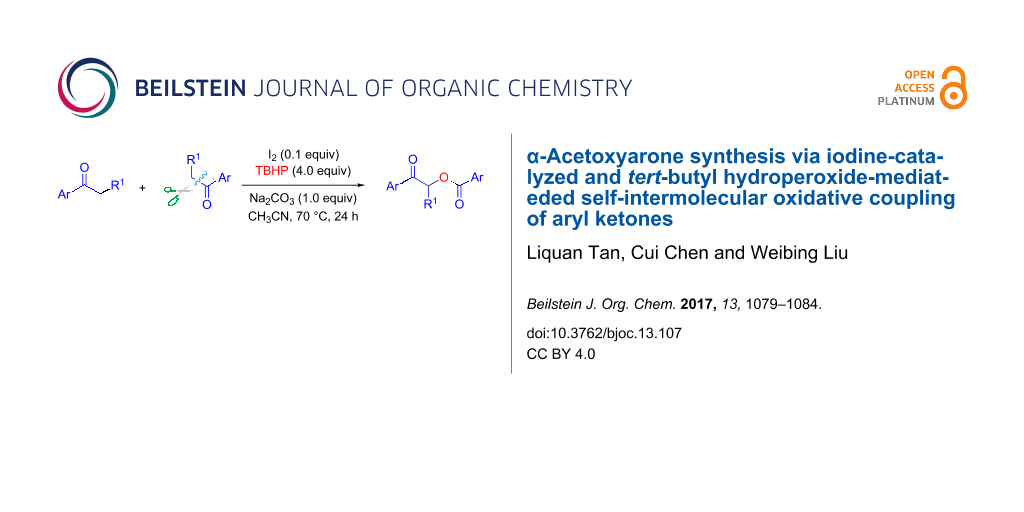
Abstract
We present a metal-free method for α-acetoxyarone synthesis by self-intermolecular oxidative coupling of aryl ketones using I2−tert-butyl hydroperoxide (TBHP). Under the optimum conditions, various aryl ketones gave the corresponding products in moderate to excellent yields. A series of control experiments were performed; the results suggest the involvement of radical pathways. Multiple radical intermediates were generated in situ and the overall process involved several different reactions, which proceeded self-sequentially in a single reactor. A labeling experiment using 18O-labeled H2O confirmed that the oxygen in the product was derived from TBHP, not from H2O in the TBHP solvent.

Graphical Abstract
Introduction
In recent years, α-acetoxyaryl ketones have attracted considerable interest because this structural motif is found in a variety of biologically active natural products and pharmaceuticals, and α-acetoxyaryl ketones are widely used as synthetic intermediates [1-5]. Traditional methods for the preparation of α-acyloxy ketones focus on the substitution reactions of α-halo carbonyl compounds with alkaline carboxylates or carboxylic acids [6,7], and transition-metal-catalyzed direct oxidative coupling reactions of carbonyl compounds with carboxylic acids (or their surrogates) [8,9]. Recently, robust approaches using organohypervalent iodine reagents and peroxide-mediated oxidative coupling have been developed [10,11]. Although impressive progress has been made [12], examples of the synthesis of α-acetoxyaryl ketones through self-intermolecular oxidative coupling of aryl ketones are still rare. Yan and coworkers reported the preparation of α-acyloxyaryl ketones from aryl ketones using a Pybox-copper(II) catalyst [13]. However, the substrate scope was limited to α-substituted aryl ketones, and acetophenones were unsuitable for this conversion. In addition, this method requires harsh catalytic conditions, using scarce iron and copper complexes. The development of novel metal-free methods for the preparation of α-acetoxyaryl ketones is therefore an attractive target for organic chemists. Simple, inexpensive, and metal-free methods [14,15], involving safe and clean oxidation procedures, need to be developed. Here, we report a metal-free, novel, and efficient self-intermolecular oxidative coupling procedure for the synthesis of α-acetoxyaryl ketones from aryl ketones using I2 and tert-butyl hydroperoxide (TBHP) [16-18] (Scheme 1). Several oxidative cross-coupling methods have been developed for the synthesis of α-acetoxy ketones from ketone derivatives and carboxylic acids [10], benzylic alcohols [19], toluene derivatives [20,21] and alkenes [22,23] using TBHP as the oxidant (Scheme 1). However, to the best of our knowledge, this is the first example of the use of TBHP as the oxidant for the construction of α-acetoxyaryl ketones from aryl ketones via self-intermolecular oxidative coupling.
![[1860-5397-13-107-i1]](/bjoc/content/inline/1860-5397-13-107-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Previous and present approaches.
Scheme 1: Previous and present approaches.
Results and Discussion
In our first attempt, the reaction of acetophenone (1a) in the presence of an I2–TBHP system gave the desired product 2a in 46% yield. The yield increased to 71% when the reaction time was prolonged to 24 h (Table 1, entries 1–3). The reaction did not occur in the absence of I2 or Na2CO3, indicating that these species both play important roles in this reaction (Table 1, entries 4 and 5). The reaction was almost unaffected by the solvent (Table 1, entries 3, 6−8). Acetonitrile was slightly more effective than the other solvents tested. An increase in the amount of TBHP from 2.0 equiv to 4.0 equiv significantly affected the reaction efficiency, leading to a pronounced increase in the yield (Table 1, entry 9). Further increasing the TBHP loading did not have any beneficial effect (Table 1, entry 10). An increase in the amount of I2 from 0.1 equiv to 0.5 equiv did not affect product formation (Table 1, entry 11). However, decreasing the amount of Na2CO3 from 1.0 equiv to 0.1 equiv significantly decreased the product yield. The effects of other peroxides, i.e., di-tert-butyl peroxide (DTBP), benzoyl peroxide, dicumyl peroxide (DCP), cumene hydroperoxide (CHP), potassium hydrogen persulfate, and 3-chloroperoxybenzoic acid (m-CPBA), on the reaction were investigated. All these peroxides gave sluggish reactions with poor yields, except m-CPBA, which gave the desired product 2a in 81% yield (Table 1, entries 13–18). Finally, we investigated the effect of reaction temperature to this transformation, which indicated that the optimum reaction temperature is: 70 °C (Table 1, entries 19 and 20).
Table 1: Optimization studiesa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-107-i5.svg?max-width=637&scale=1.0)
|
||||
| Entry | peroxide (2.0 equiv) | solvent | Time (h) | Yield%b |
|---|---|---|---|---|
| 1 | TBHP | CH3CN | 12 | 46 |
| 2 | TBHP | CH3CN | 24 | 71 |
| 3 | TBHP | CH3CN | 36 | 71 |
| 4c | TBHP | CH3CN | 24 | 0 |
| 5d | TBHP | CH3CN | 24 | 0 |
| 6 | TBHP | dioxane | 24 | 70 |
| 7 | TBHP | DCE | 24 | 68 |
| 8 | TBHP | cyclohexane | 24 | 63 |
| 9 | TBHP (4.0) | CH3CN | 24 | 84 |
| 10 | TBHP (6.0) | CH3CN | 24 | 84 |
| 11e | TBHP (4.0) | CH3CN | 24 | 84 |
| 12f | TBHP (4.0) | CH3CN | 24 | 33 |
| 13 | DTBP (4.0) | CH3CN | 24 | 23 |
| 14 | benzoyl peroxide (4.0) | CH3CN | 24 | 47 |
| 15 | DCP (4.0) | CH3CN | 24 | 29 |
| 16 | CHP (4.0) | CH3CN | 24 | 11 |
| 17 | K2S2O8 (4.0) | CH3CN | 24 | 11 |
| 18 | m-CPBA (4.0) | CH3CN | 24 | 81 |
| 19g | TBHP (4.0) | CH3CN | 24 | trace |
| 20h | TBHP (4.0) | CH3CN | 24 | 84 |
aReaction conditions: 1a (0.5 mmol), I2 (0.1 equiv), TBHP (2.0 equiv), Na2CO3 (1.0 equiv), solvent (2.0 mL); bGC yield; cwithout I2; dwithout Na2CO3; eI2: 0.5 equiv; fNa2CO3: 0.1 equiv; greaction temperature: rt; hreflux.
After the optimization study, the generality of the optimum conditions with various substituted aryl ketones was investigated (Scheme 2). Initially, acetophenone derivatives 1a–h were used; various electron-donating (i.e., methyl and methoxy) and electron-withdrawing (i.e., F−, Br−, and Cl−) substituents were well-tolerated under our reaction conditions. Acetophenones bearing electron-withdrawing substituents performed slightly better in this reaction than those bearing electron-donating substituents, and afforded the desired product in relatively high yields (2f, 2g, and 2h).
![[1860-5397-13-107-i2]](/bjoc/content/inline/1860-5397-13-107-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope. (All of these reactions were carried out on a 2.0 mmol scale using CH3CN (2.0 mL) as a solvent.)
Scheme 2: Substrate scope. (All of these reactions were carried out on a 2.0 mmol scale using CH3CN (2.0 mL) ...
The position of a given substituent on the phenyl ring of acetophenone affected the reaction slightly, and para-substituted acetophenones gave better results than ortho- and meta-substituted acetophenones (2b, 2c, and 2d). The scope of this reaction was extended by varying the aliphatic part of the arone (1j–n); for example, propiophenones and butyrophenones all reacted as anticipated to give the desired α-acetoxyaryl ketones 2j–n in moderate yields. In addition, different substituents on the phenyl ring had no discernible impact on the outcome. 1-(Thiophen-2-yl)ethanone (1i), which has a heteroaryl functionality, gave 2i in 83% isolated yield.
A series of control experiments were performed to clarify the reaction mechanism (Scheme 3). When the reactions were performed in the presence of an excess of the free-radical scavenger 2,2,6,6-tetramethylpiperidine-N-oxyl, product formation was completely suppressed (Scheme 3, reaction 1), indicating that a radical pathway may be involved in this reaction. The oxygen source was identified by performing the reaction with excess 18O-labeled H218O; 2a was obtained in 79% yield, with no 18O in the product; this excludes the possibility of the oxygen being derived from H2O in the TBHP solvent (Scheme 3, reaction 2).When 2-iodo-1-phenylethanone was used as a surrogate of 1a under the optimum conditions or in the absence of I2, 2a was isolated in 91% and 87% yields, respectively (Scheme 3, reactions 3 and 4). We also observed that 2a was obtained in almost quantitative yields when 1a was reacted with tert-butylperoxybenzoate (TBPB) or benzoic acid under the standard conditions (Scheme 3, reactions 5 and 6). These results suggest that 2-iodo-1-phenyl ketone, TBPB, and benzoic acid are generated in situ from 1a as intermediates.
![[1860-5397-13-107-i3]](/bjoc/content/inline/1860-5397-13-107-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Control reactions for clarifying the mechanism.
Scheme 3: Control reactions for clarifying the mechanism.
The mechanism has not yet been clarified in detail. A probable catalytic cycle is proposed in Scheme 4 based on the above experimental results and previous literature reports. The process begins with the formation of α-iodoaryl ketones 5 and 6 via iodination of aryl ketones with I2 and TBHP [24,25]. An I−/I2 redox cycle promotes tert-butoxyl and tert-butylperoxyl radical formation from TBHP [26-28]. In the presence of TBHP and I2, α-iodoaryl ketones 5 and 6 are oxidized to a 1,2-diketone intermediate 7 and an α-carbonyl radical 9, which can be further transformed to tert-butyl perester 8 and cation 11 [22]. The I− anion can be reoxidized by tert-butyl perester 8 to regenerate I2, a tert-butoxyl radical, and an aromatic acid anion under alkaline conditions. Finally, the reactions between intermediates 8 and 9, 10 and 11 or 5 all afford the final product, according to previous reports [22,29].
![[1860-5397-13-107-i4]](/bjoc/content/inline/1860-5397-13-107-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, we have developed an efficient, novel, and metal-free synthesis of α-acetoxyaryl ketones from aryl ketones using I2−TBHP. A facile α-acylation reaction involving self-intermolecular oxidative coupling of aryl ketones was observed for the first time in the presence of I2−TBHP. Multiple radical intermediates are generated in situ, and the overall process involves several different reactions, which proceed self-sequentially in a single reactor. The reaction conditions are mild and the substrate scope is broad. This method has good potential applications in organic synthesis and medicinal chemistry. The inside of the reaction mixture has not been studied in depth, but we have begun mechanistic studies.
Supporting Information
| Supporting Information File 1: Full experimental details and copies of NMR spectral data. | ||
| Format: PDF | Size: 2.7 MB | Download |
References
-
Shindo, M.; Yoshimura, Y.; Hayashi, M.; Soejima, H.; Yoshikawa, T.; Matsumoto, K.; Shishido, K. Org. Lett. 2007, 9, 1963. doi:10.1021/ol0705200
Return to citation in text: [1] -
Loner, C. M.; Luzzio, F. A.; Demuth, D. R. Tetrahedron Lett. 2012, 53, 5641. doi:10.1016/j.tetlet.2012.08.032
Return to citation in text: [1] -
Murahashi, S., Susumu.; Saito, T.; Hanaoka, H.; Murakami, Y.; Naota, T.; Kumobayashi, H.; Akutagawa, S. J. Org. Chem. 1993, 58, 2929. doi:10.1021/jo00063a002
Return to citation in text: [1] -
Green, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis. Wiley: New York, 1991; p 87.
Return to citation in text: [1] -
Akita, H.; Enoki, Y.; Yamada, H.; Oishi, T. Chem. Pharm. Bull. 1989, 37, 2876. doi:10.1248/cpb.37.2876
Return to citation in text: [1] -
Levine, P. A.; Walti, A. Org. Synth. Coll. Vol. II 1943, 5.
Return to citation in text: [1] -
Clark, J. H.; Miller, J. M. Tetrahedron Lett. 1977, 18, 599. doi:10.1016/S0040-4039(01)92703-0
Return to citation in text: [1] -
Malova Krizkova, P.; Hammerschmidt, F. Eur. J. Org. Chem. 2013, 5143. doi:10.1002/ejoc.201300439
Return to citation in text: [1] -
Cadierno, V.; Francos, J.; Gimeno, J. Organometallics 2011, 30, 852. doi:10.1021/om1010325
Return to citation in text: [1] -
Uyanik, M.; Suzuki, D.; Yasui, T.; Ishihara, K. Angew. Chem. 2011, 123, 5443. doi:10.1002/ange.201101522
Return to citation in text: [1] [2] -
Ochiai, M.; Takeuchi, Y.; Katayama, T.; Sueda, T.; Miyamoto, K. J. Am. Chem. Soc. 2005, 127, 12244. doi:10.1021/ja0542800
Return to citation in text: [1] -
Reddi,, R. N.; Gontala, A.; Prasad, P. K.; Sudalai, A. Asian J. Org. Chem. 2016, 5, 48. doi:10.1002/ajoc.201500355
Return to citation in text: [1] -
Jia, W.-G.; Zhang, H.; Li, D.-D.; Yan, L.-Q. RSC Adv. 2016, 6, 27590. doi:10.1039/C6RA02186G
Return to citation in text: [1] -
Nagano, T.; Jia, Z.; Li, X.; Yan, M.; Lu, G.; Chan, A. S. C.; Hayashi, T. Chem. Lett. 2010, 39, 929. doi:10.1246/cl.2010.929
Return to citation in text: [1] -
Uyanik, M.; Suzuki, D.; Watanabe, M.; Tanaka, H.; Furukawa, K.; Ishihara, K. Chem. Lett. 2015, 44, 387. doi:10.1246/cl.141110
Return to citation in text: [1] -
Roch, S. Science 2010, 327, 1376. doi:10.1126/science.1182300
Return to citation in text: [1] -
Uyanik, M.; Suzuki, D.; Yasui, T.; Ishihara, K. Angew. Chem., Int. Ed. 2011, 50, 5331. doi:10.1002/anie.201101522
Return to citation in text: [1] -
Uyanik, M.; Hayashi, H.; Ishihara, K. Science 2014, 345, 291. doi:10.1126/science.1254976
Return to citation in text: [1] -
Guo, S.; Yu, J.-T.; Dai, Q.; Yang, H.; Cheng, J. Chem. Commun. 2014, 50, 6240. doi:10.1039/c4cc01652a
Return to citation in text: [1] -
Reddi, R. N.; Prasad, P. K.; Sudalai, A. Org. Lett. 2014, 16, 5674. doi:10.1021/ol5027393
Return to citation in text: [1] -
Chen, C.; Liu, W.; Zhou, P.; Liu, H. L. RSC Adv. 2017, 7, 20394. doi:10.1039/C7RA02298K
Return to citation in text: [1] -
Mondal, B.; Sahoo, S. C.; Pan, S. C. Eur. J. Org. Chem. 2015, 3135. doi:10.1002/ejoc.201500233
Return to citation in text: [1] [2] [3] -
Zhu, F.; Wang, Z.-X. Tetrahedron 2014, 70, 9819. doi:10.1016/j.tet.2014.11.002
Return to citation in text: [1] -
Li, H.-Z.; Xue, W.-J.; Wu, A.-X. Tetrahedron 2014, 70, 4645. doi:10.1016/j.tet.2014.05.045
Return to citation in text: [1] -
Mupparapu, N.; Vishwakarma, R. A.; Ahmed, Q. N. Tetrahedron 2015, 71, 3417. doi:10.1016/j.tet.2015.03.088
Return to citation in text: [1] -
Lai, J.; Chang, L.; Yuan, G. Org. Lett. 2016, 18, 3194. doi:10.1021/acs.orglett.6b01412
Return to citation in text: [1] -
Uyanik, M.; Ishihara, K. ChemCatChem 2012, 4, 177. doi:10.1002/cctc.201100352
Return to citation in text: [1] -
Finkbeiner, P.; Nachtsheim, B. J. Synthesis 2013, 45, 979. doi:10.1055/s-0032-1318330
Return to citation in text: [1] -
Zhu, M.; Wei, W.; Yang, D.; Cui, H.; Cui, H.; Sun, X.; Wang, H. Org. Biomol. Chem. 2016, 14, 10998. doi:10.1039/C6OB02173E
Return to citation in text: [1]
| 1. | Shindo, M.; Yoshimura, Y.; Hayashi, M.; Soejima, H.; Yoshikawa, T.; Matsumoto, K.; Shishido, K. Org. Lett. 2007, 9, 1963. doi:10.1021/ol0705200 |
| 2. | Loner, C. M.; Luzzio, F. A.; Demuth, D. R. Tetrahedron Lett. 2012, 53, 5641. doi:10.1016/j.tetlet.2012.08.032 |
| 3. | Murahashi, S., Susumu.; Saito, T.; Hanaoka, H.; Murakami, Y.; Naota, T.; Kumobayashi, H.; Akutagawa, S. J. Org. Chem. 1993, 58, 2929. doi:10.1021/jo00063a002 |
| 4. | Green, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis. Wiley: New York, 1991; p 87. |
| 5. | Akita, H.; Enoki, Y.; Yamada, H.; Oishi, T. Chem. Pharm. Bull. 1989, 37, 2876. doi:10.1248/cpb.37.2876 |
| 12. | Reddi,, R. N.; Gontala, A.; Prasad, P. K.; Sudalai, A. Asian J. Org. Chem. 2016, 5, 48. doi:10.1002/ajoc.201500355 |
| 22. | Mondal, B.; Sahoo, S. C.; Pan, S. C. Eur. J. Org. Chem. 2015, 3135. doi:10.1002/ejoc.201500233 |
| 10. | Uyanik, M.; Suzuki, D.; Yasui, T.; Ishihara, K. Angew. Chem. 2011, 123, 5443. doi:10.1002/ange.201101522 |
| 11. | Ochiai, M.; Takeuchi, Y.; Katayama, T.; Sueda, T.; Miyamoto, K. J. Am. Chem. Soc. 2005, 127, 12244. doi:10.1021/ja0542800 |
| 22. | Mondal, B.; Sahoo, S. C.; Pan, S. C. Eur. J. Org. Chem. 2015, 3135. doi:10.1002/ejoc.201500233 |
| 29. | Zhu, M.; Wei, W.; Yang, D.; Cui, H.; Cui, H.; Sun, X.; Wang, H. Org. Biomol. Chem. 2016, 14, 10998. doi:10.1039/C6OB02173E |
| 8. | Malova Krizkova, P.; Hammerschmidt, F. Eur. J. Org. Chem. 2013, 5143. doi:10.1002/ejoc.201300439 |
| 9. | Cadierno, V.; Francos, J.; Gimeno, J. Organometallics 2011, 30, 852. doi:10.1021/om1010325 |
| 24. | Li, H.-Z.; Xue, W.-J.; Wu, A.-X. Tetrahedron 2014, 70, 4645. doi:10.1016/j.tet.2014.05.045 |
| 25. | Mupparapu, N.; Vishwakarma, R. A.; Ahmed, Q. N. Tetrahedron 2015, 71, 3417. doi:10.1016/j.tet.2015.03.088 |
| 6. | Levine, P. A.; Walti, A. Org. Synth. Coll. Vol. II 1943, 5. |
| 7. | Clark, J. H.; Miller, J. M. Tetrahedron Lett. 1977, 18, 599. doi:10.1016/S0040-4039(01)92703-0 |
| 26. | Lai, J.; Chang, L.; Yuan, G. Org. Lett. 2016, 18, 3194. doi:10.1021/acs.orglett.6b01412 |
| 27. | Uyanik, M.; Ishihara, K. ChemCatChem 2012, 4, 177. doi:10.1002/cctc.201100352 |
| 28. | Finkbeiner, P.; Nachtsheim, B. J. Synthesis 2013, 45, 979. doi:10.1055/s-0032-1318330 |
| 10. | Uyanik, M.; Suzuki, D.; Yasui, T.; Ishihara, K. Angew. Chem. 2011, 123, 5443. doi:10.1002/ange.201101522 |
| 20. | Reddi, R. N.; Prasad, P. K.; Sudalai, A. Org. Lett. 2014, 16, 5674. doi:10.1021/ol5027393 |
| 21. | Chen, C.; Liu, W.; Zhou, P.; Liu, H. L. RSC Adv. 2017, 7, 20394. doi:10.1039/C7RA02298K |
| 16. | Roch, S. Science 2010, 327, 1376. doi:10.1126/science.1182300 |
| 17. | Uyanik, M.; Suzuki, D.; Yasui, T.; Ishihara, K. Angew. Chem., Int. Ed. 2011, 50, 5331. doi:10.1002/anie.201101522 |
| 18. | Uyanik, M.; Hayashi, H.; Ishihara, K. Science 2014, 345, 291. doi:10.1126/science.1254976 |
| 22. | Mondal, B.; Sahoo, S. C.; Pan, S. C. Eur. J. Org. Chem. 2015, 3135. doi:10.1002/ejoc.201500233 |
| 23. | Zhu, F.; Wang, Z.-X. Tetrahedron 2014, 70, 9819. doi:10.1016/j.tet.2014.11.002 |
| 14. | Nagano, T.; Jia, Z.; Li, X.; Yan, M.; Lu, G.; Chan, A. S. C.; Hayashi, T. Chem. Lett. 2010, 39, 929. doi:10.1246/cl.2010.929 |
| 15. | Uyanik, M.; Suzuki, D.; Watanabe, M.; Tanaka, H.; Furukawa, K.; Ishihara, K. Chem. Lett. 2015, 44, 387. doi:10.1246/cl.141110 |
| 13. | Jia, W.-G.; Zhang, H.; Li, D.-D.; Yan, L.-Q. RSC Adv. 2016, 6, 27590. doi:10.1039/C6RA02186G |
| 19. | Guo, S.; Yu, J.-T.; Dai, Q.; Yang, H.; Cheng, J. Chem. Commun. 2014, 50, 6240. doi:10.1039/c4cc01652a |
© 2017 Tan et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)