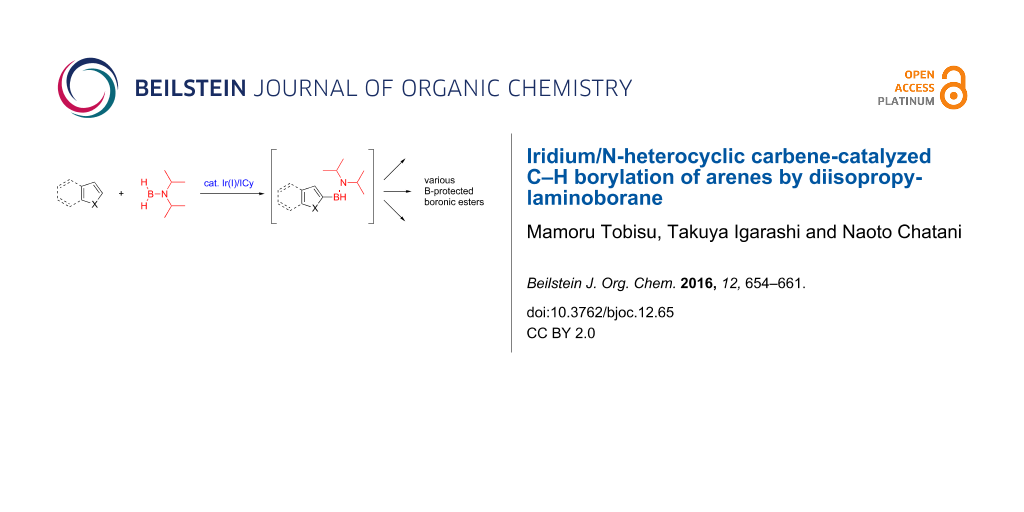
Abstract
Catalytic C–H borylation of arenes has been widely used in organic synthesis because it allows the introduction of a versatile boron functionality directly onto simple, unfunctionalized arenes. We report herein the use of diisopropylaminoborane as a boron source in C–H borylation of arenes. An iridium(I) complex with 1,3-dicyclohexylimidazol-2-ylidene is found to efficiently catalyze the borylation of arenes and heteroarenes. The resulting aminoborylated products can be converted to the corresponding boronic acid derivatives simply by treatment with suitable diols or diamines.

Graphical Abstract
Introduction
Catalytic C–H borylation of arenes has become an essential tool in organic synthesis [1]. The eminent features of this methodology include 1) no directing group is needed, allowing the direct functionalization of simple arenes; 2) the regioselectivity is readily predictable based on steric factors; 3) the resulting boryl group is versatile and can be converted into a variety of carbon- or heteroatom-based substituents. An iridium complex in conjunction with 4,4’-di-tert-butylbipyridine (dtbpy) developed by Ishiyama, Miyaura and Hartwig has served as the state-of-the-art catalyst for C–H borylation of arenes [2]. In addition to the Ir/dtbpy system, various other catalytic systems have also been developed. For example, base metals such as Fe [3-6], Co [7] and Ni [8,9] have been shown to be viable metal centers for the use as C–H borylation catalysts. We also reported the first use of a Pt catalyst that enables C–H borylation of simple hindered arenes such as mesitylene [10,11]. Metal-free C–H borylation has also been reported [12]. Ligand modification has been used to control the regioselectivity of C–H borylation reactions; for example, in the ortho-selective C–H borylation of arenes containing a directing group [13], and to improve the meta/para-selectivity of monosubstituted benzenes [14,15]. Despite considerable progress in the C–H borylation reaction, the scope of the boryl group that can be introduced is relatively limited (Scheme 1). The most commonly used boron sources are pinacolborane (HBpin, 1a) and bis(pinacolato)diboron (B2pin2, 1b), which form pinacol esters of arylboronic acids. Although the pinacol ester products prepared in these reactions are amenable to a range of transformations, their reactivity is generally lower than that of the corresponding boronic acids. Because of this lower reactivity, several transformations require deprotection of a pinacol ester under oxidative conditions (e.g., NaIO4) [16]. Hartwig reported that the Ir/dtbpy system is also able to introduce more reactive neopentyl and hexylene glycolate esters and catecolates using the corresponding diboron reagents 1c, 1d, and 1e, respectively [17,18]. Suginome reported that an Ir/1,2-diphenylphosphinoethane catalyst can promote C–H borylation using 1,8-naphthalenediaminatoborane (1f) [19]. The resulting diaminonaphthalene (dan)-protected arylboronic acid is synthetically useful because it can be readily deprotected with a dilute aqueous acid [20].
![[1860-5397-12-65-i1]](/bjoc/content/inline/1860-5397-12-65-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Catalytic C–H borylation of arenes and related reported boron sources.
Scheme 1: Catalytic C–H borylation of arenes and related reported boron sources.
We envisioned that diisopropylaminoborane (1g) [21] could be a useful boron source because the resulting aminoborylated products are sufficiently labile to be converted into various boron derivatives by treatment with protecting groups in a one-pot reaction sequence. The reactivity of 1g has previously been well-exploited in catalytic borylation of aryl halides [22-27]. Herein, we report the C–H borylation of arenes using 1g catalyzed by an Ir/N-heterocyclic carbene (NHC) system.
Results and Discussion
On the basis of a superior reactivity of indoles in several C–H borylation reactions [7-9], we initially examined the borylation of indole 2 with aminoborane 1g using an iridium catalyst under forcing conditions (140 °C, 15 h). Although all the attempts to isolate an initially formed aminoborylated product 3 were not successful, its formation was confirmed by 11B NMR (δ = 40.7 ppm in cyclohexane-d12). The crude reaction mixture was treated with pinacol and the yield of the product was estimated by 1H NMR spectroscopy. Using dtbpy, the common ligand for iridium-catalyzed C–H borylation [2], the reaction failed to give 2-B under these conditions (Table 1, entry 1). Several mono- and diphosphine ligands were found to be active for the formation of 2-B, but the best yield was only 21% (Table 1, entries 2–6). Our success in C–H borylation using NHC ligands [8,10] led us to investigate a series of NHC ligands for this process. Among the NHC ligands examined, 1,3-dicyclohexylimidazol-2-ylidene (ICy) [28-33] was found to be most effective, giving 2-B in 33% yield with a 2-/3-borylation ratio of 88:12 (Table 1, entry 9). It should be noted that [Ir(cod)(ICy)2](CF3CO2) was previously reported to promote C–H borylation of arenes using HBpin [34]. Further optimization using an ICy ligand determined that decreasing the reaction temperature to 110 °C and shortening the reaction time to 4 h markedly improved the yield of 2-B (72%) with near complete regioselectivity (99:1) (Table 1, entry 12).
Table 1: Effect of the ligand on the Ir-catalyzed borylation of 2 with 1g.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-65-i3.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Ligand | Base | Temperature [°C] | NMR yieldb [%] | 2-Isomer/3-Isomer |
|---|---|---|---|---|---|
| 1 | dtbpy | none | 140 | trace | – |
| 2 | PPh3 | none | 140 | 21 | 57/43 |
| 3 | PCy3 | none | 140 | 3 | >99/1 |
| 4 | dppe | none | 140 | 2 | >99/1 |
| 5 | xantphos | none | 140 | 18 | 56/44 |
| 6 | Xphos | none | 140 | 19 | 71/29 |
| 7 | IMes·HCl | NaOt-Bu | 140 | 5 | >99/1 |
| 8 | IPr·HCl | NaOt-Bu | 140 | 3 | >99/1 |
| 9 | ICy·HCl | NaOt-Bu | 140 | 33 | 88/12 |
| 10 | It-Bu·HCl | NaOt-Bu | 140 | 0 | – |
| 11 | ICy·HCl | NaOt-Bu | 110 | 58 | 95/5 |
| 12 | ICy·HCl | NaOt-Bu | 110 | 72 (65)c | 99/1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-65-i4.svg?max-width=637&scale=1.0)
|
|||||
aReaction conditions: 2 (0.50 mmol), 1g (1.0 mmol), [Ir(OMe)(cod)]2 (0.050 mmol), ligand (0.10 mmol), NaOt-Bu (0.20 mmol) in methylcyclohexane (1.0 mL) at 140 °C for 15 h. After treatment with pinacol (2.0 mmol), the borylated product was converted to the corresponding pinacolate. bThe yield refers to a combined NMR yield of 2- and 3-borylated products. cIsolated yield.
Having optimized the conditions, we next explored the scope of Ir/ICy-catalyzed borylation of heteroarene substrates using 1g (Table 2). Functionalized indoles, such as those containing methoxy, fluoro, bromo and chloro substituents, all underwent the borylation to form the corresponding 2-borylated products 4-B, 5-B, 6-B and 7-B, respectively (Table 2, entries 1–4). When 1,4-dimethylindole (8) was used, 2-borylated product 8-B was formed exclusively with no borylation occurring at the benzylic position (Table 2, entry 5) [8,35,36]. Benzothiophenes readily gave 2-borylated products using our system, as exemplified by the high yields obtained from 9 and 10 (Table 2, entries 6 and 7). Although benzofuran 11 was borylated at the 2-position successfully, the isolated yield was somewhat lower than the yield calculated from the 1H NMR data, probably because of the instability of 11-B during isolation (Table 2, entry 8). Our protocol was able to borylate non-benzofused five-membered heteroarenes. Pyrrole 12 was much less reactive than indoles, and required neat conditions to obtain a modest yield of the borylated product 12-B (Table 2, entry 9). Thiophene (13) afforded a 1.1:1 mixture of 2-borylated and 2,5-diborylated products under our standard conditions (Table 2, entry 10). 2-Substituted thiophenes 14 and 15 and furan 16 were borylated successfully at the 5-positions (Table 2, entries 11–13). Electron-deficient heteroarenes such as pyridine and quinolone failed to form the borylated product under the current conditions.
Table 2: Scope of the heteroarene substrates.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-65-i5.svg?max-width=637&scale=1.0)
|
|||
| Entry | Heteroarene | Product | Isolated yield (NMR yield) [%] |
|---|---|---|---|
| 1 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-65-i6.svg?max-width=637&scale=1.0)
4 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-12-65-i7.svg?max-width=637&scale=1.0)
4-B |
48 (51) |
| 2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-12-65-i8.svg?max-width=637&scale=1.0)
5 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-12-65-i9.svg?max-width=637&scale=1.0)
5-B |
75 (82) |
| 3 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-12-65-i10.svg?max-width=637&scale=1.0)
6 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-12-65-i11.svg?max-width=637&scale=1.0)
6-B |
66 (66) |
| 4 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-12-65-i12.svg?max-width=637&scale=1.0)
7 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-12-65-i13.svg?max-width=637&scale=1.0)
7-B |
50 (55)b |
| 5 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-12-65-i14.svg?max-width=637&scale=1.0)
8 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-12-65-i15.svg?max-width=637&scale=1.0)
8-B |
51 (58) |
| 6 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-12-65-i16.svg?max-width=637&scale=1.0)
9 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-12-65-i17.svg?max-width=637&scale=1.0)
9-B |
94 (>99) |
| 7 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-12-65-i18.svg?max-width=637&scale=1.0)
10 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-12-65-i19.svg?max-width=637&scale=1.0)
10-B |
91 (99) |
| 8 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-12-65-i20.svg?max-width=637&scale=1.0)
11 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-12-65-i21.svg?max-width=637&scale=1.0)
11-B |
65 (87) |
| 9 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-12-65-i22.svg?max-width=637&scale=1.0)
12 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-12-65-i23.svg?max-width=637&scale=1.0)
12-B |
0
50 (52)c |
| 10 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-12-65-i24.svg?max-width=637&scale=1.0)
13 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-12-65-i25.svg?max-width=637&scale=1.0)
|
71 (91)
13-B:13-2B = 1.1:1 |
| 11 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-12-65-i26.svg?max-width=637&scale=1.0)
14 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-12-65-i27.svg?max-width=637&scale=1.0)
14-B |
96 (>99) |
| 12 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-12-65-i28.svg?max-width=637&scale=1.0)
15 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-12-65-i29.svg?max-width=637&scale=1.0)
15-B |
91 (>99) |
| 13 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-12-65-i30.svg?max-width=637&scale=1.0)
16 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-12-65-i31.svg?max-width=637&scale=1.0)
16-B |
68 (92) |
aReaction conditions: heteroarene (0.50 mmol), 1g (1.0 mmol), [Ir(OMe)(cod)]2 (0.050 mmol), ICy·HCl (0.10 mmol), NaOt-Bu (0.20 mmol) in methylcyclohexane (1.0 mL) at 110 °C for 4 h. After treatment with pinacol (2.0 mmol), the borylated product was converted to the corresponding pinacolate. In cases where NMR yield is modest, the recovered starting heteroarene can account for the material balance, unless otherwise noted. bDebrominative borylation also occurred with a yield of 6%. cRun using 1.0 mL of N-methylpyrrole instead of methylcyclohexane.
We next turned our attention to the borylation of benzene derivatives. Unfortunately, benzene derivatives proved to be much less reactive than heteroarenes when borylated with 1g. For example, Ir/ICy-catalyzed borylation of benzene with 1g afforded 17-B in 48% isolated yield even when the reaction was conducted under neat conditions (Table 3, entry 1) (see the Supporting Information File 1 for details on the optimization for the borylation of benzene). Borylation was relatively independent of the electronic nature of the arene substrates, as indicated by the similar yields and regioselectivity observed with toluene, anisole and trifluoromethylbenzene (Table 3, entries 2–4). Similar to the reported C–H borylation using other boron sources, 1,3-disubstituted benzenes were borylated at the 5-position in a regioselective manner (Table 3, entry 5). Naphthalene also underwent borylation with 1g at the less hindered 2-position (Table 3, entry 6).
Table 3: Scope of the arene substrates.a
![[Graphic 30]](/bjoc/content/inline/1860-5397-12-65-i32.svg?max-width=637&scale=1.0)
|
||||
| Entry | Arene | Product | Isolated yield [%] | Ratio of o/m/p isomers |
|---|---|---|---|---|
| 1 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-12-65-i33.svg?max-width=637&scale=1.0)
17 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-12-65-i34.svg?max-width=637&scale=1.0)
17-B |
48 | – |
| 2 |
![[Graphic 33]](/bjoc/content/inline/1860-5397-12-65-i35.svg?max-width=637&scale=1.0)
18 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-12-65-i36.svg?max-width=637&scale=1.0)
18-B |
42 | 0/64/36 |
| 3 |
![[Graphic 35]](/bjoc/content/inline/1860-5397-12-65-i37.svg?max-width=637&scale=1.0)
19 |
![[Graphic 36]](/bjoc/content/inline/1860-5397-12-65-i38.svg?max-width=637&scale=1.0)
19-B |
35 | 0/60/40 |
| 4 |
![[Graphic 37]](/bjoc/content/inline/1860-5397-12-65-i39.svg?max-width=637&scale=1.0)
20 |
![[Graphic 38]](/bjoc/content/inline/1860-5397-12-65-i40.svg?max-width=637&scale=1.0)
20-B |
49 | 0/76/24 |
| 5 |
![[Graphic 39]](/bjoc/content/inline/1860-5397-12-65-i41.svg?max-width=637&scale=1.0)
21 |
![[Graphic 40]](/bjoc/content/inline/1860-5397-12-65-i42.svg?max-width=637&scale=1.0)
21-B |
31 | – |
| 6b |
![[Graphic 41]](/bjoc/content/inline/1860-5397-12-65-i43.svg?max-width=637&scale=1.0)
22 |
![[Graphic 42]](/bjoc/content/inline/1860-5397-12-65-i44.svg?max-width=637&scale=1.0)
22-B |
50 | – |
aReaction conditions: arene (1.0 mL), 1g (0.50 mmol), [Ir(OMe)(cod)]2 (0.050 mmol), ICy·HCl (0.10 mmol), NaOt-Bu (0.20 mmol) at 110 °C for 15 h. After treatment with pinacol (2.0 mmol), the borylated product was converted to the corresponding pinacolate. bNaphthalene (3.0 mmol) was used in methylcyclohexane (1.0 mL).
Our protocol was performed on a gram scale without any difficulty using a lower loading of the iridium catalyst (Scheme 2, top). Using 1g as the boron source in C–H borylation reactions has the synthetic advantage of allowing various substituents to be introduced onto the boron atom during the work-up stage simply by changing the reagents added. For example, addition of different diols delivered the corresponding boronic esters 10-Bnep and 10-Bmep (Scheme 2, bottom). It was also possible to introduce Suginome’s dan group, which allows us to use the borylated products in more elaborate manner, such as iterative cross-coupling reactions [20].
![[1860-5397-12-65-i2]](/bjoc/content/inline/1860-5397-12-65-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scalability and derivatization.
Scheme 2: Scalability and derivatization.
Conclusion
We have developed a C–H borylation of arenes and heteroarenes using diisopropylaminoborane as a borylating reagent. An iridium(I) complex bearing an ICy ligand was the most efficient catalyst. The initially formed aminoborylated products can readily be converted to the corresponding organoboron compounds bearing various boron-protecting groups.
Experimental
Procedure for the Ir-catalyzed borylation of heteroarenes using 1g
In a glovebox filled with nitrogen, [Ir(OMe)(cod)]2 (33.1 mg, 0.050 mmol, 0.10 equiv), ICy·HCl (26.2 mg, 0.10 mmol, 0.20 equiv), NaOt-Bu (19.2 mg, 0.20 mmol, 0.40 equiv) and methylcyclohexane (1.0 mL) were added to a 10 mL sample vial with a Teflon-sealed screwcap, and stirred for 5 min at room temperature. A heteroarene (0.50 mmol, 1.0 equiv) and 1g (113.1 mg, 2.0 equiv) were added, and then the cap was screwed on seal the vial. The vial was stirred at 110 °C for 4 h. The reaction mixture was cooled to room temperature. Pinacol (236 mg, 2.0 mmol) in THF (2.0 mL) was added and the reaction mixture was stirred under N2 at room temperature for 1.5 h. The crude mixture was filtered through a pad of Celite and eluted with EtOAc. The filtrate was concentrated in vacuo and sampled for analysis by 1H NMR spectroscopy using 1,2-dichloroethane as an internal standard. The residue was purified by flash column chromatography over silica gel eluting with hexane/EtOAc. Product-containing fractions were concentrated in vacuo to give a pure borylated product.
Supporting Information
| Supporting Information File 1: Experimental procedures, data for optimization studies and copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 6.3 MB | Download |
References
-
Mkhalid, I. A. I.; Barnard, J. H.; Marder, T. B.; Murphy, J. M.; Hartwig, J. F. Chem. Rev. 2010, 110, 890. doi:10.1021/cr900206p
Return to citation in text: [1] -
Ishiyama, T.; Takagi, J.; Ishida, K.; Miyaura, N.; Anastasi, N. R.; Hartwig, J. F. J. Am. Chem. Soc. 2002, 124, 390. doi:10.1021/ja0173019
Return to citation in text: [1] [2] -
Hatanaka, T.; Ohki, Y.; Tatsumi, K. Chem. – Asian J. 2010, 5, 1657. doi:10.1002/asia.201000140
Return to citation in text: [1] -
Yan, G.; Jiang, Y.; Kuang, C.; Wang, S.; Liu, H.; Zhang, Y.; Wang, J. Chem. Commun. 2010, 46, 3170. doi:10.1039/b926945b
Return to citation in text: [1] -
Dombray, T.; Werncke, C. G.; Jiang, S.; Grellier, M.; Vendier, L.; Bontemps, S.; Sortais, J.-B.; Sabo-Etienne, S.; Darcel, C. J. Am. Chem. Soc. 2015, 137, 4062. doi:10.1021/jacs.5b00895
Return to citation in text: [1] -
Mazzacano, T. J.; Mankad, N. P. J. Am. Chem. Soc. 2013, 135, 17258. doi:10.1021/ja408861p
See for Cu–Fe mixed catalyst.
Return to citation in text: [1] -
Obligacion, J. V.; Semproni, S. P.; Chirik, P. J. J. Am. Chem. Soc. 2014, 136, 4133. doi:10.1021/ja500712z
Return to citation in text: [1] [2] -
Furukawa, T.; Tobisu, M.; Chatani, N. Chem. Commun. 2015, 51, 6508. doi:10.1039/C5CC01378J
Return to citation in text: [1] [2] [3] [4] -
Zhang, H.; Hagihara, S.; Itami, K. Chem. Lett. 2015, 44, 779. doi:10.1246/cl.150154
Return to citation in text: [1] [2] -
Furukawa, T.; Tobisu, M.; Chatani, N. J. Am. Chem. Soc. 2015, 137, 12211. doi:10.1021/jacs.5b07677
Return to citation in text: [1] [2] -
Takaya, J.; Ito, S.; Nomoto, H.; Saito, N.; Kirai, N.; Iwasawa, N. Chem. Commun. 2015, 51, 17662. doi:10.1039/C5CC07263H
Return to citation in text: [1] -
Légaré, M.-A.; Courtemanche, M.-A.; Rochette, É.; Fontaine, F.-G. Science 2015, 349, 513. doi:10.1126/science.aab3591
Return to citation in text: [1] -
Ros, A.; Fernández, R.; Lassaletta, J. M. Chem. Soc. Rev. 2014, 43, 3229. doi:10.1039/c3cs60418g
See for a review.
Return to citation in text: [1] -
Kuninobu, Y.; Ida, H.; Nishi, M.; Kanai, M. Nat. Chem. 2015, 7, 712. doi:10.1038/nchem.2322
See for directed meta-borylation.
Return to citation in text: [1] -
Saito, Y.; Segawa, Y.; Itami, K. J. Am. Chem. Soc. 2015, 137, 5193. doi:10.1021/jacs.5b02052
Return to citation in text: [1] -
Tzschucke, C. C.; Murphy, J. M.; Hartwig, J. F. Org. Lett. 2007, 9, 761. doi:10.1021/ol062902w
Return to citation in text: [1] -
Liskey, C. W.; Hartwig, J. F. Synthesis 2013, 45, 1837. doi:10.1055/s-0033-1338863
Return to citation in text: [1] -
Liskey, C. W.; Wei, C. S.; Pahls, D. R.; Hartwig, J. F. Chem. Commun. 2009, 5603. doi:10.1039/b913949d
Return to citation in text: [1] -
Iwadate, N.; Suginome, M. J. Organomet. Chem. 2009, 694, 1713. doi:10.1016/j.jorganchem.2008.11.068
Return to citation in text: [1] -
Noguchi, H.; Hojo, K.; Suginome, M. J. Am. Chem. Soc. 2007, 129, 758. doi:10.1021/ja067975p
Return to citation in text: [1] [2] -
Köster, R.; Bellut, H.; Hattori, S. Justus Liebigs Ann. Chem. 1968, 720, 1. doi:10.1002/jlac.19687200102
Return to citation in text: [1] -
Euzenat, L.; Horhant, D.; Ribourdouille, Y.; Duriez, C.; Alcaraz, G.; Vaultier, M. Chem. Commun. 2003, 2280. doi:10.1039/B306874A
Return to citation in text: [1] -
Pasumansky, L.; Haddenham, D.; Clary, J. W.; Fisher, G. B.; Goralski, C. T.; Singaram, B. J. Org. Chem. 2008, 73, 1898. doi:10.1021/jo702271c
Return to citation in text: [1] -
Marciasini, L.; Richy, N.; Vaultier, M.; Pucheault, M. Chem. Commun. 2012, 48, 1553. doi:10.1039/C1CC14605J
Return to citation in text: [1] -
Marciasini, L. D.; Richy, N.; Vaultier, M.; Pucheault, M. Adv. Synth. Catal. 2013, 355, 1083. doi:10.1002/adsc.201200942
Return to citation in text: [1] -
Guerrand, H. D. S.; Marciasini, L. D.; Jousseaume, M.; Vaultier, M.; Pucheault, M. Chem. – Eur. J. 2014, 20, 5573. doi:10.1002/chem.201304861
Return to citation in text: [1] -
Marciasini, L.; Cacciuttolo, B.; Vaultier, M.; Pucheault, M. Org. Lett. 2015, 17, 3532. doi:10.1021/acs.orglett.5b01620
Return to citation in text: [1] -
Tobisu, M.; Yasutome, A.; Kinuta, H.; Nakamura, K.; Chatani, N. Org. Lett. 2014, 16, 5572. doi:10.1021/ol502583h
Return to citation in text: [1] -
Tobisu, M.; Takahira, T.; Ohtsuki, A.; Chatani, N. Org. Lett. 2015, 17, 680. doi:10.1021/ol503707m
Return to citation in text: [1] -
Tobisu, M.; Takahira, T.; Chatani, N. Org. Lett. 2015, 17, 4352. doi:10.1021/acs.orglett.5b02200
Return to citation in text: [1] -
Tobisu, M.; Morioka, T.; Ohtsuki, A.; Chatani, N. Chem. Sci. 2015, 6, 3410. doi:10.1039/C5SC00305A
Return to citation in text: [1] -
Morioka, T.; Nishizawa, A.; Nakamura, K.; Tobisu, M.; Chatani, N. Chem. Lett. 2015, 44, 1729. doi:10.1246/cl.150936
Return to citation in text: [1] -
Nakamura, K.; Tobisu, M.; Chatani, N. Org. Lett. 2015, 17, 6142. doi:10.1021/acs.orglett.5b03151
Return to citation in text: [1] -
Frey, G. D.; Rentzsch, C. F.; von Preysing, D.; Scherg, T.; Mühlhofer, M.; Herdtweck, E.; Herrmann, W. A. J. Organomet. Chem. 2006, 691, 5725. doi:10.1016/j.jorganchem.2006.08.099
Return to citation in text: [1] -
Ishiyama, T.; Ishida, K.; Takagi, J.; Miyaura, N. Chem. Lett. 2001, 30, 1082. doi:10.1246/cl.2001.1082
Return to citation in text: [1] -
Larsen, M. A.; Wilson, C. V.; Hartwig, J. F. J. Am. Chem. Soc. 2015, 137, 8633. doi:10.1021/jacs.5b04899
Return to citation in text: [1]
| 28. | Tobisu, M.; Yasutome, A.; Kinuta, H.; Nakamura, K.; Chatani, N. Org. Lett. 2014, 16, 5572. doi:10.1021/ol502583h |
| 29. | Tobisu, M.; Takahira, T.; Ohtsuki, A.; Chatani, N. Org. Lett. 2015, 17, 680. doi:10.1021/ol503707m |
| 30. | Tobisu, M.; Takahira, T.; Chatani, N. Org. Lett. 2015, 17, 4352. doi:10.1021/acs.orglett.5b02200 |
| 31. | Tobisu, M.; Morioka, T.; Ohtsuki, A.; Chatani, N. Chem. Sci. 2015, 6, 3410. doi:10.1039/C5SC00305A |
| 32. | Morioka, T.; Nishizawa, A.; Nakamura, K.; Tobisu, M.; Chatani, N. Chem. Lett. 2015, 44, 1729. doi:10.1246/cl.150936 |
| 33. | Nakamura, K.; Tobisu, M.; Chatani, N. Org. Lett. 2015, 17, 6142. doi:10.1021/acs.orglett.5b03151 |
| 2. | Ishiyama, T.; Takagi, J.; Ishida, K.; Miyaura, N.; Anastasi, N. R.; Hartwig, J. F. J. Am. Chem. Soc. 2002, 124, 390. doi:10.1021/ja0173019 |
| 8. | Furukawa, T.; Tobisu, M.; Chatani, N. Chem. Commun. 2015, 51, 6508. doi:10.1039/C5CC01378J |
| 10. | Furukawa, T.; Tobisu, M.; Chatani, N. J. Am. Chem. Soc. 2015, 137, 12211. doi:10.1021/jacs.5b07677 |
| 1. | Mkhalid, I. A. I.; Barnard, J. H.; Marder, T. B.; Murphy, J. M.; Hartwig, J. F. Chem. Rev. 2010, 110, 890. doi:10.1021/cr900206p |
| 8. | Furukawa, T.; Tobisu, M.; Chatani, N. Chem. Commun. 2015, 51, 6508. doi:10.1039/C5CC01378J |
| 9. | Zhang, H.; Hagihara, S.; Itami, K. Chem. Lett. 2015, 44, 779. doi:10.1246/cl.150154 |
| 22. | Euzenat, L.; Horhant, D.; Ribourdouille, Y.; Duriez, C.; Alcaraz, G.; Vaultier, M. Chem. Commun. 2003, 2280. doi:10.1039/B306874A |
| 23. | Pasumansky, L.; Haddenham, D.; Clary, J. W.; Fisher, G. B.; Goralski, C. T.; Singaram, B. J. Org. Chem. 2008, 73, 1898. doi:10.1021/jo702271c |
| 24. | Marciasini, L.; Richy, N.; Vaultier, M.; Pucheault, M. Chem. Commun. 2012, 48, 1553. doi:10.1039/C1CC14605J |
| 25. | Marciasini, L. D.; Richy, N.; Vaultier, M.; Pucheault, M. Adv. Synth. Catal. 2013, 355, 1083. doi:10.1002/adsc.201200942 |
| 26. | Guerrand, H. D. S.; Marciasini, L. D.; Jousseaume, M.; Vaultier, M.; Pucheault, M. Chem. – Eur. J. 2014, 20, 5573. doi:10.1002/chem.201304861 |
| 27. | Marciasini, L.; Cacciuttolo, B.; Vaultier, M.; Pucheault, M. Org. Lett. 2015, 17, 3532. doi:10.1021/acs.orglett.5b01620 |
| 7. | Obligacion, J. V.; Semproni, S. P.; Chirik, P. J. J. Am. Chem. Soc. 2014, 136, 4133. doi:10.1021/ja500712z |
| 7. | Obligacion, J. V.; Semproni, S. P.; Chirik, P. J. J. Am. Chem. Soc. 2014, 136, 4133. doi:10.1021/ja500712z |
| 8. | Furukawa, T.; Tobisu, M.; Chatani, N. Chem. Commun. 2015, 51, 6508. doi:10.1039/C5CC01378J |
| 9. | Zhang, H.; Hagihara, S.; Itami, K. Chem. Lett. 2015, 44, 779. doi:10.1246/cl.150154 |
| 3. | Hatanaka, T.; Ohki, Y.; Tatsumi, K. Chem. – Asian J. 2010, 5, 1657. doi:10.1002/asia.201000140 |
| 4. | Yan, G.; Jiang, Y.; Kuang, C.; Wang, S.; Liu, H.; Zhang, Y.; Wang, J. Chem. Commun. 2010, 46, 3170. doi:10.1039/b926945b |
| 5. | Dombray, T.; Werncke, C. G.; Jiang, S.; Grellier, M.; Vendier, L.; Bontemps, S.; Sortais, J.-B.; Sabo-Etienne, S.; Darcel, C. J. Am. Chem. Soc. 2015, 137, 4062. doi:10.1021/jacs.5b00895 |
| 6. |
Mazzacano, T. J.; Mankad, N. P. J. Am. Chem. Soc. 2013, 135, 17258. doi:10.1021/ja408861p
See for Cu–Fe mixed catalyst. |
| 20. | Noguchi, H.; Hojo, K.; Suginome, M. J. Am. Chem. Soc. 2007, 129, 758. doi:10.1021/ja067975p |
| 2. | Ishiyama, T.; Takagi, J.; Ishida, K.; Miyaura, N.; Anastasi, N. R.; Hartwig, J. F. J. Am. Chem. Soc. 2002, 124, 390. doi:10.1021/ja0173019 |
| 21. | Köster, R.; Bellut, H.; Hattori, S. Justus Liebigs Ann. Chem. 1968, 720, 1. doi:10.1002/jlac.19687200102 |
| 14. |
Kuninobu, Y.; Ida, H.; Nishi, M.; Kanai, M. Nat. Chem. 2015, 7, 712. doi:10.1038/nchem.2322
See for directed meta-borylation. |
| 15. | Saito, Y.; Segawa, Y.; Itami, K. J. Am. Chem. Soc. 2015, 137, 5193. doi:10.1021/jacs.5b02052 |
| 17. | Liskey, C. W.; Hartwig, J. F. Synthesis 2013, 45, 1837. doi:10.1055/s-0033-1338863 |
| 18. | Liskey, C. W.; Wei, C. S.; Pahls, D. R.; Hartwig, J. F. Chem. Commun. 2009, 5603. doi:10.1039/b913949d |
| 20. | Noguchi, H.; Hojo, K.; Suginome, M. J. Am. Chem. Soc. 2007, 129, 758. doi:10.1021/ja067975p |
| 13. |
Ros, A.; Fernández, R.; Lassaletta, J. M. Chem. Soc. Rev. 2014, 43, 3229. doi:10.1039/c3cs60418g
See for a review. |
| 19. | Iwadate, N.; Suginome, M. J. Organomet. Chem. 2009, 694, 1713. doi:10.1016/j.jorganchem.2008.11.068 |
| 12. | Légaré, M.-A.; Courtemanche, M.-A.; Rochette, É.; Fontaine, F.-G. Science 2015, 349, 513. doi:10.1126/science.aab3591 |
| 34. | Frey, G. D.; Rentzsch, C. F.; von Preysing, D.; Scherg, T.; Mühlhofer, M.; Herdtweck, E.; Herrmann, W. A. J. Organomet. Chem. 2006, 691, 5725. doi:10.1016/j.jorganchem.2006.08.099 |
| 10. | Furukawa, T.; Tobisu, M.; Chatani, N. J. Am. Chem. Soc. 2015, 137, 12211. doi:10.1021/jacs.5b07677 |
| 11. | Takaya, J.; Ito, S.; Nomoto, H.; Saito, N.; Kirai, N.; Iwasawa, N. Chem. Commun. 2015, 51, 17662. doi:10.1039/C5CC07263H |
| 16. | Tzschucke, C. C.; Murphy, J. M.; Hartwig, J. F. Org. Lett. 2007, 9, 761. doi:10.1021/ol062902w |
| 8. | Furukawa, T.; Tobisu, M.; Chatani, N. Chem. Commun. 2015, 51, 6508. doi:10.1039/C5CC01378J |
| 35. | Ishiyama, T.; Ishida, K.; Takagi, J.; Miyaura, N. Chem. Lett. 2001, 30, 1082. doi:10.1246/cl.2001.1082 |
| 36. | Larsen, M. A.; Wilson, C. V.; Hartwig, J. F. J. Am. Chem. Soc. 2015, 137, 8633. doi:10.1021/jacs.5b04899 |
© 2016 Tobisu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)