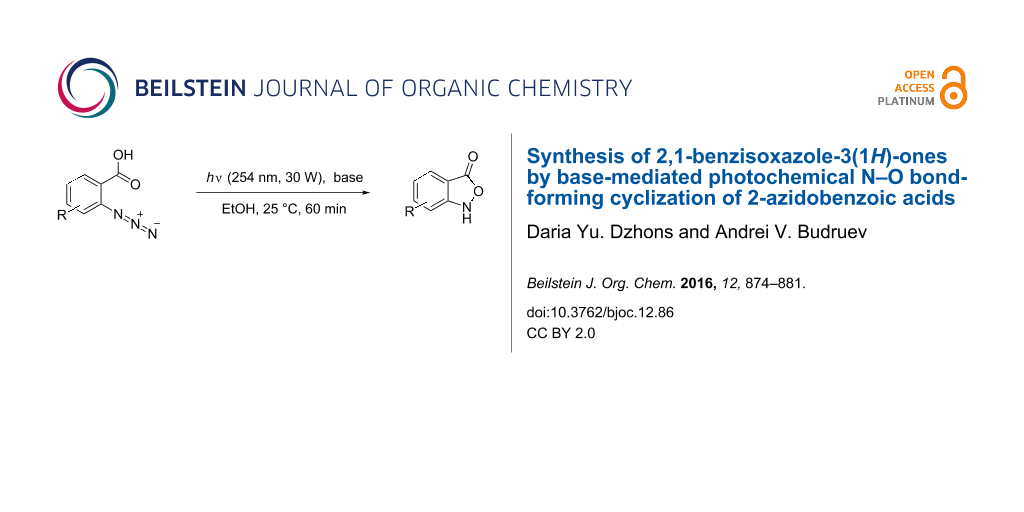
Abstract
The base-mediated photochemical cyclization of 2-azidobenzoic acids with the formation of 2,1-benzisoxazole-3(1H)-ones is reported. The optimization and scope of this cyclization reaction is discussed. It is shown that an essential step of the ring closure of 2-azidobenzoic acids is the formation and photolysis of 2-azidobenzoate anions.

Graphical Abstract
Introduction
Substituted 2,1-benzisoxazoles display diverse biological activity [1-6] (Figure 1) and are widely used as starting materials for the synthesis of important heterocyclic pharmacophores, such as acridines [7,8], quinolines [9-13] and quinazolines [14-16]. Therefore, the search for new methods leading to 2,1-benzisoxazoles is of great interest.
![[1860-5397-12-86-1]](/bjoc/content/figures/1860-5397-12-86-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected examples of biologically active, fused 2,1-isoxazole derivatives.
Figure 1: Selected examples of biologically active, fused 2,1-isoxazole derivatives.
For the preparative synthesis of 2,1-benzisoxazoles, in addition to the traditional method based on the reductive heterocyclization of ortho-substituted nitro compounds [17-21], two other routes are available: the annulation of nitroso compounds [22,23] and the thermal [24], catalytic [25-27] or photochemical cyclization of aryl azides [28-31].
However, the presence of electron-withdrawing substituents in the 3-, 5- and 7-position of the benzisoxazole dramatically reduces the thermal stability of these compounds. Indeed, these compounds are the most labile representatives of several isomeric oxazoles [32], and they begin to decompose at temperatures slightly above 30 °C, which limits the number of methods for their preparation. Thus the search for new methods for the synthesis of substituted 2,1-benzisoxazoles under mild reaction conditions is required.
Previously, in the investigation of the photochemical cyclization of 2-azidobenzoic acid (1a) using aqueous organic solvent mixtures (Scheme 1), the formation of the cyclization products such as 2,1-benzisoxazole-3(1H)-one (2a) and 2-oxo-3-carboxy-3H-azepine (3a) has been reported [30]. The structure of benzisoxazole 2a was determined by IR, 1H and 13C NMR spectroscopy and by comparison of its mass spectrum with the corresponding spectrum from the NIST library (NIST: 37717). In addition, the structure was confirmed by the alternative synthesis of 2a through the heterocyclization of 2-nitrobenzoic acid [28,29] and X-ray structure analysis [33].
![[1860-5397-12-86-i1]](/bjoc/content/inline/1860-5397-12-86-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Photochemical cyclization of substituted 2-azidobenzoic acids and possible reaction mechanism [34-59].
Scheme 1: Photochemical cyclization of substituted 2-azidobenzoic acids and possible reaction mechanism [34-59].
The authors observed that the yields of both 2a and 3a increased with increasing amount of water as nucleophilic solvent in the reaction mixture [30] and obtained a maximum yield of 20% and 50%, respectively, at a water content of 50%. Under these conditions, no formation of the primary amine 4 (a product of the typical triplet nitrene reaction (Scheme 1, intermediate D)) was detected. The replacement of the aprotic solvent dioxane with acetonitrile or THF did not affect the yield of the cyclization products. The photolysis of 1a (Scheme 1, intermediate A) resulted in low yields of 2a because of the competitive formation of reaction products.
In the past, research has been focused on the formation of 2-substituted 3H-azepines 3 as products of the photolysis or thermolysis of aromatic azides [34,35]. The proposed mechanism for their formation was confirmed by identification of the reaction intermediates using low-temperature and time-resolved spectroscopy [36-43]. It is currently believed that azepines are formed through the singlet nitrogen pathway of the reaction (Scheme 1, path: 1 → A → B → C → 3) [44-49].
These nitrenes form benzazirines [19] (Scheme 1, intermediate B) that rearrange into cyclic ketenimines – 1,2-didehydroazepines (Scheme 1, intermediate C) [50]. According to quantum-chemical calculations, the energy barrier of this rearrangement is approximately 40 kJ mol−1 [51], with the limiting step being the formation of B. Therefore, the direction of this reaction solely depends on the conversion of A and formation of C.
Dyall et al. [52,53] have proposed a pericyclic mechanism for the formation of heterocyclic compounds in the pyrolysis of aryl azides with unsaturated ortho-substituents. A possible reaction mechanism for the photochemical formation of 2 (Scheme 1, path: 1 → A → 2) based on the report by Platz et al. [54] includes the benzofuroxan formation by photolysis of 2-azidonitrobenzene through the intermediate singlet nitrene A (Scheme 1) without the formation of other intermediates.
The formation of substituted 2,1-benzisoxazoles from aryl azides was reported for the first time by Smith et al. [24] in the synthesis of 3-phenyl-2,1-benzisoxazole (3-phenylanthranil) by thermolysis of 2-azidobenzophenone. In another work [55], the photochemical formation of 3-amino-6-nitro-2,1-benzisoxazole starting from 2-azido-4-nitrobenzamide was observed. The authors subsequently investigated the multiplicity of the involved nitrene by repeating the reaction in the presence of isoprene as a triplet nitrene quencher. The addition of isoprene lead to a significantly increased yield of 3-amino-6-nitro-2,1-benzisoxazole and an insignificant decrease of the primary amine yield. Thus it was demonstrated that the formation of 3-amino-6-nitro-2,1-benzisoxazole goes through an intermediate singlet nitrene.
Possibly, similar to the benzofuroxan and 3-amino-6-nitro-2,1-benzisoxazole formation, the carboxylate group of A (Lewis base) donates an electron lone pair to the electron-deficient singlet nitrene fragment of A (Lewis acid) with formation of the N–O bond in 2 through a 1,5-electrocyclization reaction [56,57].
Nonreacted singlet nitrenes A may undergo intersystem crossing (ISC) into the less reactive triplet state (Scheme 1, intermediate D). Although a multiplicity change is a spin-forbidden transition, it can be partially allowed in some cases. According to another report [39], the major products formed from triplet nitrenes are primary amines 4 through hydrogen-atom abstraction [58], secondary amines, 1,2-arylhydrazides and azo compounds 5, which are obtained by recombination of radicals [59]. Moreover, it was shown that yields of primary amines increased in the photolysis reactions of aryl azides without participating ortho-substituents in hydrocarbons as the solvents [39].
In the present research it is demonstrated that using ethanol as the solvent for the photolysis reaction leads to benzisoxazole 2a with an increased yield of 35% and the yield can be further improved to 40% by the addition of a base. Thus, the optimization of the reaction conditions of the base-mediated photochemical synthesis of substituted 2,1-benzisoxazole-3(1H)-ones 2 has been performed.
Results and Discussion
For optimizing the reaction conditions for the synthesis of 2a by photolysis of 1a, the reaction was performed in different solvents in the absence or presence of a base. As solvents, alcohols and aqueous organic solvent mixtures were tested and alkali metal hydroxides, carbonates or acetates were screened as the base. All reactions were carried out by irradiating the base suspended in the solution of 1a with a mercury low-pressure quartz lamp (254 nm) in a quartz reactor with intensive stirring.
It was found that the yields of 2a after photolysis of 1a substantially increased in the presence of a base. No dependency on the nature of the base could be observed and the yield did not improve further when more than 1 equivalent of the base was used (Table 1). Without irradiation, the reaction did not proceed at all. Replacing EtOH as the solvent with iPrOH did not change the yield of 2a. A chromatographic separation of the reaction mixture obtained by photolysis of 1a in alcohols showed that 2a had formed as the sole product. In this case, neither the formation of 3a nor the corresponding 2-ethoxy- or 2-isopropoxy-substituted azepines could be detected.
Table 1: Optimization of conditions for the synthesis of substituted 2,1-benzisoxazole-3(1H)-ones.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-86-i3.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Base (equiv) | Solvent | Yield (2a)b | Yield (3a)b | t, minc |
|---|---|---|---|---|---|
| 1 | – | 1,4-dioxane | – | – | 60 |
| 2 | – | EtOH | 38% | 1% | 60 |
| 3 | K2CO3 (0.5) | EtOH | 63% | – | 60 |
| 4 | K2CO3 (1) | EtOH | 75% | – | 60 |
| 5 | K2CO3 (1) | iPrOH | 75% | – | 60 |
| 6 | K2CO3 (5) | 1,4-dioxane/water 1:1 (v/v) | 43% | 8% | 60 |
| 7 | K2CO3 (3) | EtOH | 75% | – | 60 |
| 8 | Na2CO3 (0.5) | EtOH | 65% | – | 60 |
| 9 | Na2CO3 (1) | EtOH | 73% | – | 60 |
| 10 | NaHCO3 (0.5) | EtOH | 70% | – | 60 |
| 11 | NaHCO3 (1) | EtOH | 70% | – | 60 |
| 12 | KOH (1) | EtOH | 60% | – | 60 |
| 13 | NaOH (1) | Water | 30% | 12% | 60 |
| 14 | NaOAc (1) | EtOH | 75% | – | 60 |
| 15 | NaOAc (1) | 1,4-dioxane/water 1:1 (v/v) | 63% | 20% | 60 |
aReaction conditions: 1a (0.78 mmol), solvent (15.0 mL); UV light (2 × 15 W Hg low-pressure lamp (254 nm), UV intensity was approximately 7 mW/cm2). bYields were determined by HPLC analysis using an external standard. cIrradiation time: the degree of conversion of 1a was 100%.
The photolysis in a mixture of 1,4-dioxane/water led to a decreased yield of 2a with a simultaneous increase in the yield of 3a. In our opinion, this effect of the solvent or base on the yield of 2a may be explained by the formation of 2a through a heterocyclization of 2-azidobenzoate anions. Therefore, the role of the base in the reaction is the in situ generation of the 2-azidobenzoate anion and the solvent efficiently supports this formation.
The best yield of 2a was obtained by photolysis of 1a in ethanol in the presence of sodium acetate (Table 1, entry 14).
With the optimal conditions at hand, we next investigated the scope of the cyclization using differently substituted 2-azidobenzoic acids (Table 2). The desired products 2b–f were isolated in moderate to high yields (Table 2). Both electron-withdrawing groups (Cl, Br, I) and the electron-donating group (triphenylmethyl) were well-tolerated.
Table 2: Substrate scope for the heterocyclization of 2-azidobenzoic acid.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-86-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | R1 | R2 | Yield (2)b | t, minc |
|---|---|---|---|---|
| 1 (1a) | H | H | 75% (2a) | 60 |
| 2 (1b) | Cl | Cl | 92% (2b) | 60 |
| 3 (1c) | Br | H | 68% (2c) | 60 |
| 4 (1d) | Br | Br | 62% (2d) | 60 |
| 5 (1e)d | I | H | 51% (2e) | 60 |
| 6 (1f) | Tre | H | 39% (2f) | 60 |
aReaction conditions: 1 (0.78 mmol), NaOAc (1 equiv), EtOH 96% (15.0 mL); UV light (2 × 15 W Hg low-pressure lamp (254 nm), UV intensity was approximately 7 mW/cm2). bYield of the isolated product. cIrradiation time: the degree of conversion of 1a–f was 100%. dFor the optimal conditions for synthesis of 2e see Table 4, entry 10. eTr – triphenylmethyl group (trityl group).
The thermal stability of the products of 2 decreases in the series: 2f > 2a > 2e > 2d > 2c > 2b. Halogen-substituted compounds 2b–e decompose at room temperature within about 5–30 min and the products 2a,f are stable for a couple of hours.
In general, azides with electron-withdrawing groups tend to give higher yields than those substituted with electron-donating groups (2b−e vs 2f, Table 2, entries 2–5 vs entry 6). However, the yield of 2 decreased in the series Cl → Br → I (2b–e, Table 2, entries 2–5), which was probably a manifestation of the internal photochemical heavy-atom effect [60]. The presence of bromo- or iodo-substituents in the substrates 1c–e increases the probability of intersystem crossing for the initially formed singlet nitrenes (Scheme 1, intermediate A) to the triplet state (Scheme 1, intermediate D), thus resulting in decreased yields of the cyclization products 2c–e. In these cases, the formation of the triplet nitrene D should lead to increased yields of primary amines. However, as can be seen from Table 1, the formation of the latter was not observed when ethanol was used as the solvent. To further test the solvent effect on the outcome of the reaction, we repeated the photolysis of azides 1a–f under the optimized conditions but in aqueous dioxane instead of ethanol. As can be seen in Table 3, the photolysis of 1c,d in a 1,4-dioxane/water mixture under these conditions lead to decreased yields of 2c,d and to the formation of azepine 3c and primary amines 4c,d with significant yields. An explanation for the decreased yields observed for isoxazoles 2c,d may be the photo-induced breaking of the C–Br bond that completely changed the way of the reaction. However, this possibility does not explain the formation of primary amines 4c,d.
Table 3: Scope for photo-induced heterocyclization of 2-azidobenzoic acids in 1,4-dioxane/water mixture in the presence of potassium carbonate.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-86-i5.svg?max-width=637&scale=1.0)
|
||||||
| Entry | R1 | R2 | Yield (2)b | Yield (3)b | Yield (4)b | t, minc |
|---|---|---|---|---|---|---|
| 1 (1a) | H | H | 25% (2a) | 20% (3a) | – (4a) | 90 |
| 2 (1b) | Cl | Cl | 92% (2b) | – (3b) | – (4b) | 60 |
| 3 (1c) | Br | H | 34% (2c) | 18% (3c) | 10% (4c) | 180 |
| 4 (1d) | Br | Br | 62% (2d) | – (3d) | 4% (4d) | 60 |
| 5 (1e) | I | H | 5% (2e) | – (3e) | – (4e) | 120 |
| 6 (1f) | Tr | H | 40 % (2f) | – (3f) | – (4f) | 60 |
aReaction conditions: 1 (0.78 mmol), K2CO3 (1 equiv), 1,4-dioxane/water 1:1 (v/v) (15.0 mL); UV light (2 × 15 W Hg low-pressure lamp (254 nm), UV intensity was approximately 7 mW/cm2). bYields determined by HPLC analysis using an external standard. cIrradiation time: the degree of conversion of 1a–f was 100%.
It should be noted that the photolysis of double ortho-substituted azides (Table 3, entries 2, 4, R2 ≠ H) did not lead to the formation of azepines 3. This confirms the suggestions of previous reports [61,62] about the impossibility of ring expansion of such aryl azides.
Interestingly, under these conditions (Table 3), the yields for 3H-azepine 3a and 3c were determined as 20 and 18%, respectively. We wondered if the yields of these compounds could be improved to allow a preparative synthesis of 3a and 3c. For this reason, the reaction conditions were optimized towards azepines 3a and 3c. According to previously published reports [30,63,64], the yields of the corresponding 3Н-azepines could be increased by increasing the amount of the nucleophile (water) present in the reaction mixture. Thus, we attempted the preparative synthesis of 3a based on our technique described earlier [30]. After irradiating a solution of azide 1a in acetonitrile/water (3:7, v/v) for 24 h in a quartz reactor, a complete conversion of starting compound 1a according to HPLC monitoring was observed. Following work-up and preparative column chromatography azepine 3a could be isolated in 50% yield. For the preparative synthesis of 3c the method had to be slightly modified: In this case the reaction was performed in 1,4-dioxane/water (1:10, v/v) solution and irradiated for 1.5 h in a quartz reactor. After this, the conversion of 1c (according to HPLC monitoring) was found to be 100% and azepine 3c was isolated after preparative column chromatography in 50% yield (see Supporting Information File 1).
As is also shown in Table 3, the high photochemical sensitivity of both the C–I bond and the azide group present in 1e, unlike the others, complicates the synthesis of benzisoxazolone 2e. The photolysis of 1e under these conditions resulted in the formation of several products in low yields. Therefore, the synthesis towards benzisoxazolone 2e was reoptimized (Table 4). It was found that increasing the amount and strength of the base resulted in an increased selectivity and reaction rate. Indeed, using 10 equiv of sodium hydroxide as the base in the reaction resulted in a 51% yield of benzisoxazole 2e at complete photolysis of 1e (Table 4, entry 10). Under these conditions, in addition to compound 2e, the formation of 2a together with some other unidentified products was observed (albeit in low yields). A similar observation has been previously reported by Platz et al. [65].
Table 4: Optimization of conditions for the synthesis of 5-iodo-2,1-benzisoxazole-3(1H)-one (2e).a
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-86-i6.svg?max-width=637&scale=1.0)
|
||||
| Entry | Base (equiv) | Solvent | Yield (2e)b | t, minc |
|---|---|---|---|---|
| 1 | – | 1,4-dioxane/water 1:1 (v/v) | 6% | 60 |
| 2 | – | EtOH | 12% | 140 |
| 3 | NaOAc (1) | EtOH | 16% | 60 |
| 4 | NaOAc (1) | EtOH | 10% | 160 |
| 5 | NaOAc (1) | 1,4-dioxane/water 1:1 (v/v) | 5% | 60 |
| 6 | NaOAc (1.4) | EtOH | 14% | 60 |
| 7 | NaOAc (1.2) | EtOH | 13% | 90 |
| 8 | KOH (2.8) | EtOH | 30% | 90 |
| 9 | KOH (2.8) | EtOH | 18% | 110 |
| 10 | KOH (10) | EtOH | 51% | 60 |
| 11 | KOH (10) | EtOH | 34% | 90 |
aReaction conditions: 1e (0.78 mmol), solvent (15.0 mL); UV light (2 ×15 W Hg low-pressure lamp (254 nm), UV intensity was approximately 7 mW/cm2). bYields determined by HPLC analysis using an external standard. cIrradiation time: the degree of conversion of 1e was 100%.
Based on the results mentioned above and described in the related reports (Scheme 1), a possible reaction mechanism for the formation of 2 was proposed (Scheme 2). At first, 1 produced a salt of 2-azidobenzoic acid 1 (Scheme 2, 1-anion) by neutralization of a base. Next, the salt was decomposed by irradiation and the singlet nitrene A (Scheme 2, intermediate A) was formed. Finally, the electron pair of the carboxylic group (Scheme 2, intermediate A) was joined by 1,5-electrocyclization to the electron-deficient singlet nitrene A with formation of 2-anion (see Scheme 2), which was neutralized by water (path I). Thus, the first path of cyclization of 1 was realized.
![[1860-5397-12-86-i2]](/bjoc/content/inline/1860-5397-12-86-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed reaction mechanism of the base-mediated photochemical cyclization of 2-azidobenzoic acids.
Scheme 2: Proposed reaction mechanism of the base-mediated photochemical cyclization of 2-azidobenzoic acids.
Meanwhile a molecular form of 1 produced azepine 3 (path II) by irradiation. The detailed mechanism of the formation of 3 is shown in Scheme 2.
Thus, in the photochemical reaction both ionic and molecular forms of 1 can be used. To increase the yield of 2, it is necessary to shift the equilibrium towards the ionic form 1 (in situ salt formation).
Conclusion
In summary, we have developed an effective photochemical cyclization strategy for the synthesis of functionalized 2,1-benzisoxazole-3(1H)-ones. The present work offers a method to access 2,1-benzisoxazole-3(1H)-ones in good yields by using mild reaction conditions at room temperature. The proposed photochemical strategy permits the synthesis the high thermo-labile compounds from the class of 2,1-benzisoxazole-3(1H)-ones. Based on the results of the control experiments, it was found that an important stage of the ring closure is the formation of 2-azidobenzoate anion photolysis that results in the heterocyclization product.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization and spectral data for synthesized compounds. | ||
| Format: PDF | Size: 444.0 KB | Download |
References
-
Pierce, A. C.; Jacobs, M.; Stuver-Moody, C. J. Med. Chem. 2008, 51, 1972–1975. doi:10.1021/jm701248t
Return to citation in text: [1] -
Parish, C. A.; Smith, S. K.; Calati, K.; Zink, D.; Wilson, K.; Roemer, T.; Jiang, B.; Xu, D.; Bills, G.; Platas, G.; Peláez, F.; Díez, M. T.; Tsou, N.; McKeown, A. E.; Ball, R. G.; Powles, M. A.; Yeung, L.; Liberator, P.; Harris, G. J. Am. Chem. Soc. 2008, 130, 7060–7066. doi:10.1021/ja711209p
Return to citation in text: [1] -
Tong, Y.; Stewart, K. D.; Thomas, S.; Przytulinska, M.; Johnson, E. F.; Klinghofer, V.; Leverson, J.; McCall, O.; Soni, N. B.; Luo, Y.; Lin, N.-h.; Sowin, T. J.; Giranda, V. L.; Penning, T. D. Bioorg. Med. Chem. Lett. 2008, 18, 5206–5208. doi:10.1016/j.bmcl.2008.08.079
Return to citation in text: [1] -
Puetz, C.; Sundermann, C.; Sundermann, B.; Ijzerman, A.; Tromp, R.; Von Frijtag, D.; Kuenzel, J. Analogs of nitrobenzylthioinosine. U.S. Patent 7,358,235 B2, May 5, 2005.
Return to citation in text: [1] -
McClay, K. R.; Steffan, R. J. Antimicrobial compounds. U.S. Patent 7,973,065 B2, July 5, 2011.
Return to citation in text: [1] -
Bräse, S.; Gläser, F.; Kramer, C.; Lindner, S.; Linsenmeier, A. M.; Masters, K.-S.; Meister, A. C.; Ruff, B. M.; Zhong, S. The chemistry of mycotoxins; Springer: Berlin, Germany, 2013; Vol. 97. doi:10.1007/978-3-7091-1312-7
Return to citation in text: [1] -
Rahimizadeh, M.; Pordel, M.; Bakavoli, M.; Bakhtiarpoor, Z.; Orafaie, A. Monatsh. Chem. 2009, 140, 633–638. doi:10.1007/s00706-009-0109-7
Return to citation in text: [1] -
Sadeghian, A.; Pordel, M.; Safdari, H.; Fahmidekar, M. A.; Sadeghian, H. Med. Chem. Res. 2012, 21, 3897–3901. doi:10.1007/s00044-011-9933-5
Return to citation in text: [1] -
Grimberg, B. T.; Jaworska, M. M.; Hough, L. B.; Zimmerman, P. A.; Phillips, J. G. Bioorg. Med. Chem. Lett. 2009, 19, 5452–5457. doi:10.1016/j.bmcl.2009.07.095
Return to citation in text: [1] -
Fan, X.; Zhang, Y. Tetrahedron Lett. 2002, 43, 7001–7003. doi:10.1016/S0040-4039(02)01583-6
Return to citation in text: [1] -
Li, Q.; Woods, K. W.; Wang, W.; Lin, N.-H.; Claiborne, A.; Gu, W.-z.; Cohen, J.; Stoll, V. S.; Hutchins, C.; Frost, D.; Rosenberg, S. H.; Sham, H. L. Bioorg. Med. Chem. Lett. 2005, 15, 2033–2039. doi:10.1016/j.bmcl.2005.02.062
Return to citation in text: [1] -
Kraus, J. M.; Verlinde, C. L. M. J.; Karimi, M.; Lepesheva, G. I.; Gelb, M. H.; Buckner, F. S. J. Med. Chem. 2009, 52, 1639–1647. doi:10.1021/jm801313t
Return to citation in text: [1] -
Kraus, J. M.; Tatipaka, H. B.; McGuffin, S. A.; Chennamaneni, N. K.; Karimi, M.; Arif, J.; Verlinde, C. L. M. J.; Buckner, F. S.; Gelb, M. H. J. Med. Chem. 2010, 53, 3887–3898. doi:10.1021/jm9013136
Return to citation in text: [1] -
Chattopadhyaya, J.; Upadhayaya, R. S. Quinoline, naphthalene and conformationally constrained quinoline or naphthalene derivates as anti-mycobacterial agents. WO Patent WO2009091324 A1, Jan 9, 2009.
Return to citation in text: [1] -
Lyssikatos, J. P.; Greca, S. D. L.; Yang, B. V. Quinolin-2-one derivatives useful as anticancer agents. U.S. Patent 6,495,564 B1, Dec 17, 2002.
Return to citation in text: [1] -
Massarotti, A.; Theeramunkong, S.; Mesenzani, O.; Caldarelli, A.; Genazzani, A. A.; Tron, G. C. Chem. Biol. Drug Des. 2011, 78, 913–922. doi:10.1111/j.1747-0285.2011.01245.x
Return to citation in text: [1] -
Bamberger, E.; Pyman, F. L. Ber. Dtsch. Chem. Ges. 1909, 42, 2297–2330. doi:10.1002/cber.190904202129
Return to citation in text: [1] -
Wierenga, W.; Harrison, A. W.; Evans, B. R.; Chidester, C. G. J. Org. Chem. 1984, 49, 438–442. doi:10.1021/jo00177a010
Return to citation in text: [1] -
Wierenga, W.; Evans, B. R.; Zurenko, G. E. J. Med. Chem. 1984, 27, 1212–1215. doi:10.1021/jm00375a022
Return to citation in text: [1] [2] -
Chauhan, J.; Fletcher, S. Tetrahedron Lett. 2012, 53, 4951–4954. doi:10.1016/j.tetlet.2012.07.006
Return to citation in text: [1] -
Kim, B. H.; Jin, Y.; Jun, Y. M.; Han, R.; Baik, W.; Lee, B. M. Tetrahedron Lett. 2000, 41, 2137–2140. doi:10.1016/S0040-4039(00)00098-8
Return to citation in text: [1] -
Wiecław, M.; Bobin, M.; Kwast, A.; Bujok, R.; Wróbel, Z.; Wojciechowski, K. Mol. Diversity 2015, 19, 807–816. doi:10.1007/s11030-015-9627-x
Return to citation in text: [1] -
Otley, K. D.; Ellman, J. A. J. Org. Chem. 2014, 79, 8296–8303. doi:10.1021/jo5015432
Return to citation in text: [1] -
Smith, P. A. S.; Brown, B. B.; Putney, R. K.; Reinisch, R. F. J. Am. Chem. Soc. 1953, 75, 6335–6337. doi:10.1021/ja01120a534
Return to citation in text: [1] [2] -
Stokes, B. J.; Vogel, C. V.; Urnezis, L. K.; Pan, M.; Driver, T. G. Org. Lett. 2010, 12, 2884–2887. doi:10.1021/ol101040p
Return to citation in text: [1] -
Anand, D.; Patel, O. P. S.; Maurya, R. K.; Kant, R.; Yadav, P. P. J. Org. Chem. 2015, 80, 12410–12419. doi:10.1021/acs.joc.5b02276
Return to citation in text: [1] -
Kotov, A. D.; Prokaznikov, M. A.; Antonova, E. A.; Rusakov, A. I. Chem. Heterocycl. Compd. 2014, 50, 647–657. doi:10.1007/s10593-014-1517-0
Return to citation in text: [1] -
Budruev, A. V.; Karyakina, L. N.; Oleinik, A. V. High Energy Chem. 2003, 37, 29–33. doi:10.1023/A:1021961526268
Return to citation in text: [1] [2] -
Budruev, A. V.; Karyakina, L. N.; Oleinik, A. V. High Energy Chem. 2004, 38, 20–24. doi:10.1023/B:HIEC.0000012059.38366.8e
Return to citation in text: [1] [2] -
Sinjagina, D. Yu.; Budruev, A. V. High Energy Chem. 2013, 47, 162–165. doi:10.1134/S0018143913040127
Return to citation in text: [1] [2] [3] [4] [5] -
Chainikova, E.; Teregulova, A.; Lobov, A.; Erastov, A.; Safiullin, R. Tetrahedron Lett. 2015, 56, 1332–1334. doi:10.1016/j.tetlet.2015.02.014
Return to citation in text: [1] -
Kabanda, M. M.; Ebenso, E. E. J. Theor. Comput. Chem. 2013, 12, 1350070. doi:10.1142/S0219633613500703
Return to citation in text: [1] -
Mitsumori, T.; Sekine, A.; Uekusa, H.; Ohashi, Y. Acta Crystallogr., Sect. B: Struct. Sci. 2010, 66, 647–661. doi:10.1107/S010876811003661X
Return to citation in text: [1] -
Doering, W. von E.; Odum, R. A. Tetrahedron 1966, 22, 81–93. doi:10.1016/0040-4020(66)80104-7
Return to citation in text: [1] [2] -
Splitter, J. S.; Calvin, M. Tetrahedron Lett. 1968, 9, 1445–1448. doi:10.1016/S0040-4039(01)98975-0
Return to citation in text: [1] [2] -
Sundberg, R. J.; Suter, S. R.; Brenner, M. J. Am. Chem. Soc. 1972, 94, 513–520. doi:10.1021/ja00757a032
Return to citation in text: [1] [2] -
Inui, H.; Sawada, K.; Oishi, S.; Ushida, K.; McMahon, R. J. J. Am. Chem. Soc. 2013, 135, 10246–10249. doi:10.1021/ja404172s
Return to citation in text: [1] [2] -
Reiser, A.; Wagner, H. M. The azido group. In The Chemistry of the Azido Group; Patai, S., Ed.; John Wiley & Sons: Chichester, United Kingdom, 1971; pp 441–501. doi:10.1002/9780470771266.ch8
Return to citation in text: [1] [2] -
Smith, P. A. S. Aryl and Heteroaryl Azides and Nitrenes. In Azides and nitrenes, reactivity and utility; Scriven, E. F. V., Ed.; Academic Press: New York, NY, U.S.A., 1984; pp 95–204. doi:10.1016/B978-0-12-633480-7.50007-9
Return to citation in text: [1] [2] [3] [4] -
Scriven, E. F. V.; Turnbull, K. Chem. Rev. 1988, 88, 297–368. doi:10.1021/cr00084a001
Return to citation in text: [1] [2] -
Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Angew. Chem., Int. Ed. 2005, 44, 5188–5240. doi:10.1002/anie.200400657
Angew. Chem., Int. Ed. 2005, 117, 5320–5374. doi:10.1002/ange.200400657
Return to citation in text: [1] [2] -
Bucher, G. Photochemical Reactivity of Azides. In CRC Handbook of Organic Photochemistry and Photobiology; Horspool, W. M.; Lenci, F., Eds.; CRC Press: Boca Raton, FL, U.S.A., 2010; Vol. 1 & 2, pp 1–31.
Return to citation in text: [1] [2] -
Albini, A.; Fagnoni, M. Photogeneration of carbenes and nitrenes. Photochemically Generated Intermediates in Synthesis; John Wiley & Sons: Hoboken, NJ, U.S.A., 2013; pp 302–327. doi:10.1002/9781118689202.ch07
Return to citation in text: [1] [2] -
Lwowski, W., Ed. Nitrenes; Interscience: New York, NY, U.S.A., 1970; pp 47–98.
Return to citation in text: [1] [2] -
Li, Y.-Z.; Kirby, J. P.; George, M. W.; Poliakoff, M.; Schuster, G. B. J. Am. Chem. Soc. 1988, 110, 8092–8098. doi:10.1021/ja00232a022
Return to citation in text: [1] [2] -
Hayes, J. C.; Sheridan, R. S. J. Am. Chem. Soc. 1990, 112, 5879–5881. doi:10.1021/ja00171a038
Return to citation in text: [1] [2] -
Donnelly, T.; Dunkin, I. R.; Norwood, D. S. D.; Prentice, A.; Shields, C. J.; Thomson, P. C. P. J. Chem. Soc., Perkin Trans. 2 1985, 307–310. doi:10.1039/p29850000307
Return to citation in text: [1] [2] -
Budyka, M. F.; Kantor, M. M.; Alfimov, M. V. Russ. Chem. Rev. 1992, 61, 25–39. doi:10.1070/RC1992v061n01ABEH000929
Return to citation in text: [1] [2] -
Gritsan, N.; Platz, M. Photochemistry of Azides: The Azide/Nitrene Interface. In Organic Azides: Syntheses and Applications; Bräse, S.; Banert, K., Eds.; John Wiley & Sons: Hoboken, NJ, U.S.A., 2010; pp 311–372.
Return to citation in text: [1] [2] -
Schrock, A. K.; Schuster, G. B. J. Am. Chem. Soc. 1984, 106, 5228–5234. doi:10.1021/ja00330a032
Return to citation in text: [1] [2] -
Platz, M. S. Nitrenes. In Reactive Intermediate Chemistry; Moss, R. A.; Platz, M. S.; Jones, M., Jr., Eds.; John Wiley & Sons: Hoboken, NJ, U.S.A., 2004; pp 501–559.
Return to citation in text: [1] [2] -
Dyall, L. K.; Kemp, J. E. J. Chem. Soc. B 1968, 976–979. doi:10.1039/j29680000976
Return to citation in text: [1] [2] -
Dyall, L. K. Aust. J. Chem. 1977, 30, 2669–2678. doi:10.1071/CH9772669
Return to citation in text: [1] [2] -
McCulla, R. D.; Burdzinski, G.; Platz, M. S. Org. Lett. 2006, 8, 1637–1640. doi:10.1021/ol0602452
Return to citation in text: [1] [2] -
Nakayama, H.; Nozawa, M.; Kanaoka, Y. Chem. Pharm. Bull. 1979, 27, 2775–2780. doi:10.1248/cpb.27.2775
Return to citation in text: [1] [2] -
Hudlicky, T. Organic Synthesis: Theory and Applications; JAI Press: Amsterdam, Netherlands, 1996; Vol. 3, pp 149–229.
Return to citation in text: [1] [2] -
Eswaran, S. V. 2,1-Benzisoxazoles. In The Chemistry of Heterocyclic Compounds: Isoxazoles; Grünanger, P.; Vita-Fenzi, P.; Dowling, J. E., Eds.; John Wiley & Sons: New York, NY, U.S.A., 1999; Vol. 49, pp 143–148. doi:10.1002/9780470187364.ch2
Return to citation in text: [1] [2] -
Hall, J. H.; Hill, J. W.; Fargher, J. M. J. Am. Chem. Soc. 1968, 90, 5313. doi:10.1021/ja01021a069
Return to citation in text: [1] [2] -
Reiser, A.; Willets, F. W.; Terry, G. C.; Williams, V.; Marley, R. Trans. Faraday Soc. 1968, 64, 3265. doi:10.1039/tf9686403265
Return to citation in text: [1] [2] -
Koziar, J. C.; Cowan, D. O. Acc. Chem. Res. 1978, 11, 334–341. doi:10.1021/ar50129a003
Return to citation in text: [1] -
Abramovitch, R. A.; Challand, S. R.; Scriven, E. F. V. J. Am. Chem. Soc. 1972, 94, 1374–1376. doi:10.1021/ja00759a066
Return to citation in text: [1] -
Banks, R. E.; Sparkes, G. R. J. Chem. Soc., Perkin Trans. 1 1972, 2964–2970. doi:10.1039/p19720002964
Return to citation in text: [1] -
Lamara, K.; Smalley, R. K. Tetrahedron 1991, 47, 2277. doi:10.1016/S0040-4020(01)96138-1
Return to citation in text: [1] -
Bou-Hamdan, F. R.; Lévesque, F.; O’Brien, A. G.; Seeberger, P. H. Beilstein J. Org. Chem. 2011, 7, 1124. doi:10.3762/bjoc.7.129
Return to citation in text: [1] -
Watt, D. S.; Kawada, K.; Leyva, E.; Platz, M. S. Tetrahedron Lett. 1989, 30, 899–902. doi:10.1016/S0040-4039(00)95273-0
Return to citation in text: [1]
| 58. | Hall, J. H.; Hill, J. W.; Fargher, J. M. J. Am. Chem. Soc. 1968, 90, 5313. doi:10.1021/ja01021a069 |
| 59. | Reiser, A.; Willets, F. W.; Terry, G. C.; Williams, V.; Marley, R. Trans. Faraday Soc. 1968, 64, 3265. doi:10.1039/tf9686403265 |
| 39. | Smith, P. A. S. Aryl and Heteroaryl Azides and Nitrenes. In Azides and nitrenes, reactivity and utility; Scriven, E. F. V., Ed.; Academic Press: New York, NY, U.S.A., 1984; pp 95–204. doi:10.1016/B978-0-12-633480-7.50007-9 |
| 1. | Pierce, A. C.; Jacobs, M.; Stuver-Moody, C. J. Med. Chem. 2008, 51, 1972–1975. doi:10.1021/jm701248t |
| 2. | Parish, C. A.; Smith, S. K.; Calati, K.; Zink, D.; Wilson, K.; Roemer, T.; Jiang, B.; Xu, D.; Bills, G.; Platas, G.; Peláez, F.; Díez, M. T.; Tsou, N.; McKeown, A. E.; Ball, R. G.; Powles, M. A.; Yeung, L.; Liberator, P.; Harris, G. J. Am. Chem. Soc. 2008, 130, 7060–7066. doi:10.1021/ja711209p |
| 3. | Tong, Y.; Stewart, K. D.; Thomas, S.; Przytulinska, M.; Johnson, E. F.; Klinghofer, V.; Leverson, J.; McCall, O.; Soni, N. B.; Luo, Y.; Lin, N.-h.; Sowin, T. J.; Giranda, V. L.; Penning, T. D. Bioorg. Med. Chem. Lett. 2008, 18, 5206–5208. doi:10.1016/j.bmcl.2008.08.079 |
| 4. | Puetz, C.; Sundermann, C.; Sundermann, B.; Ijzerman, A.; Tromp, R.; Von Frijtag, D.; Kuenzel, J. Analogs of nitrobenzylthioinosine. U.S. Patent 7,358,235 B2, May 5, 2005. |
| 5. | McClay, K. R.; Steffan, R. J. Antimicrobial compounds. U.S. Patent 7,973,065 B2, July 5, 2011. |
| 6. | Bräse, S.; Gläser, F.; Kramer, C.; Lindner, S.; Linsenmeier, A. M.; Masters, K.-S.; Meister, A. C.; Ruff, B. M.; Zhong, S. The chemistry of mycotoxins; Springer: Berlin, Germany, 2013; Vol. 97. doi:10.1007/978-3-7091-1312-7 |
| 17. | Bamberger, E.; Pyman, F. L. Ber. Dtsch. Chem. Ges. 1909, 42, 2297–2330. doi:10.1002/cber.190904202129 |
| 18. | Wierenga, W.; Harrison, A. W.; Evans, B. R.; Chidester, C. G. J. Org. Chem. 1984, 49, 438–442. doi:10.1021/jo00177a010 |
| 19. | Wierenga, W.; Evans, B. R.; Zurenko, G. E. J. Med. Chem. 1984, 27, 1212–1215. doi:10.1021/jm00375a022 |
| 20. | Chauhan, J.; Fletcher, S. Tetrahedron Lett. 2012, 53, 4951–4954. doi:10.1016/j.tetlet.2012.07.006 |
| 21. | Kim, B. H.; Jin, Y.; Jun, Y. M.; Han, R.; Baik, W.; Lee, B. M. Tetrahedron Lett. 2000, 41, 2137–2140. doi:10.1016/S0040-4039(00)00098-8 |
| 30. | Sinjagina, D. Yu.; Budruev, A. V. High Energy Chem. 2013, 47, 162–165. doi:10.1134/S0018143913040127 |
| 14. | Chattopadhyaya, J.; Upadhayaya, R. S. Quinoline, naphthalene and conformationally constrained quinoline or naphthalene derivates as anti-mycobacterial agents. WO Patent WO2009091324 A1, Jan 9, 2009. |
| 15. | Lyssikatos, J. P.; Greca, S. D. L.; Yang, B. V. Quinolin-2-one derivatives useful as anticancer agents. U.S. Patent 6,495,564 B1, Dec 17, 2002. |
| 16. | Massarotti, A.; Theeramunkong, S.; Mesenzani, O.; Caldarelli, A.; Genazzani, A. A.; Tron, G. C. Chem. Biol. Drug Des. 2011, 78, 913–922. doi:10.1111/j.1747-0285.2011.01245.x |
| 34. | Doering, W. von E.; Odum, R. A. Tetrahedron 1966, 22, 81–93. doi:10.1016/0040-4020(66)80104-7 |
| 35. | Splitter, J. S.; Calvin, M. Tetrahedron Lett. 1968, 9, 1445–1448. doi:10.1016/S0040-4039(01)98975-0 |
| 9. | Grimberg, B. T.; Jaworska, M. M.; Hough, L. B.; Zimmerman, P. A.; Phillips, J. G. Bioorg. Med. Chem. Lett. 2009, 19, 5452–5457. doi:10.1016/j.bmcl.2009.07.095 |
| 10. | Fan, X.; Zhang, Y. Tetrahedron Lett. 2002, 43, 7001–7003. doi:10.1016/S0040-4039(02)01583-6 |
| 11. | Li, Q.; Woods, K. W.; Wang, W.; Lin, N.-H.; Claiborne, A.; Gu, W.-z.; Cohen, J.; Stoll, V. S.; Hutchins, C.; Frost, D.; Rosenberg, S. H.; Sham, H. L. Bioorg. Med. Chem. Lett. 2005, 15, 2033–2039. doi:10.1016/j.bmcl.2005.02.062 |
| 12. | Kraus, J. M.; Verlinde, C. L. M. J.; Karimi, M.; Lepesheva, G. I.; Gelb, M. H.; Buckner, F. S. J. Med. Chem. 2009, 52, 1639–1647. doi:10.1021/jm801313t |
| 13. | Kraus, J. M.; Tatipaka, H. B.; McGuffin, S. A.; Chennamaneni, N. K.; Karimi, M.; Arif, J.; Verlinde, C. L. M. J.; Buckner, F. S.; Gelb, M. H. J. Med. Chem. 2010, 53, 3887–3898. doi:10.1021/jm9013136 |
| 33. | Mitsumori, T.; Sekine, A.; Uekusa, H.; Ohashi, Y. Acta Crystallogr., Sect. B: Struct. Sci. 2010, 66, 647–661. doi:10.1107/S010876811003661X |
| 65. | Watt, D. S.; Kawada, K.; Leyva, E.; Platz, M. S. Tetrahedron Lett. 1989, 30, 899–902. doi:10.1016/S0040-4039(00)95273-0 |
| 7. | Rahimizadeh, M.; Pordel, M.; Bakavoli, M.; Bakhtiarpoor, Z.; Orafaie, A. Monatsh. Chem. 2009, 140, 633–638. doi:10.1007/s00706-009-0109-7 |
| 8. | Sadeghian, A.; Pordel, M.; Safdari, H.; Fahmidekar, M. A.; Sadeghian, H. Med. Chem. Res. 2012, 21, 3897–3901. doi:10.1007/s00044-011-9933-5 |
| 34. | Doering, W. von E.; Odum, R. A. Tetrahedron 1966, 22, 81–93. doi:10.1016/0040-4020(66)80104-7 |
| 35. | Splitter, J. S.; Calvin, M. Tetrahedron Lett. 1968, 9, 1445–1448. doi:10.1016/S0040-4039(01)98975-0 |
| 36. | Sundberg, R. J.; Suter, S. R.; Brenner, M. J. Am. Chem. Soc. 1972, 94, 513–520. doi:10.1021/ja00757a032 |
| 37. | Inui, H.; Sawada, K.; Oishi, S.; Ushida, K.; McMahon, R. J. J. Am. Chem. Soc. 2013, 135, 10246–10249. doi:10.1021/ja404172s |
| 38. | Reiser, A.; Wagner, H. M. The azido group. In The Chemistry of the Azido Group; Patai, S., Ed.; John Wiley & Sons: Chichester, United Kingdom, 1971; pp 441–501. doi:10.1002/9780470771266.ch8 |
| 39. | Smith, P. A. S. Aryl and Heteroaryl Azides and Nitrenes. In Azides and nitrenes, reactivity and utility; Scriven, E. F. V., Ed.; Academic Press: New York, NY, U.S.A., 1984; pp 95–204. doi:10.1016/B978-0-12-633480-7.50007-9 |
| 40. | Scriven, E. F. V.; Turnbull, K. Chem. Rev. 1988, 88, 297–368. doi:10.1021/cr00084a001 |
| 41. |
Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Angew. Chem., Int. Ed. 2005, 44, 5188–5240. doi:10.1002/anie.200400657
Angew. Chem., Int. Ed. 2005, 117, 5320–5374. doi:10.1002/ange.200400657 |
| 42. | Bucher, G. Photochemical Reactivity of Azides. In CRC Handbook of Organic Photochemistry and Photobiology; Horspool, W. M.; Lenci, F., Eds.; CRC Press: Boca Raton, FL, U.S.A., 2010; Vol. 1 & 2, pp 1–31. |
| 43. | Albini, A.; Fagnoni, M. Photogeneration of carbenes and nitrenes. Photochemically Generated Intermediates in Synthesis; John Wiley & Sons: Hoboken, NJ, U.S.A., 2013; pp 302–327. doi:10.1002/9781118689202.ch07 |
| 44. | Lwowski, W., Ed. Nitrenes; Interscience: New York, NY, U.S.A., 1970; pp 47–98. |
| 45. | Li, Y.-Z.; Kirby, J. P.; George, M. W.; Poliakoff, M.; Schuster, G. B. J. Am. Chem. Soc. 1988, 110, 8092–8098. doi:10.1021/ja00232a022 |
| 46. | Hayes, J. C.; Sheridan, R. S. J. Am. Chem. Soc. 1990, 112, 5879–5881. doi:10.1021/ja00171a038 |
| 47. | Donnelly, T.; Dunkin, I. R.; Norwood, D. S. D.; Prentice, A.; Shields, C. J.; Thomson, P. C. P. J. Chem. Soc., Perkin Trans. 2 1985, 307–310. doi:10.1039/p29850000307 |
| 48. | Budyka, M. F.; Kantor, M. M.; Alfimov, M. V. Russ. Chem. Rev. 1992, 61, 25–39. doi:10.1070/RC1992v061n01ABEH000929 |
| 49. | Gritsan, N.; Platz, M. Photochemistry of Azides: The Azide/Nitrene Interface. In Organic Azides: Syntheses and Applications; Bräse, S.; Banert, K., Eds.; John Wiley & Sons: Hoboken, NJ, U.S.A., 2010; pp 311–372. |
| 50. | Schrock, A. K.; Schuster, G. B. J. Am. Chem. Soc. 1984, 106, 5228–5234. doi:10.1021/ja00330a032 |
| 51. | Platz, M. S. Nitrenes. In Reactive Intermediate Chemistry; Moss, R. A.; Platz, M. S.; Jones, M., Jr., Eds.; John Wiley & Sons: Hoboken, NJ, U.S.A., 2004; pp 501–559. |
| 52. | Dyall, L. K.; Kemp, J. E. J. Chem. Soc. B 1968, 976–979. doi:10.1039/j29680000976 |
| 53. | Dyall, L. K. Aust. J. Chem. 1977, 30, 2669–2678. doi:10.1071/CH9772669 |
| 54. | McCulla, R. D.; Burdzinski, G.; Platz, M. S. Org. Lett. 2006, 8, 1637–1640. doi:10.1021/ol0602452 |
| 55. | Nakayama, H.; Nozawa, M.; Kanaoka, Y. Chem. Pharm. Bull. 1979, 27, 2775–2780. doi:10.1248/cpb.27.2775 |
| 56. | Hudlicky, T. Organic Synthesis: Theory and Applications; JAI Press: Amsterdam, Netherlands, 1996; Vol. 3, pp 149–229. |
| 57. | Eswaran, S. V. 2,1-Benzisoxazoles. In The Chemistry of Heterocyclic Compounds: Isoxazoles; Grünanger, P.; Vita-Fenzi, P.; Dowling, J. E., Eds.; John Wiley & Sons: New York, NY, U.S.A., 1999; Vol. 49, pp 143–148. doi:10.1002/9780470187364.ch2 |
| 58. | Hall, J. H.; Hill, J. W.; Fargher, J. M. J. Am. Chem. Soc. 1968, 90, 5313. doi:10.1021/ja01021a069 |
| 59. | Reiser, A.; Willets, F. W.; Terry, G. C.; Williams, V.; Marley, R. Trans. Faraday Soc. 1968, 64, 3265. doi:10.1039/tf9686403265 |
| 28. | Budruev, A. V.; Karyakina, L. N.; Oleinik, A. V. High Energy Chem. 2003, 37, 29–33. doi:10.1023/A:1021961526268 |
| 29. | Budruev, A. V.; Karyakina, L. N.; Oleinik, A. V. High Energy Chem. 2004, 38, 20–24. doi:10.1023/B:HIEC.0000012059.38366.8e |
| 30. | Sinjagina, D. Yu.; Budruev, A. V. High Energy Chem. 2013, 47, 162–165. doi:10.1134/S0018143913040127 |
| 31. | Chainikova, E.; Teregulova, A.; Lobov, A.; Erastov, A.; Safiullin, R. Tetrahedron Lett. 2015, 56, 1332–1334. doi:10.1016/j.tetlet.2015.02.014 |
| 30. | Sinjagina, D. Yu.; Budruev, A. V. High Energy Chem. 2013, 47, 162–165. doi:10.1134/S0018143913040127 |
| 30. | Sinjagina, D. Yu.; Budruev, A. V. High Energy Chem. 2013, 47, 162–165. doi:10.1134/S0018143913040127 |
| 63. | Lamara, K.; Smalley, R. K. Tetrahedron 1991, 47, 2277. doi:10.1016/S0040-4020(01)96138-1 |
| 64. | Bou-Hamdan, F. R.; Lévesque, F.; O’Brien, A. G.; Seeberger, P. H. Beilstein J. Org. Chem. 2011, 7, 1124. doi:10.3762/bjoc.7.129 |
| 25. | Stokes, B. J.; Vogel, C. V.; Urnezis, L. K.; Pan, M.; Driver, T. G. Org. Lett. 2010, 12, 2884–2887. doi:10.1021/ol101040p |
| 26. | Anand, D.; Patel, O. P. S.; Maurya, R. K.; Kant, R.; Yadav, P. P. J. Org. Chem. 2015, 80, 12410–12419. doi:10.1021/acs.joc.5b02276 |
| 27. | Kotov, A. D.; Prokaznikov, M. A.; Antonova, E. A.; Rusakov, A. I. Chem. Heterocycl. Compd. 2014, 50, 647–657. doi:10.1007/s10593-014-1517-0 |
| 28. | Budruev, A. V.; Karyakina, L. N.; Oleinik, A. V. High Energy Chem. 2003, 37, 29–33. doi:10.1023/A:1021961526268 |
| 29. | Budruev, A. V.; Karyakina, L. N.; Oleinik, A. V. High Energy Chem. 2004, 38, 20–24. doi:10.1023/B:HIEC.0000012059.38366.8e |
| 30. | Sinjagina, D. Yu.; Budruev, A. V. High Energy Chem. 2013, 47, 162–165. doi:10.1134/S0018143913040127 |
| 24. | Smith, P. A. S.; Brown, B. B.; Putney, R. K.; Reinisch, R. F. J. Am. Chem. Soc. 1953, 75, 6335–6337. doi:10.1021/ja01120a534 |
| 60. | Koziar, J. C.; Cowan, D. O. Acc. Chem. Res. 1978, 11, 334–341. doi:10.1021/ar50129a003 |
| 22. | Wiecław, M.; Bobin, M.; Kwast, A.; Bujok, R.; Wróbel, Z.; Wojciechowski, K. Mol. Diversity 2015, 19, 807–816. doi:10.1007/s11030-015-9627-x |
| 23. | Otley, K. D.; Ellman, J. A. J. Org. Chem. 2014, 79, 8296–8303. doi:10.1021/jo5015432 |
| 32. | Kabanda, M. M.; Ebenso, E. E. J. Theor. Comput. Chem. 2013, 12, 1350070. doi:10.1142/S0219633613500703 |
| 61. | Abramovitch, R. A.; Challand, S. R.; Scriven, E. F. V. J. Am. Chem. Soc. 1972, 94, 1374–1376. doi:10.1021/ja00759a066 |
| 62. | Banks, R. E.; Sparkes, G. R. J. Chem. Soc., Perkin Trans. 1 1972, 2964–2970. doi:10.1039/p19720002964 |
| 19. | Wierenga, W.; Evans, B. R.; Zurenko, G. E. J. Med. Chem. 1984, 27, 1212–1215. doi:10.1021/jm00375a022 |
| 36. | Sundberg, R. J.; Suter, S. R.; Brenner, M. J. Am. Chem. Soc. 1972, 94, 513–520. doi:10.1021/ja00757a032 |
| 37. | Inui, H.; Sawada, K.; Oishi, S.; Ushida, K.; McMahon, R. J. J. Am. Chem. Soc. 2013, 135, 10246–10249. doi:10.1021/ja404172s |
| 38. | Reiser, A.; Wagner, H. M. The azido group. In The Chemistry of the Azido Group; Patai, S., Ed.; John Wiley & Sons: Chichester, United Kingdom, 1971; pp 441–501. doi:10.1002/9780470771266.ch8 |
| 39. | Smith, P. A. S. Aryl and Heteroaryl Azides and Nitrenes. In Azides and nitrenes, reactivity and utility; Scriven, E. F. V., Ed.; Academic Press: New York, NY, U.S.A., 1984; pp 95–204. doi:10.1016/B978-0-12-633480-7.50007-9 |
| 40. | Scriven, E. F. V.; Turnbull, K. Chem. Rev. 1988, 88, 297–368. doi:10.1021/cr00084a001 |
| 41. |
Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Angew. Chem., Int. Ed. 2005, 44, 5188–5240. doi:10.1002/anie.200400657
Angew. Chem., Int. Ed. 2005, 117, 5320–5374. doi:10.1002/ange.200400657 |
| 42. | Bucher, G. Photochemical Reactivity of Azides. In CRC Handbook of Organic Photochemistry and Photobiology; Horspool, W. M.; Lenci, F., Eds.; CRC Press: Boca Raton, FL, U.S.A., 2010; Vol. 1 & 2, pp 1–31. |
| 43. | Albini, A.; Fagnoni, M. Photogeneration of carbenes and nitrenes. Photochemically Generated Intermediates in Synthesis; John Wiley & Sons: Hoboken, NJ, U.S.A., 2013; pp 302–327. doi:10.1002/9781118689202.ch07 |
| 44. | Lwowski, W., Ed. Nitrenes; Interscience: New York, NY, U.S.A., 1970; pp 47–98. |
| 45. | Li, Y.-Z.; Kirby, J. P.; George, M. W.; Poliakoff, M.; Schuster, G. B. J. Am. Chem. Soc. 1988, 110, 8092–8098. doi:10.1021/ja00232a022 |
| 46. | Hayes, J. C.; Sheridan, R. S. J. Am. Chem. Soc. 1990, 112, 5879–5881. doi:10.1021/ja00171a038 |
| 47. | Donnelly, T.; Dunkin, I. R.; Norwood, D. S. D.; Prentice, A.; Shields, C. J.; Thomson, P. C. P. J. Chem. Soc., Perkin Trans. 2 1985, 307–310. doi:10.1039/p29850000307 |
| 48. | Budyka, M. F.; Kantor, M. M.; Alfimov, M. V. Russ. Chem. Rev. 1992, 61, 25–39. doi:10.1070/RC1992v061n01ABEH000929 |
| 49. | Gritsan, N.; Platz, M. Photochemistry of Azides: The Azide/Nitrene Interface. In Organic Azides: Syntheses and Applications; Bräse, S.; Banert, K., Eds.; John Wiley & Sons: Hoboken, NJ, U.S.A., 2010; pp 311–372. |
| 56. | Hudlicky, T. Organic Synthesis: Theory and Applications; JAI Press: Amsterdam, Netherlands, 1996; Vol. 3, pp 149–229. |
| 57. | Eswaran, S. V. 2,1-Benzisoxazoles. In The Chemistry of Heterocyclic Compounds: Isoxazoles; Grünanger, P.; Vita-Fenzi, P.; Dowling, J. E., Eds.; John Wiley & Sons: New York, NY, U.S.A., 1999; Vol. 49, pp 143–148. doi:10.1002/9780470187364.ch2 |
| 39. | Smith, P. A. S. Aryl and Heteroaryl Azides and Nitrenes. In Azides and nitrenes, reactivity and utility; Scriven, E. F. V., Ed.; Academic Press: New York, NY, U.S.A., 1984; pp 95–204. doi:10.1016/B978-0-12-633480-7.50007-9 |
| 24. | Smith, P. A. S.; Brown, B. B.; Putney, R. K.; Reinisch, R. F. J. Am. Chem. Soc. 1953, 75, 6335–6337. doi:10.1021/ja01120a534 |
| 55. | Nakayama, H.; Nozawa, M.; Kanaoka, Y. Chem. Pharm. Bull. 1979, 27, 2775–2780. doi:10.1248/cpb.27.2775 |
| 52. | Dyall, L. K.; Kemp, J. E. J. Chem. Soc. B 1968, 976–979. doi:10.1039/j29680000976 |
| 53. | Dyall, L. K. Aust. J. Chem. 1977, 30, 2669–2678. doi:10.1071/CH9772669 |
| 54. | McCulla, R. D.; Burdzinski, G.; Platz, M. S. Org. Lett. 2006, 8, 1637–1640. doi:10.1021/ol0602452 |
| 50. | Schrock, A. K.; Schuster, G. B. J. Am. Chem. Soc. 1984, 106, 5228–5234. doi:10.1021/ja00330a032 |
| 51. | Platz, M. S. Nitrenes. In Reactive Intermediate Chemistry; Moss, R. A.; Platz, M. S.; Jones, M., Jr., Eds.; John Wiley & Sons: Hoboken, NJ, U.S.A., 2004; pp 501–559. |
© 2016 Dzhons and Budruev; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)