach”.
![[1860-5397-12-285-4]](/bjoc/content/figures/1860-5397-12-285-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: General structure of the acylhydrazino-peptomers synthesized in this study.
Figure 4: General structure of the acylhydrazino-peptomers synthesized in this study.
Results and Discussion
Our approach involves the use of two multicomponent reactions (Scheme 1): the hydrazino-Ugi four-component reaction (HU-4CR) and the classical Ugi reaction (U-4CR). The strategy was based on the formation of an acylhydrazino-peptomer via an initial hydrazino-Ugi reaction followed by a hydrazinolysis reaction (or ester hydrolysis) and a subsequent hydrazino-Ugi reaction (or a classical Ugi reaction).
![[1860-5397-12-285-i1]](/bjoc/content/inline/1860-5397-12-285-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
The hydrazides 3a–c used in the first MCR were prepared by the reaction of glycine-derived esters 2, 5 and 7 with hydrazine monohydrate (hydrazinolysis), following a known procedure [63,64] (Scheme 2).
![[1860-5397-12-285-i2]](/bjoc/content/inline/1860-5397-12-285-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of hydrazides 3a–c. Reagents and conditions: (i) CH3I, NaHCO3, DMF, rt, 46 h; (ii) N2H4.H2O, EtOH, reflux, 2–3 h; (iii) (Boc)2O, NaOH, dioxane/H2O, overnight.
Scheme 2: Synthesis of hydrazides 3a–c. Reagents and conditions: (i) CH3I, NaHCO3, DMF, rt, 46 h; (ii) N2H4.H2...
The obtained hydrazides were then reacted with isobutyraldehyde 8a/acetone 8b, carboxylic acids 10a–c (formic, acetic or propionic acid) and ethyl isocyanoacetate 9 (Scheme 3). The reactions were conducted at room temperature in trifluoroethanol (TFE) for 1–2 days to yield the acylhydrazino-peptomers 11a–f in moderate to good yields (59–90%). Aliphatic aldehydes, ketones and carboxylic acids succeeded in the hydrazino-Ugi reaction, except for paraformaldehyde, fatty acids and aromatic substrates, which led to formation of a complex mixture. These results limited the obtention of a greater variety of acylhydrazino-peptomers by this method. The choice of TFE as the solvent for the hydrazino-Ugi reaction was important because the same reaction carried out in methanol, often the solvent of choice for Ugi reactions, provided the formation of a complex mixture. This fact has already been reported [34].
![[1860-5397-12-285-i3]](/bjoc/content/inline/1860-5397-12-285-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of acylhydrazino-peptomers 11a–f.
Scheme 3: Synthesis of acylhydrazino-peptomers 11a–f.
To further functionalize the acylhydrazino-peptomers structures, a second Ugi reaction was carried out. Hence, some acylhydrazino-peptomers were subjected to hydrazinolysis reaction or ester hydrolysis to give the corresponding hydrazides 12a,b (Scheme 4) or acids 13a–c (Scheme 5), respectively, which were used in the following step (hydrazino-Ugi reaction or classical Ugi reaction) to yield the corresponding acylhydrazino-peptomers 14a,b or 15a–c in moderate to good yields (47–90%). All compounds were fully characterized by 1H and 13C NMR and HRMS giving data consistent with the proposed structures.
![[1860-5397-12-285-i4]](/bjoc/content/inline/1860-5397-12-285-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of acylhydrazino-peptomers 14a,b.
Scheme 4: Synthesis of acylhydrazino-peptomers 14a,b.
![[1860-5397-12-285-i5]](/bjoc/content/inline/1860-5397-12-285-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of acylhydrazino-peptomers 15a–c.
Scheme 5: Synthesis of acylhydrazino-peptomers 15a–c.
Functionalization on both termini of compounds 11, 14 and 15 (ester hydrolysis and/or Boc deprotection) allows subsequent Ugi or hydrazino-Ugi reactions to further elongate the peptomers main chain.
Conclusion
In summary, we have developed a concise protocol for the synthesis of functionalized acylhydrazino-peptomers by consecutive Ugi reactions. The general route allows an easy access to highly functionalized peptomers in good yields and a reduced number of steps. This method may also be employed to obtain new classes of peptidomimetics with potential biological activity.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures, NMR and mass spectra of all compounds. | ||
| Format: PDF | Size: 1.9 MB | Download |
References
-
Zuckermann, R. N.; Kodadek, T. Curr. Opin. Mol. Ther. 2009, 11, 299–307.
Return to citation in text: [1] -
Simpson, L. S.; Burdine, L.; Dutta, A. K.; Feranchak, A. P.; Kodadek, T. J. Am. Chem. Soc. 2009, 131, 5760–5762. doi:10.1021/ja900852k
Return to citation in text: [1] -
Mora, P.; Masip, I.; Cortés, N.; Marquina, R.; Merino, R.; Merino, J.; Carbonell, T.; Mingarro, I.; Messeguer, A.; Pérez-Payá, E. J. Med. Chem. 2005, 48, 1265–1268. doi:10.1021/jm040834i
Return to citation in text: [1] -
Fowler, S. A.; Stacy, D. M.; Blackwell, H. E. Org. Lett. 2008, 10, 2329–2332. doi:10.1021/ol800908h
Return to citation in text: [1] -
Wenger, R. M.; Payne, T. Prog. Clin. Biol. Res. 1989, 291, 301–305.
Return to citation in text: [1] -
Hamy, F.; Felder, E. R.; Heizmann, G.; Lazdins, J.; Aboul-ela, F.; Varani, G.; Karn, J.; Klimkait, T. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 3548–3553. doi:10.1073/pnas.94.8.3548
Return to citation in text: [1] -
Stawikowski, M.; Stawikowska, R.; Jaśkiewicz, A.; Zabłotna, E.; Rolka, K. ChemBioChem 2005, 6, 1057–1061. doi:10.1002/cbic.200400412
Return to citation in text: [1] -
Panda, S. S.; El-Nachef, C.; Bajaj, K.; Katritzky, A. R. Eur. J. Org. Chem. 2013, 4156–4162. doi:10.1002/ejoc.201201731
Return to citation in text: [1] -
Kessler, H. Angew. Chem., Int. Ed. Engl. 1993, 32, 543–544. doi:10.1002/anie.199305431
Return to citation in text: [1] -
Yoo, B.; Kirshenbaum, K. Curr. Opin. Chem. Biol. 2008, 12, 714–721. doi:10.1016/j.cbpa.2008.08.015
Return to citation in text: [1] -
Zuckermann, R. N. Pept. Sci. 2011, 96, 545–555. doi:10.1002/bip.21573
Return to citation in text: [1] -
Simon, R. J.; Kania, R. S.; Zuckermann, R. N.; Huebner, V. D.; Jewell, D. A.; Banville, S.; Ng, S.; Wang, L.; Rosenberg, S.; Marlowe, C. K.; Spellmeyer, D. C.; Tan, R.; Frankel, A. D.; Santi, D. V.; Cohen, F. E.; Bartlett, P. A. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 9367–9371. doi:10.1073/pnas.89.20.9367
Return to citation in text: [1] -
Udugamasooriya, D. G.; Dineen, S. P.; Brekken, R. A.; Kodadek, T. J. Am. Chem. Soc. 2008, 130, 5744–5752. doi:10.1021/ja711193x
Return to citation in text: [1] -
Wrenn, S. J.; Weisinger, R. M.; Halpin, D. R.; Harbury, P. B. J. Am. Chem. Soc. 2007, 129, 13137–13143. doi:10.1021/ja073993a
Return to citation in text: [1] -
Zuckermann, R. N.; Martin, E. J.; Spellmeyer, D. C.; Stauber, G. B.; Shoemaker, K. R.; Kerr, J. M.; Figliozzi, G. M.; Goff, D. A.; Siani, M. A.; Simon, R. J.; Banville, S. C.; Brown, E. G.; Wang, L.; Richter, L. S.; Moss, W. H. J. Med. Chem. 1994, 37, 2678–2685. doi:10.1021/jm00043a007
Return to citation in text: [1] -
Rezai, T.; Bock, J. E.; Zhou, M. V.; Kalyanaraman, C.; Lokey, R. S.; Jacobson, M. P. J. Am. Chem. Soc. 2006, 128, 14073–14080. doi:10.1021/ja063076p
Return to citation in text: [1] -
Ovadia, O.; Linde, Y.; Haskell-Luevano, C.; Dirain, M. L.; Sheynis, T.; Jelinek, R.; Gilon, C.; Hoffman, A. Bioorg. Med. Chem. 2010, 18, 580–589. doi:10.1016/j.bmc.2009.12.010
Return to citation in text: [1] -
Gangloff, N.; Ulbricht, J.; Lorson, T.; Schlaad, H.; Luxenhofer, R. Chem. Rev. 2016, 116, 1753–1802. doi:10.1021/acs.chemrev.5b00201
Return to citation in text: [1] -
Sun, J.; Zuckermann, R. N. ACS Nano 2013, 7, 4715–4732. doi:10.1021/nn4015714
Return to citation in text: [1] -
Sarma, B. K.; Kodadek, T. ACS Comb. Sci. 2012, 14, 558–564. doi:10.1021/co3000852
Return to citation in text: [1] -
Proulx, C.; Sabatino, D.; Hopewell, R.; Spiegel, J.; Ramos, Y. G.; Lubell, W. D. Future Med. Chem. 2011, 3, 1139–1164. doi:10.4155/fmc.11.74
Return to citation in text: [1] [2] -
Zega, A. Curr. Med. Chem. 2005, 12, 589–597. doi:10.2174/0929867053362802
Return to citation in text: [1] [2] -
Gante, J. Synthesis 1989, 405–413. doi:10.1055/s-1989-27269
Return to citation in text: [1] [2] -
Gante, J. Angew. Chem., Int. Ed. Engl. 1970, 9, 813. doi:10.1002/anie.197008131
Return to citation in text: [1] -
Bailey, M. D.; Halmos, T.; Goudreau, N.; Lescop, E.; Llinàs-Brunet, M. J. Med. Chem. 2004, 47, 3788–3799. doi:10.1021/jm049864b
Return to citation in text: [1] -
Randolph, J. T.; Zhang, X.; Huang, P. P.; Klein, L. L.; Kurtz, K. A.; Konstantinidis, A. K.; He, W.; Kati, W. M.; Kempf, D. J. Bioorg. Med. Chem. Lett. 2008, 18, 2745–2750. doi:10.1016/j.bmcl.2008.02.053
Return to citation in text: [1] -
Magrath, J.; Abeles, R. H. J. Med. Chem. 1992, 35, 4279–4283. doi:10.1021/jm00101a004
Return to citation in text: [1] -
Baggio, R.; Shi, Y.-Q.; Wu, Y.-q.; Abeles, R. H. Biochemistry 1996, 35, 3351–3353. doi:10.1021/bi952879e
Return to citation in text: [1] -
Xing, R.; Hanzlik, R. P. J. Med. Chem. 1998, 41, 1344–1351. doi:10.1021/jm970802d
Return to citation in text: [1] -
Ekici, Ö. D.; Li, Z. Z.; Campbell, A. J.; James, K. E.; Asgian, J. L.; Mikolajczyk, J.; Salvesen, G. S.; Ganesan, R.; Jelakovic, S.; Grütter, M. G.; Powers, J. C. J. Med. Chem. 2006, 49, 5728–5749. doi:10.1021/jm0601405
Return to citation in text: [1] -
Guy, L.; Vidal, J.; Collet, A.; Amour, A.; Reboud-Ravaux, M. J. Med. Chem. 1998, 41, 4833–4843. doi:10.1021/jm980419o
Return to citation in text: [1] -
Amour, A.; Collet, A.; Dubar, C.; Reboud-Ravaux, M. Int. J. Pept. Protein Res. 1994, 43, 297–304. doi:10.1111/j.1399-3011.1994.tb00394.x
Return to citation in text: [1] -
Bushkova, E.; Parchinsky, V.; Krasavin, M. Mol. Diversity 2010, 14, 493–499. doi:10.1007/s11030-009-9200-6
Return to citation in text: [1] [2] -
Krasavin, M.; Bushkova, E.; Parchinsky, V.; Shumsky, A. Synthesis 2010, 933–942. doi:10.1055/s-0029-1219274
Return to citation in text: [1] [2] [3] -
Lakontseva, E.; Krasavin, M. Tetrahedron Lett. 2010, 51, 4095–4099. doi:10.1016/j.tetlet.2010.05.133
Return to citation in text: [1] -
Bonnet, D.; Samson, F.; Rommens, C.; Gras-masse, H.; Melnyk, O. J. Pept. Res. 1999, 54, 270–278. doi:10.1034/j.1399-3011.1999.00105.x
Return to citation in text: [1] -
Bordessa, A.; Keita, M.; Maréchal, X.; Formicola, L.; Lagarde, N.; Rodrigo, J.; Bernadat, G.; Bauvais, C.; Soulier, J.-L.; Dufau, L.; Milcent, T.; Crousse, B.; Reboud-Ravaux, M.; Ongeri, S. Eur. J. Med. Chem. 2013, 70, 505–524. doi:10.1016/j.ejmech.2013.09.059
Return to citation in text: [1] -
Laurencin, M.; Legrand, B.; Duval, E.; Henry, J.; Baudy-Floc'h, M.; Zatylny-Gaudin, C.; Bondon, A. J. Med. Chem. 2012, 55, 2025–2034. doi:10.1021/jm2011595
Return to citation in text: [1] -
Niedrich, H.; Köller, G. J. Prakt. Chem. 1974, 316, 729–740. doi:10.1002/prac.19743160504
Return to citation in text: [1] [2] -
Sarma, B. K.; Liu, X.; Kodadek, T. Bioorg. Med. Chem. 2016, 24, 3953–3963. doi:10.1016/j.bmc.2016.04.047
Return to citation in text: [1] -
Cheguillaume, A.; Lehardy, F.; Bouget, K.; Baudy-Floc'h, M.; Le Grel, P. J. Org. Chem. 1999, 64, 2924–2927. doi:10.1021/jo981487l
Return to citation in text: [1] [2] -
Bouget, K.; Aubin, S.; Delcros, J.-G.; Arlot-Bonnemains, Y.; Baudy-Floc'h, M. Bioorg. Med. Chem. 2003, 11, 4881–4889. doi:10.1016/j.bmc.2003.09.018
Return to citation in text: [1] -
Aubin, S.; Martin, B.; Delcros, J.-G.; Arlot-Bonnemains, Y.; Baudy-Floc'h, M. J. Med. Chem. 2005, 48, 330–334. doi:10.1021/jm049455f
Return to citation in text: [1] -
Lee, J.; Huang, W.; Broering, J. M.; Barron, A. E.; Seo, J. Bioorg. Med. Chem. Lett. 2015, 25, 2849–2852. doi:10.1016/j.bmcl.2015.04.092
Return to citation in text: [1] -
Østergaard, S.; Holm, A. Mol. Diversity 1997, 3, 17–27. doi:10.1023/A:1009698507588
Return to citation in text: [1] -
Zuckermann, R. N.; Kerr, J. M.; Kent, S. B. H.; Moos, W. H. J. Am. Chem. Soc. 1992, 114, 10646–10647. doi:10.1021/ja00052a076
Return to citation in text: [1] -
Sarma, B. K.; Yousufuddin, M.; Kodadek, T. Chem. Commun. 2011, 47, 10590–10592. doi:10.1039/c1cc12750k
Return to citation in text: [1] -
Fara, M. A.; Diaz-Mochón, J. J.; Bradley, M. Tetrahedron Lett. 2006, 47, 1011–1014. doi:10.1016/j.tetlet.2005.11.127
Return to citation in text: [1] -
Dömling, A.; Ugi, I. Angew. Chem., Int. Ed. 2000, 39, 3168–3210. doi:10.1002/1521-3773(20000915)39:18<3168::AID-ANIE3168>3.0.CO;2-U
Return to citation in text: [1] -
Wessjohann, L. A.; Rivera, D. G.; Vercillo, O. E. Chem. Rev. 2009, 109, 796–814. doi:10.1021/cr8003407
Return to citation in text: [1] -
Dömling, A. Chem. Rev. 2006, 106, 17–89. doi:10.1021/cr0505728
Return to citation in text: [1] -
Dömling, A.; Beck, B.; Eichelberger, U.; Sakamuri, S.; Menon, S.; Chen, Q.-Z.; Lu, Y.; Wessjohann, L. A. Angew. Chem., Int. Ed. 2006, 45, 7235–7239. doi:10.1002/anie.200601259
Return to citation in text: [1] -
Barreto, A. F. S.; Vercillo, O. E.; Birkett, M. A.; Caulfied, J. C.; Wessjohann, L. A.; Andrade, C. K. Z. Org. Biomol. Chem. 2011, 9, 5024–5027. doi:10.1039/c1ob05471f
Return to citation in text: [1] [2] -
Pando, O.; Stark, S.; Denkert, A.; Porzel, A.; Preusentanz, R.; Wessjohann, L. A. J. Am. Chem. Soc. 2011, 133, 7692–7695. doi:10.1021/ja2022027
Return to citation in text: [1] -
Neves Filho, R. A. W.; Westermann, B.; Wessjohann, L. A. Beilstein J. Org. Chem. 2011, 7, 1504–1507. doi:10.3762/bjoc.7.175
Return to citation in text: [1] -
Vercillo, O. E.; Andrade, C. K. Z.; Wessjohann, L. A. Org. Lett. 2008, 10, 205–208. doi:10.1021/ol702521g
Return to citation in text: [1] [2] -
Barreto, A. F. S.; Vercillo, O. E.; Wessjohann, L. A.; Andrade, C. K. Z. Beilstein J. Org. Chem. 2014, 10, 1017–1022. doi:10.3762/bjoc.10.101
Return to citation in text: [1] [2] -
Constabel, F.; Ugi, I. Tetrahedron 2001, 57, 5785–5789. doi:10.1016/S0040-4020(01)00516-6
Return to citation in text: [1] -
Dömling, A. Nucleosides Nucleotides 1998, 17, 1667–1670. doi:10.1080/07328319808004699
Return to citation in text: [1] -
Xu, P.; Zhang, T.; Wang, W.; Zou, X.; Zhang, X.; Fu, Y. Synthesis 2003, 1171–1176. doi:10.1055/s-2003-39391
Return to citation in text: [1] -
Zarganes-Tzitzikas, T.; Patil, P.; Khoury, K.; Herdtweck, E.; Domling, A. Eur. J. Org. Chem. 2015, 51–55. doi:10.1002/ejoc.201403401
Return to citation in text: [1] -
Brauch, S.; Gabriel, L.; Westermann, B. Chem. Commun. 2010, 46, 3387–3389. doi:10.1039/b927388c
Return to citation in text: [1] -
Yale, H. L.; Losee, K.; Martins, J.; Holsing, M.; Perry, F. M.; Bernstein, J. J. Am. Chem. Soc. 1953, 75, 1933–1942. doi:10.1021/ja01104a046
Return to citation in text: [1] -
Bruice, T. C.; Benkovic, S. J. J. Am. Chem. Soc. 1964, 86, 418–426. doi:10.1021/ja01057a026
Return to citation in text: [1]
| 49. | Dömling, A.; Ugi, I. Angew. Chem., Int. Ed. 2000, 39, 3168–3210. doi:10.1002/1521-3773(20000915)39:18<3168::AID-ANIE3168>3.0.CO;2-U |
| 50. | Wessjohann, L. A.; Rivera, D. G.; Vercillo, O. E. Chem. Rev. 2009, 109, 796–814. doi:10.1021/cr8003407 |
| 51. | Dömling, A. Chem. Rev. 2006, 106, 17–89. doi:10.1021/cr0505728 |
| 52. | Dömling, A.; Beck, B.; Eichelberger, U.; Sakamuri, S.; Menon, S.; Chen, Q.-Z.; Lu, Y.; Wessjohann, L. A. Angew. Chem., Int. Ed. 2006, 45, 7235–7239. doi:10.1002/anie.200601259 |
| 53. | Barreto, A. F. S.; Vercillo, O. E.; Birkett, M. A.; Caulfied, J. C.; Wessjohann, L. A.; Andrade, C. K. Z. Org. Biomol. Chem. 2011, 9, 5024–5027. doi:10.1039/c1ob05471f |
| 54. | Pando, O.; Stark, S.; Denkert, A.; Porzel, A.; Preusentanz, R.; Wessjohann, L. A. J. Am. Chem. Soc. 2011, 133, 7692–7695. doi:10.1021/ja2022027 |
| 55. | Neves Filho, R. A. W.; Westermann, B.; Wessjohann, L. A. Beilstein J. Org. Chem. 2011, 7, 1504–1507. doi:10.3762/bjoc.7.175 |
| 33. | Bushkova, E.; Parchinsky, V.; Krasavin, M. Mol. Diversity 2010, 14, 493–499. doi:10.1007/s11030-009-9200-6 |
| 34. | Krasavin, M.; Bushkova, E.; Parchinsky, V.; Shumsky, A. Synthesis 2010, 933–942. doi:10.1055/s-0029-1219274 |
| 1. | Zuckermann, R. N.; Kodadek, T. Curr. Opin. Mol. Ther. 2009, 11, 299–307. |
| 2. | Simpson, L. S.; Burdine, L.; Dutta, A. K.; Feranchak, A. P.; Kodadek, T. J. Am. Chem. Soc. 2009, 131, 5760–5762. doi:10.1021/ja900852k |
| 3. | Mora, P.; Masip, I.; Cortés, N.; Marquina, R.; Merino, R.; Merino, J.; Carbonell, T.; Mingarro, I.; Messeguer, A.; Pérez-Payá, E. J. Med. Chem. 2005, 48, 1265–1268. doi:10.1021/jm040834i |
| 4. | Fowler, S. A.; Stacy, D. M.; Blackwell, H. E. Org. Lett. 2008, 10, 2329–2332. doi:10.1021/ol800908h |
| 5. | Wenger, R. M.; Payne, T. Prog. Clin. Biol. Res. 1989, 291, 301–305. |
| 6. | Hamy, F.; Felder, E. R.; Heizmann, G.; Lazdins, J.; Aboul-ela, F.; Varani, G.; Karn, J.; Klimkait, T. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 3548–3553. doi:10.1073/pnas.94.8.3548 |
| 7. | Stawikowski, M.; Stawikowska, R.; Jaśkiewicz, A.; Zabłotna, E.; Rolka, K. ChemBioChem 2005, 6, 1057–1061. doi:10.1002/cbic.200400412 |
| 8. | Panda, S. S.; El-Nachef, C.; Bajaj, K.; Katritzky, A. R. Eur. J. Org. Chem. 2013, 4156–4162. doi:10.1002/ejoc.201201731 |
| 15. | Zuckermann, R. N.; Martin, E. J.; Spellmeyer, D. C.; Stauber, G. B.; Shoemaker, K. R.; Kerr, J. M.; Figliozzi, G. M.; Goff, D. A.; Siani, M. A.; Simon, R. J.; Banville, S. C.; Brown, E. G.; Wang, L.; Richter, L. S.; Moss, W. H. J. Med. Chem. 1994, 37, 2678–2685. doi:10.1021/jm00043a007 |
| 31. | Guy, L.; Vidal, J.; Collet, A.; Amour, A.; Reboud-Ravaux, M. J. Med. Chem. 1998, 41, 4833–4843. doi:10.1021/jm980419o |
| 32. | Amour, A.; Collet, A.; Dubar, C.; Reboud-Ravaux, M. Int. J. Pept. Protein Res. 1994, 43, 297–304. doi:10.1111/j.1399-3011.1994.tb00394.x |
| 33. | Bushkova, E.; Parchinsky, V.; Krasavin, M. Mol. Diversity 2010, 14, 493–499. doi:10.1007/s11030-009-9200-6 |
| 34. | Krasavin, M.; Bushkova, E.; Parchinsky, V.; Shumsky, A. Synthesis 2010, 933–942. doi:10.1055/s-0029-1219274 |
| 35. | Lakontseva, E.; Krasavin, M. Tetrahedron Lett. 2010, 51, 4095–4099. doi:10.1016/j.tetlet.2010.05.133 |
| 36. | Bonnet, D.; Samson, F.; Rommens, C.; Gras-masse, H.; Melnyk, O. J. Pept. Res. 1999, 54, 270–278. doi:10.1034/j.1399-3011.1999.00105.x |
| 14. | Wrenn, S. J.; Weisinger, R. M.; Halpin, D. R.; Harbury, P. B. J. Am. Chem. Soc. 2007, 129, 13137–13143. doi:10.1021/ja073993a |
| 37. | Bordessa, A.; Keita, M.; Maréchal, X.; Formicola, L.; Lagarde, N.; Rodrigo, J.; Bernadat, G.; Bauvais, C.; Soulier, J.-L.; Dufau, L.; Milcent, T.; Crousse, B.; Reboud-Ravaux, M.; Ongeri, S. Eur. J. Med. Chem. 2013, 70, 505–524. doi:10.1016/j.ejmech.2013.09.059 |
| 13. | Udugamasooriya, D. G.; Dineen, S. P.; Brekken, R. A.; Kodadek, T. J. Am. Chem. Soc. 2008, 130, 5744–5752. doi:10.1021/ja711193x |
| 25. | Bailey, M. D.; Halmos, T.; Goudreau, N.; Lescop, E.; Llinàs-Brunet, M. J. Med. Chem. 2004, 47, 3788–3799. doi:10.1021/jm049864b |
| 26. | Randolph, J. T.; Zhang, X.; Huang, P. P.; Klein, L. L.; Kurtz, K. A.; Konstantinidis, A. K.; He, W.; Kati, W. M.; Kempf, D. J. Bioorg. Med. Chem. Lett. 2008, 18, 2745–2750. doi:10.1016/j.bmcl.2008.02.053 |
| 9. | Kessler, H. Angew. Chem., Int. Ed. Engl. 1993, 32, 543–544. doi:10.1002/anie.199305431 |
| 10. | Yoo, B.; Kirshenbaum, K. Curr. Opin. Chem. Biol. 2008, 12, 714–721. doi:10.1016/j.cbpa.2008.08.015 |
| 11. | Zuckermann, R. N. Pept. Sci. 2011, 96, 545–555. doi:10.1002/bip.21573 |
| 12. | Simon, R. J.; Kania, R. S.; Zuckermann, R. N.; Huebner, V. D.; Jewell, D. A.; Banville, S.; Ng, S.; Wang, L.; Rosenberg, S.; Marlowe, C. K.; Spellmeyer, D. C.; Tan, R.; Frankel, A. D.; Santi, D. V.; Cohen, F. E.; Bartlett, P. A. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 9367–9371. doi:10.1073/pnas.89.20.9367 |
| 27. | Magrath, J.; Abeles, R. H. J. Med. Chem. 1992, 35, 4279–4283. doi:10.1021/jm00101a004 |
| 28. | Baggio, R.; Shi, Y.-Q.; Wu, Y.-q.; Abeles, R. H. Biochemistry 1996, 35, 3351–3353. doi:10.1021/bi952879e |
| 29. | Xing, R.; Hanzlik, R. P. J. Med. Chem. 1998, 41, 1344–1351. doi:10.1021/jm970802d |
| 30. | Ekici, Ö. D.; Li, Z. Z.; Campbell, A. J.; James, K. E.; Asgian, J. L.; Mikolajczyk, J.; Salvesen, G. S.; Ganesan, R.; Jelakovic, S.; Grütter, M. G.; Powers, J. C. J. Med. Chem. 2006, 49, 5728–5749. doi:10.1021/jm0601405 |
| 20. | Sarma, B. K.; Kodadek, T. ACS Comb. Sci. 2012, 14, 558–564. doi:10.1021/co3000852 |
| 24. | Gante, J. Angew. Chem., Int. Ed. Engl. 1970, 9, 813. doi:10.1002/anie.197008131 |
| 63. | Yale, H. L.; Losee, K.; Martins, J.; Holsing, M.; Perry, F. M.; Bernstein, J. J. Am. Chem. Soc. 1953, 75, 1933–1942. doi:10.1021/ja01104a046 |
| 64. | Bruice, T. C.; Benkovic, S. J. J. Am. Chem. Soc. 1964, 86, 418–426. doi:10.1021/ja01057a026 |
| 19. | Sun, J.; Zuckermann, R. N. ACS Nano 2013, 7, 4715–4732. doi:10.1021/nn4015714 |
| 21. | Proulx, C.; Sabatino, D.; Hopewell, R.; Spiegel, J.; Ramos, Y. G.; Lubell, W. D. Future Med. Chem. 2011, 3, 1139–1164. doi:10.4155/fmc.11.74 |
| 22. | Zega, A. Curr. Med. Chem. 2005, 12, 589–597. doi:10.2174/0929867053362802 |
| 23. | Gante, J. Synthesis 1989, 405–413. doi:10.1055/s-1989-27269 |
| 34. | Krasavin, M.; Bushkova, E.; Parchinsky, V.; Shumsky, A. Synthesis 2010, 933–942. doi:10.1055/s-0029-1219274 |
| 18. | Gangloff, N.; Ulbricht, J.; Lorson, T.; Schlaad, H.; Luxenhofer, R. Chem. Rev. 2016, 116, 1753–1802. doi:10.1021/acs.chemrev.5b00201 |
| 53. | Barreto, A. F. S.; Vercillo, O. E.; Birkett, M. A.; Caulfied, J. C.; Wessjohann, L. A.; Andrade, C. K. Z. Org. Biomol. Chem. 2011, 9, 5024–5027. doi:10.1039/c1ob05471f |
| 56. | Vercillo, O. E.; Andrade, C. K. Z.; Wessjohann, L. A. Org. Lett. 2008, 10, 205–208. doi:10.1021/ol702521g |
| 57. | Barreto, A. F. S.; Vercillo, O. E.; Wessjohann, L. A.; Andrade, C. K. Z. Beilstein J. Org. Chem. 2014, 10, 1017–1022. doi:10.3762/bjoc.10.101 |
| 16. | Rezai, T.; Bock, J. E.; Zhou, M. V.; Kalyanaraman, C.; Lokey, R. S.; Jacobson, M. P. J. Am. Chem. Soc. 2006, 128, 14073–14080. doi:10.1021/ja063076p |
| 17. | Ovadia, O.; Linde, Y.; Haskell-Luevano, C.; Dirain, M. L.; Sheynis, T.; Jelinek, R.; Gilon, C.; Hoffman, A. Bioorg. Med. Chem. 2010, 18, 580–589. doi:10.1016/j.bmc.2009.12.010 |
| 21. | Proulx, C.; Sabatino, D.; Hopewell, R.; Spiegel, J.; Ramos, Y. G.; Lubell, W. D. Future Med. Chem. 2011, 3, 1139–1164. doi:10.4155/fmc.11.74 |
| 22. | Zega, A. Curr. Med. Chem. 2005, 12, 589–597. doi:10.2174/0929867053362802 |
| 23. | Gante, J. Synthesis 1989, 405–413. doi:10.1055/s-1989-27269 |
| 56. | Vercillo, O. E.; Andrade, C. K. Z.; Wessjohann, L. A. Org. Lett. 2008, 10, 205–208. doi:10.1021/ol702521g |
| 57. | Barreto, A. F. S.; Vercillo, O. E.; Wessjohann, L. A.; Andrade, C. K. Z. Beilstein J. Org. Chem. 2014, 10, 1017–1022. doi:10.3762/bjoc.10.101 |
| 58. | Constabel, F.; Ugi, I. Tetrahedron 2001, 57, 5785–5789. doi:10.1016/S0040-4020(01)00516-6 |
| 59. | Dömling, A. Nucleosides Nucleotides 1998, 17, 1667–1670. doi:10.1080/07328319808004699 |
| 60. | Xu, P.; Zhang, T.; Wang, W.; Zou, X.; Zhang, X.; Fu, Y. Synthesis 2003, 1171–1176. doi:10.1055/s-2003-39391 |
| 61. | Zarganes-Tzitzikas, T.; Patil, P.; Khoury, K.; Herdtweck, E.; Domling, A. Eur. J. Org. Chem. 2015, 51–55. doi:10.1002/ejoc.201403401 |
| 62. | Brauch, S.; Gabriel, L.; Westermann, B. Chem. Commun. 2010, 46, 3387–3389. doi:10.1039/b927388c |
| 40. | Sarma, B. K.; Liu, X.; Kodadek, T. Bioorg. Med. Chem. 2016, 24, 3953–3963. doi:10.1016/j.bmc.2016.04.047 |
| 38. | Laurencin, M.; Legrand, B.; Duval, E.; Henry, J.; Baudy-Floc'h, M.; Zatylny-Gaudin, C.; Bondon, A. J. Med. Chem. 2012, 55, 2025–2034. doi:10.1021/jm2011595 |
| 39. | Niedrich, H.; Köller, G. J. Prakt. Chem. 1974, 316, 729–740. doi:10.1002/prac.19743160504 |
| 46. | Zuckermann, R. N.; Kerr, J. M.; Kent, S. B. H.; Moos, W. H. J. Am. Chem. Soc. 1992, 114, 10646–10647. doi:10.1021/ja00052a076 |
| 47. | Sarma, B. K.; Yousufuddin, M.; Kodadek, T. Chem. Commun. 2011, 47, 10590–10592. doi:10.1039/c1cc12750k |
| 48. | Fara, M. A.; Diaz-Mochón, J. J.; Bradley, M. Tetrahedron Lett. 2006, 47, 1011–1014. doi:10.1016/j.tetlet.2005.11.127 |
| 45. | Østergaard, S.; Holm, A. Mol. Diversity 1997, 3, 17–27. doi:10.1023/A:1009698507588 |
| 41. | Cheguillaume, A.; Lehardy, F.; Bouget, K.; Baudy-Floc'h, M.; Le Grel, P. J. Org. Chem. 1999, 64, 2924–2927. doi:10.1021/jo981487l |
| 39. | Niedrich, H.; Köller, G. J. Prakt. Chem. 1974, 316, 729–740. doi:10.1002/prac.19743160504 |
| 44. | Lee, J.; Huang, W.; Broering, J. M.; Barron, A. E.; Seo, J. Bioorg. Med. Chem. Lett. 2015, 25, 2849–2852. doi:10.1016/j.bmcl.2015.04.092 |
| 41. | Cheguillaume, A.; Lehardy, F.; Bouget, K.; Baudy-Floc'h, M.; Le Grel, P. J. Org. Chem. 1999, 64, 2924–2927. doi:10.1021/jo981487l |
| 42. | Bouget, K.; Aubin, S.; Delcros, J.-G.; Arlot-Bonnemains, Y.; Baudy-Floc'h, M. Bioorg. Med. Chem. 2003, 11, 4881–4889. doi:10.1016/j.bmc.2003.09.018 |
| 43. | Aubin, S.; Martin, B.; Delcros, J.-G.; Arlot-Bonnemains, Y.; Baudy-Floc'h, M. J. Med. Chem. 2005, 48, 330–334. doi:10.1021/jm049455f |
© 2016 Barreto et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)