
Abstract
Novel 13α-estrone derivatives were synthesized by Sonogashira coupling. Transformations of 2- or 4-iodo regioisomers of 13α-estrone and its 3-methyl ether were carried out under different conditions in a microwave reactor. The 2-iodo isomers were reacted with para-substituted phenylacetylenes using Pd(PPh3)4 as catalyst and CuI as a cocatalyst. Coupling reactions of 4-iodo derivatives could be achieved by changing the catalyst to Pd(PPh3)2Cl2. The product phenethynyl derivatives were partially or fully saturated. Compounds bearing a phenolic OH group furnished benzofurans under the conditions used for the partial saturation. The inhibitory effects of the compounds on human placental 17β-hydroxysteroid dehydrogenase type 1 isozyme (17β-HSD1) were investigated by an in vitro radiosubstrate incubation method. Certain 3-hydroxy-2-phenethynyl or -phenethyl derivatives proved to be potent 17β-HSD1 inhibitors, displaying submicromolar IC50 values.
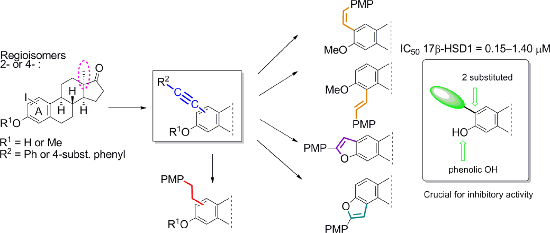
Graphical Abstract
Introduction
Synthetic modifications of the naturally occurring female prehormone estrone may lead to compounds with diverse biological activities, for example with antitumor effect [1]. One of the main requirements of estrone anticancer derivatives is the lack of their hormonal activity. Several core-modified estrones have recently been produced and diversified in order to get selectively acting compounds [2-4]. One opportunity for that is the inversion of the configuration at C-13, which is accompanied by drastic conformational change for the overall molecule resulting from the cis junction of rings C and D [2]. The influence of inversion of the configuration at C-13 in 3,17-estradiols on their in vivo and in vitro estrogenic activity was shown by Poirier et al. [5]. They demonstrated that 13 epimers exhibit no substantial binding affinity for the estrogen receptor alpha and no uterotropic activity. Accordingly, the 13α-estrane core may serve as fundamental moiety for the design of hormonally inactive estrone derivatives bearing promising biological activities. We recently published the syntheses and the in vitro biological evaluations of several 13α-estrone derivatives [6-9]. Certain compounds proved to be biologically active, bearing substantial antiproliferative or enzyme inhibitory potential [7,8]. Most literature data are mainly about 13α-estrones substituted in ring D, but compounds modified in ring A are rarely described [10,11]. More recently we have disclosed ring A halogenations in this series [12]. Electrophilic brominations or iodinations were carried out, furnishing 2-, 4- or 2,4-bis-halogenated compounds. All the halogenated 3-hydroxy and the 4-substituted regioisomers of 3-methyl ethers displayed substantial inhibitory activity against the 17β-hydroxysteroid dehydrogenase type 1 enzyme (17β-HSD1). Certain derivatives displayed a similar or more pronounced effect than those of their parent compounds 13α-estrone or 13α-estrone 3-methyl ether [13]. The 17β-HSD1 enzyme is responsible for the stereospecific reduction of prehormone estrone into the main estrogenic hormone 17β-estradiol [14,15]. 17β-Estradiol may enhance the proliferation of certain cancer cells [16]. The inhibition of 17β-HSD1 provokes an antitumor effect in hormone dependent cancers, hence 17β-HSD1 inhibitors could have good prospects as anti-estrogen therapeutics [17,18]. The recently synthesized halogenated 13α-estrones, in addition to their pharmacological importance, may serve as appropriate starting compounds for Pd-catalyzed C–C coupling reactions. Some Sonogashira couplings on estrane, but not on the 13α-estrane core have been performed at C-2, -3, -11, -16 and -17. To the best of our knowledge, 4-coupled regioisomers have not been synthesized to date [19]. Couplings of steroidal alkynes with small molecular halides are already described, and reactions of steroidal halides or triflates with small molecular alkynes also exist [20]. Certain phenethynyl estrone derivatives described in the literature possess substantial biological activities. Möller et al. performed the couplings of 2-iodoestrone-3-acetate with phenylacetylene using Pd(OAc)2 and CuI as catalysts [21]. They did not investigate the influence of the nature of the substituent on the phenyl ring of the acetylene on the course of the reactions. They carried out the full saturation of the C≡C bond of the 2-phenethynyl estrone with palladium on charcoal, furnishing the 2-phenethyl-substituted derivative. However, they did not study the partial saturation of the estrone alkyne moiety. The 2-phenethyl and 2-phenethynyl derivatives proved to be potent 17β-HSD1 inhibitors with the fully-saturated compound being slightly more potent.
The aim of the present study was to develop facile and effective Sonogashira coupling methods for the preparation of 2- or 4-phenethynyl derivatives in the 13α-estrone series. 2- or 4-iodo-13α-estrone and their 3-methyl ethers were chosen as starting compounds. The partial or full saturation of the C≡C bond of certain 2- or 4-regioisomeric phenethynyl compounds was also planned. We intended to investigate the potential inhibitory effects of the novel 13α-estrones toward human placental 17β-HSD1 activity in vitro.
Results and Discussion
Synthetic work
Sonogashira coupling
Iodo compounds 3–6 synthesized recently have been chosen as starting materials for the Sonogashira couplings, since the reactivity of the aryl iodides is higher than that of their bromo counterparts (Scheme 1) [22]. The optimizations of the coupling reactions were carried out using phenylacetylene (7a) as a model reagent. The optimal reaction conditions were found to differ depending on the position of the iodo substituent on the sterane skeleton (Scheme 1). Couplings at C-2 could efficiently be achieved using 0.1 equiv of Pd(PPh3)4 and CuI in tetrahydrofuran (THF) or dimethylformamide (DMF) as solvent in the presence of Et3N as a base at 50 °C for 20 min in a microwave reactor. 4-Phenylalkynyl regioisomers (10a, 11a) were obtained in high yields using 0.05 equiv of Pd(PPh3)2Cl2 and CuI in CH3CN or DMF, in the presence of Et3N as a base at 80 °C for 20 min in a microwave reactor. After establishing the most favorable reaction conditions, the Sonogashira reactions (of both regioisomers) were carried out with several para-substituted phenylacetylenes (7b–e). All the couplings resulted in the desired products (8–11) in high yields. The newly synthesized 4-phenethynyl derivatives are the first 4-substituted Sonogashira coupled estrones in the literature. The structures of the new compounds were confirmed by 1H, 13C and two-dimensional NMR measurements (see Supporting Information File 1).
![[1860-5397-13-126-i1]](/bjoc/content/inline/1860-5397-13-126-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Syntheses of 2- or 4-phenethynyl-13α-estrones (8–11) by Sonogashira coupling.
Scheme 1: Syntheses of 2- or 4-phenethynyl-13α-estrones (8–11) by Sonogashira coupling.
Full and partial saturation of the alkyne moiety
We have chosen four 4”-methoxy-substituted phenylalkynyl compounds (8c–11c) for partial or full saturation of the C≡C bond in both the 3-OH and the 3-OMe series (Scheme 2). The trans counterpart of the resulting diphenylethenyl moiety is related to the fully-methoxylated derivative of resveratrol (3,5,4’-trihydroxystilbene), a compound exhibiting diverse biological activities [23,24]. The chemo- and stereoselective semihydrogenation of internal alkynes may be achieved by two main catalytic methods: with molecular hydrogen using Lindlar’s catalyst [25,26] or by transfer hydrogenation with hydrogen donors [27,28]. Additionally, alkynes undergo reduction with diimide to produce cis-alkenes [29]. Li et al. carried out the semihydrogenation of different arylacetylenes using Pd(OAc)2 or Pd(PPh3)2Cl2 as the catalyst and DMF/KOH as a hydrogen source, under conventional heating [30]. The first catalyst afforded cis-alkenes in high yields with excellent chemo- and stereoselectivity. The latter catalyst displayed lower catalytic activity and stereoselectivity. The stereoselectivity of the semihydrogenation process may play a crucial role concerning the biological activity of the resulting alkenes, since geometrical isomers may possess different biological functions [31]. Here we performed the partial saturation of compounds 8c–11c by the modified procedure of Li et al. using Pd(OAc)2 or Pd(PPh3)2Cl2 as a catalyst, and DMF/KOH as a hydrogen source, in a microwave reactor. The cis-alkene 13 and the trans-alkene 15 were formed chemo- and stereoselectively under the applied conditions. The different stereochemical outcome of the hydrogenations of the two regioisomers presumably arose from the steric hindrance caused by the vicinity of ring B in the case of compound 15.
![[1860-5397-13-126-i2]](/bjoc/content/inline/1860-5397-13-126-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Partial or full hydrogenation of compounds 8c–11c.
Scheme 2: Partial or full hydrogenation of compounds 8c–11c.
The cis or trans orientation of the resulting geometric isomers was deduced from the vicinal coupling constants according to the literature data, because cis and trans couplings across a double bond are very reliable indicators of stereochemistry [32,33]. In the case of the 2-regioisomer 13, the signals of the vicinal olefinic protons appear as a singlet with double intensity, similar to those of 2,4’-dimethoxystilbene [32,33]. In the 1H NMR spectrum of the 4-substituted counterpart 15, the olefinic protons are shown as doublets with a large coupling constant of 12.2 Hz, which refers to their trans arrangement. Under the conditions used for the partial saturation, the ethynyl derivatives bearing a phenolic OH group (8c, 10c) furnished benzo[b]furans 12 and 14. There are literature reports about similar transition-metal-catalyzed cyclizations of o-alkynylphenols to construct benzofurans [34,35]. These heterocycles are important structural units in a variety of biologically active natural or synthetic compounds [36,37]. Full hydrogenation of the 2- or 4-phenethynyl intermediates (8c–11c) with palladium-on-charcoal furnished the 2- or 4-phenethyl-substituted derivatives (16–19).
In vitro 17β-HSD1 enzyme inhibition test
With the new compounds in hand (8–19, Table 1), we also determined their in vitro inhibitory potencies on human placental 17β-HSD1. In the 3-OH series, all the 2-phenylalkynyl regioisomers 8a–e proved to be effective inhibitors with IC50 values depending on the nature of the 4”-functional group. The most potent compound was unsubstituted 8a with an IC50 of 0.15 μM. The 4-substituted regioisomers 10a–e inhibited the enzyme scarcely, suppressing the conversion by less than 15%. The phenylalkynyl derivatives in the 3-OMe series 9a–e and 11a–e exerted weak inhibitions. Phenylalkenyl compounds 13 and 15 and benzofuran compounds 12 and 14 displayed weaker inhibitory activity than their alkynyl counterparts 8c and 10c. The full saturation (leading to compounds 16–19) did not influence the inhibitory potential markedly. The weak inhibitory activities of 9c, 10c or 11c were not improved in compounds 17, 18 or 19, whereas the good inhibitory effect of the 2-regioisomer 8c was retained in compound 16.
Table 1: 17β-HSD1 inhibition data of Sonogashira coupled compounds and their precursors (1–6) [12,13] indicated with an asterisk (*).
| Structure | Compound | R1 | R2 | Relative conversiona ± SD (%) or IC50 ± SD (μM) |
|---|---|---|---|---|
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-126-i3.svg?max-width=637&scale=1.0)
|
1 | H | H | IC50 = 1.2* |
| 3 | I | H | IC50 = 0.59* | |
| 5 | H | I | IC50 = 1.0* | |
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-126-i4.svg?max-width=637&scale=1.0)
|
2 | H | H | IC50 = 5.5* |
| 4 | I | H | IC50 > 10* | |
| 6 | H | I | IC50 = 0.56* | |
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-126-i5.svg?max-width=637&scale=1.0)
|
8a | H | H | IC50 = 0.15 ± 0.02 |
| 8b | Me | IC50 = 1.40 ± 0.78 | ||
| 8c | OMe | IC50 = 0.23 ± 0.03 | ||
| 8d | F | IC50 = 0.30 ± 0.08 | ||
| 8e | CF3 | IC50 = 0.93 ± 0.13 | ||
| 9a | Me | H | 88 ± 12 | |
| 9b | Me | 84 ± 5 | ||
| 9c | OMe | 85 ± 1 | ||
| 9d | F | 94 ± 5 | ||
| 9e | CF3 | 76 ± 1 | ||
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-126-i6.svg?max-width=637&scale=1.0)
|
10a | H | H | 92 ± 15 |
| 10b | Me | 89 ± 0.4 | ||
| 10c | OMe | 91 ± 2 | ||
| 10d | F | 96 ± 7 | ||
| 10e | CF3 | 85 ± 1 | ||
| 11a | Me | H | 92 ± 12 | |
| 11b | Me | 52 ± 12 | ||
| 11c | OMe | 83 ± 8 | ||
| 11d | F | 83 ± 1 | ||
| 11e | CF3 | 79 ± 3 | ||
![[Graphic 5]](/bjoc/content/inline/1860-5397-13-126-i7.svg?max-width=637&scale=1.0)
|
12 | – | OMe | 92 ± 2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-13-126-i8.svg?max-width=637&scale=1.0)
|
14 | 102 ± 6 | ||
![[Graphic 7]](/bjoc/content/inline/1860-5397-13-126-i9.svg?max-width=637&scale=1.0)
|
13 | Me | OMe | 70 ± 6 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-13-126-i10.svg?max-width=637&scale=1.0)
|
15 | 80 ± 12 | ||
![[Graphic 9]](/bjoc/content/inline/1860-5397-13-126-i11.svg?max-width=637&scale=1.0)
|
16 | H | OMe | IC50 = 0.47 ± 0.04 |
| 17 | Me | 63 ± 8 | ||
![[Graphic 10]](/bjoc/content/inline/1860-5397-13-126-i12.svg?max-width=637&scale=1.0)
|
18 | H | OMe | 98 ± 3 |
| 19 | Me | 94 ± 1 | ||
aAt 10 μM, non-inhibited control 100%. Reference for precursors (1–6) [12,13].
When all the inhibition data of the novel compounds and their precursors from Table 1 are taken into consideration, some valuable structure–activity relationships appear. 13α-Estrone (1) displays 17β-HSD1 inhibitory potential similar to that of the natural substrate estrone. Iodination at C-2 of 1 improves the inhibitory potential, resulting in a submicromolar IC50 for compound 3. Phenylalkynylation of the 2-iodo compound 3 retains or further improves the inhibition, depending on the nature of the substituent at C-4”. Concerning the 4-regioisomers, iodination leads to an efficiency similar to that of compound 1, whereas the inhibition is lost following C–C coupling. 13α-Estrone 3-methyl ether 2 possesses a weaker inhibitory effect than the 3-hydroxy compound 1. Iodination or phenylalkynylation at C-2 diminishes inhibition of 2. Introducing iodine onto C-4 of compound 2 leads to a 10-fold decrease in its IC50 value. 4-Phenylalkynyl derivatives 10 and 11, nevertheless, exert weak inhibitions on the estrone to 17β-estradiol conversion
The results reveal a great influence of the 2,4-regioisomerism on the inhibition potential of the iodinated 3-methyl ethers 4 and 6, the phenylalkynyl 8 and 10 and the phenylalkyl 16 and 18 3-hydroxy compounds.
Conclusion
In conclusion, we described here an efficient synthetic microwave procedure for the synthesis of novel phenylalkynyl derivatives of 13α-estrone (1) and its 3-methyl ether 2. The steroidal alkynes were chemo- and stereoselectively hydrogenated by transfer hydrogenation in a microwave reactor, furnishing alkenes or benzofurans depending on the nature of the substituent at C-3. Full hydrogenations of certain phenethynyl derivatives were also achieved. The newly-synthesized potent 17β-HSD1 inhibitors may serve as suitable tools for ligand-based enzyme studies. Further derivatizations of our compounds may provide promising candidates for drug development in order to get nanomolar inhibitors.
Supporting Information
| Supporting Information File 1: Experimental procedures for compounds 8–19 and their 1H, 13C NMR, MS, elemental analysis data. | ||
| Format: PDF | Size: 428.5 KB | Download |
References
-
Gupta, A.; Kumar, B. S.; Negi, A. S. J. Steroid Biochem. Mol. Biol. 2013, 137, 242–270. doi:10.1016/j.jsbmb.2013.05.011
Return to citation in text: [1] -
Schönecker, B.; Lange, C.; Kötteritzsch, M.; Günther, W.; Weston, J.; Anders, E.; Görls, H. J. Org. Chem. 2000, 65, 5487–5497. doi:10.1021/jo000108x
Return to citation in text: [1] [2] -
Jovanovic-Santa, S.; Petrović, J.; Andrić, S.; Kovačević, R.; Ðurendić, E.; Sakač, M.; Lazar, D.; Stanković, S. Bioorg. Chem. 2003, 31, 475–484. doi:10.1016/S0045-2068(03)00101-9
Return to citation in text: [1] -
Minorics, R.; Bózsity, N.; Wölfling, J.; Mernyák, E.; Schneider, G.; Márki, A.; Falkay, G.; Ocsovszki, I.; Zupkó, I. J. Steroid Biochem. Mol. Biol. 2012, 132, 168–175. doi:10.1016/j.jsbmb.2012.04.009
Return to citation in text: [1] -
Ayan, D.; Roy, J.; Maltais, R.; Poirier, D. J. Steroid Biochem. Mol. Biol. 2011, 127, 324–330. doi:10.1016/j.jsbmb.2011.07.009
Return to citation in text: [1] -
Szabó, J.; Jerkovics, N.; Schneider, G.; Wölfling, J.; Bózsity, N.; Minorics, R.; Zupkó, I.; Mernyák, E. Molecules 2016, 21, 611–623. doi:10.3390/molecules21050611
Return to citation in text: [1] -
Szabó, J.; Pataki, Z.; Wölfling, J.; Schneider, G.; Bózsity, N.; Minorics, R.; Zupkó, I.; Mernyák, E. Steroids 2016, 113, 14–21. doi:10.1016/j.steroids.2016.05.010
Return to citation in text: [1] [2] -
Mernyák, E.; Kovács, I.; Minorics, R.; Sere, P.; Czégány, D.; Sinka, I.; Wölfling, J.; Schneider, G.; Újfaludi, Z.; Boros, I.; Ocsovszki, I.; Varga, M.; Zupkó, I. J. Steroid Biochem. Mol. Biol. 2015, 150, 123–134. doi:10.1016/j.jsbmb.2015.04.001
Return to citation in text: [1] [2] -
Szabó, J.; Bacsa, I.; Wölfling, J.; Schneider, G.; Zupkó, I.; Varga, M.; Herman, B. E.; Kalmár, L.; Szécsi, M.; Mernyák, E. J. Enzyme Inhib. Med. Chem. 2016, 31, 574–579. doi:10.3109/14756366.2015.1050008
Return to citation in text: [1] -
Rao, P. N.; Cessac, J. W.; Tinley, T. L.; Mooberry, S. L. Steroids 2002, 67, 1079–1089. doi:10.1016/S0039-128X(02)00066-1
Return to citation in text: [1] -
Bonfilis, A.; Philibert, D. Use of aromatic steroids substituted in 3-position by a disubstituted aminoalkoxy chain for the manufacture of a medicament for the control of fertility, in particular male fertility. Eur. Pat. Appl. EP0676202 A2, Oct 11, 1995.
Return to citation in text: [1] -
Bacsa, I.; Jójárt, R.; Schneider, G.; Wölfling, J.; Maróti, P.; Herman, B. E.; Szécsi, M.; Mernyák, E. Steroids 2015, 104, 230–236. doi:10.1016/j.steroids.2015.10.008
Return to citation in text: [1] [2] [3] -
Herman, B. E.; Szabó, J.; Bacsa, I.; Wölfling, J.; Schneider, G.; Bálint, M.; Hetényi, C.; Mernyák, E.; Szécsi, M. J. Enzyme Inhib. Med. Chem. 2016, 31, 61–69. doi:10.1080/14756366.2016.1204610
Return to citation in text: [1] [2] [3] -
Möller, G.; Adamski, J. Mol. Cell. Endocrinol. 2009, 301, 7–19. doi:10.1016/j.mce.2008.10.040
Return to citation in text: [1] -
Marchais-Oberwinkler, S.; Henn, C.; Möller, G.; Klein, T.; Negri, M.; Oster, A.; Spadaro, A.; Werth, R.; Wetzel, M.; Xu, K.; Frotscher, M.; Hartmann, R. W.; Adamski, J. J. Steroid Biochem. Mol. Biol. 2011, 125, 66–82. doi:10.1016/j.jsbmb.2010.12.013
Return to citation in text: [1] -
Day, J. M.; Tutill, H. J.; Purohit, A.; Reed, M. J. Endocr.-Relat. Cancer 2008, 15, 665–692. doi:10.1677/ERC-08-0042
Return to citation in text: [1] -
Poirier, D. Expert Opin. Ther. Pat. 2010, 20, 1123–1145.
Return to citation in text: [1] -
Gobec, S.; Brozic, P.; Rizner, T. L. Curr. Med. Chem. 2008, 15, 137–150. doi:10.2174/092986708783330629
Return to citation in text: [1] -
Czajkowska-Szczykowska, D.; Morzycki, J. W.; Wojtkielewicz, A. Steroids 2015, 97, 13–44. doi:10.1016/j.steroids.2014.07.018
Return to citation in text: [1] -
Ivanov, A.; Boldt, S.; Nisa, Z. u.; Shah, S. J. A.; Ehlers, P.; Villinger, A.; Schneider, G.; Wölfling, J.; Rahman, Q.; Iqbal, J.; Langer, P. RSC Adv. 2016, 6, 11118–11127. doi:10.1039/C5RA25558A
Return to citation in text: [1] -
Möller, G.; Deluca, D.; Gege, C.; Rosinus, A.; Kowalik, D.; Peters, O.; Droescher, P.; Elger, W.; Adamski, J.; Hillisch, A. Bioorg. Med. Chem. Lett. 2009, 19, 6740–6744. doi:10.1016/j.bmcl.2009.09.113
Return to citation in text: [1] -
Molnár, Á, Ed. Palladium-Catalyzed Coupling Reactions: Practical Aspects and Future Developments; Wiley-VCH: Weinheim, 2013; pp 9 ff.
Return to citation in text: [1] -
Frémont, L. Life Sci. 2000, 66, 663–673. doi:10.1016/S0024-3205(99)00410-5
Return to citation in text: [1] -
Dong, Z. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2003, 523–524, 145–150. doi:10.1016/S0027-5107(02)00330-5
Return to citation in text: [1] -
Hauser, F. M.; Ganguly, D. J. Org. Chem. 2000, 65, 1842–1849. doi:10.1021/jo991483w
Return to citation in text: [1] -
You, Z.-W.; Jiang, Z.-X.; Wang, B.-L.; Qing, F.-L. J. Org. Chem. 2006, 71, 7261–7267. doi:10.1021/jo061012r
Return to citation in text: [1] -
Sajiki, H.; Mori, S.; Ohkubo, T.; Ikawa, T.; Kume, A.; Maegawa, T.; Monguchi, Y. Chem. – Eur. J. 2008, 14, 5109–5111. doi:10.1002/chem.200800535
Return to citation in text: [1] -
Hauwert, P.; Maestri, G.; Sprengers, J. W.; Catellani, M.; Elsevier, C. J. Angew. Chem., Int. Ed. 2008, 47, 3223–3226. doi:10.1002/anie.200705638
Return to citation in text: [1] -
Corey, E. J.; Pasto, D. J.; Mock, W. L. J. Am. Chem. Soc. 1961, 83, 2957–2958. doi:10.1021/ja01474a043
Return to citation in text: [1] -
Li, J.; Hua, R.; Liu, T. J. Org. Chem. 2010, 75, 2966–2970. doi:10.1021/jo100247a
Return to citation in text: [1] -
Parihar, S.; Kumar, A.; Chaturvedi, A. K.; Sachan, N. K.; Luqman, S.; Changkija, B.; Manohar, M.; Prakash, O.; Chanda, D.; Khan, F.; Chanotiya, C. S.; Shanker, K.; Dwivedi, A.; Konwar, R.; Negi, A. S. J. Steroid Biochem. Mol. Biol. 2013, 137, 332–344. doi:10.1016/j.jsbmb.2013.02.009
Return to citation in text: [1] -
Zhang, W.; Go, M. L. Eur. J. Med. Chem. 2007, 42, 841–850. doi:10.1016/j.ejmech.2006.12.012
Return to citation in text: [1] [2] -
Jagt, D. L. V.; Deck, L. M.; Royer, R. E.; Heidrich, J. E.; Sillerud, L. O. Substituted cis- and trans-stilbenes as therapeutic agents. U. S. Pat. Appl. US 20130178536 A1, July 11, 2013.
Return to citation in text: [1] [2] -
Arcadi, A.; Marinelli, F.; Cacchi, S. Synthesis 1986, 749–751. doi:10.1055/s-1986-31764
Return to citation in text: [1] -
Omelchuk, O. A.; Tikhomirov, A. S.; Shchekotikhin, A. E. Russ. Chem. Rev. 2016, 85, 817–835. doi:10.1070/RCR4613
Return to citation in text: [1] -
Acetylene chemistry: chemistry, biology and material science; Diederich, F.; Stang, P. J.; Tykwinski, R. R., Eds.; Wiley-VCH: Weinheim, 2005; pp 51 ff.
Return to citation in text: [1] -
McGallion, G. D. Curr. Org. Chem. 1999, 3, 67–76.
Return to citation in text: [1]
| 12. | Bacsa, I.; Jójárt, R.; Schneider, G.; Wölfling, J.; Maróti, P.; Herman, B. E.; Szécsi, M.; Mernyák, E. Steroids 2015, 104, 230–236. doi:10.1016/j.steroids.2015.10.008 |
| 13. | Herman, B. E.; Szabó, J.; Bacsa, I.; Wölfling, J.; Schneider, G.; Bálint, M.; Hetényi, C.; Mernyák, E.; Szécsi, M. J. Enzyme Inhib. Med. Chem. 2016, 31, 61–69. doi:10.1080/14756366.2016.1204610 |
| 1. | Gupta, A.; Kumar, B. S.; Negi, A. S. J. Steroid Biochem. Mol. Biol. 2013, 137, 242–270. doi:10.1016/j.jsbmb.2013.05.011 |
| 6. | Szabó, J.; Jerkovics, N.; Schneider, G.; Wölfling, J.; Bózsity, N.; Minorics, R.; Zupkó, I.; Mernyák, E. Molecules 2016, 21, 611–623. doi:10.3390/molecules21050611 |
| 7. | Szabó, J.; Pataki, Z.; Wölfling, J.; Schneider, G.; Bózsity, N.; Minorics, R.; Zupkó, I.; Mernyák, E. Steroids 2016, 113, 14–21. doi:10.1016/j.steroids.2016.05.010 |
| 8. | Mernyák, E.; Kovács, I.; Minorics, R.; Sere, P.; Czégány, D.; Sinka, I.; Wölfling, J.; Schneider, G.; Újfaludi, Z.; Boros, I.; Ocsovszki, I.; Varga, M.; Zupkó, I. J. Steroid Biochem. Mol. Biol. 2015, 150, 123–134. doi:10.1016/j.jsbmb.2015.04.001 |
| 9. | Szabó, J.; Bacsa, I.; Wölfling, J.; Schneider, G.; Zupkó, I.; Varga, M.; Herman, B. E.; Kalmár, L.; Szécsi, M.; Mernyák, E. J. Enzyme Inhib. Med. Chem. 2016, 31, 574–579. doi:10.3109/14756366.2015.1050008 |
| 21. | Möller, G.; Deluca, D.; Gege, C.; Rosinus, A.; Kowalik, D.; Peters, O.; Droescher, P.; Elger, W.; Adamski, J.; Hillisch, A. Bioorg. Med. Chem. Lett. 2009, 19, 6740–6744. doi:10.1016/j.bmcl.2009.09.113 |
| 5. | Ayan, D.; Roy, J.; Maltais, R.; Poirier, D. J. Steroid Biochem. Mol. Biol. 2011, 127, 324–330. doi:10.1016/j.jsbmb.2011.07.009 |
| 22. | Molnár, Á, Ed. Palladium-Catalyzed Coupling Reactions: Practical Aspects and Future Developments; Wiley-VCH: Weinheim, 2013; pp 9 ff. |
| 2. | Schönecker, B.; Lange, C.; Kötteritzsch, M.; Günther, W.; Weston, J.; Anders, E.; Görls, H. J. Org. Chem. 2000, 65, 5487–5497. doi:10.1021/jo000108x |
| 19. | Czajkowska-Szczykowska, D.; Morzycki, J. W.; Wojtkielewicz, A. Steroids 2015, 97, 13–44. doi:10.1016/j.steroids.2014.07.018 |
| 2. | Schönecker, B.; Lange, C.; Kötteritzsch, M.; Günther, W.; Weston, J.; Anders, E.; Görls, H. J. Org. Chem. 2000, 65, 5487–5497. doi:10.1021/jo000108x |
| 3. | Jovanovic-Santa, S.; Petrović, J.; Andrić, S.; Kovačević, R.; Ðurendić, E.; Sakač, M.; Lazar, D.; Stanković, S. Bioorg. Chem. 2003, 31, 475–484. doi:10.1016/S0045-2068(03)00101-9 |
| 4. | Minorics, R.; Bózsity, N.; Wölfling, J.; Mernyák, E.; Schneider, G.; Márki, A.; Falkay, G.; Ocsovszki, I.; Zupkó, I. J. Steroid Biochem. Mol. Biol. 2012, 132, 168–175. doi:10.1016/j.jsbmb.2012.04.009 |
| 20. | Ivanov, A.; Boldt, S.; Nisa, Z. u.; Shah, S. J. A.; Ehlers, P.; Villinger, A.; Schneider, G.; Wölfling, J.; Rahman, Q.; Iqbal, J.; Langer, P. RSC Adv. 2016, 6, 11118–11127. doi:10.1039/C5RA25558A |
| 13. | Herman, B. E.; Szabó, J.; Bacsa, I.; Wölfling, J.; Schneider, G.; Bálint, M.; Hetényi, C.; Mernyák, E.; Szécsi, M. J. Enzyme Inhib. Med. Chem. 2016, 31, 61–69. doi:10.1080/14756366.2016.1204610 |
| 16. | Day, J. M.; Tutill, H. J.; Purohit, A.; Reed, M. J. Endocr.-Relat. Cancer 2008, 15, 665–692. doi:10.1677/ERC-08-0042 |
| 12. | Bacsa, I.; Jójárt, R.; Schneider, G.; Wölfling, J.; Maróti, P.; Herman, B. E.; Szécsi, M.; Mernyák, E. Steroids 2015, 104, 230–236. doi:10.1016/j.steroids.2015.10.008 |
| 17. | Poirier, D. Expert Opin. Ther. Pat. 2010, 20, 1123–1145. |
| 18. | Gobec, S.; Brozic, P.; Rizner, T. L. Curr. Med. Chem. 2008, 15, 137–150. doi:10.2174/092986708783330629 |
| 10. | Rao, P. N.; Cessac, J. W.; Tinley, T. L.; Mooberry, S. L. Steroids 2002, 67, 1079–1089. doi:10.1016/S0039-128X(02)00066-1 |
| 11. | Bonfilis, A.; Philibert, D. Use of aromatic steroids substituted in 3-position by a disubstituted aminoalkoxy chain for the manufacture of a medicament for the control of fertility, in particular male fertility. Eur. Pat. Appl. EP0676202 A2, Oct 11, 1995. |
| 7. | Szabó, J.; Pataki, Z.; Wölfling, J.; Schneider, G.; Bózsity, N.; Minorics, R.; Zupkó, I.; Mernyák, E. Steroids 2016, 113, 14–21. doi:10.1016/j.steroids.2016.05.010 |
| 8. | Mernyák, E.; Kovács, I.; Minorics, R.; Sere, P.; Czégány, D.; Sinka, I.; Wölfling, J.; Schneider, G.; Újfaludi, Z.; Boros, I.; Ocsovszki, I.; Varga, M.; Zupkó, I. J. Steroid Biochem. Mol. Biol. 2015, 150, 123–134. doi:10.1016/j.jsbmb.2015.04.001 |
| 14. | Möller, G.; Adamski, J. Mol. Cell. Endocrinol. 2009, 301, 7–19. doi:10.1016/j.mce.2008.10.040 |
| 15. | Marchais-Oberwinkler, S.; Henn, C.; Möller, G.; Klein, T.; Negri, M.; Oster, A.; Spadaro, A.; Werth, R.; Wetzel, M.; Xu, K.; Frotscher, M.; Hartmann, R. W.; Adamski, J. J. Steroid Biochem. Mol. Biol. 2011, 125, 66–82. doi:10.1016/j.jsbmb.2010.12.013 |
| 27. | Sajiki, H.; Mori, S.; Ohkubo, T.; Ikawa, T.; Kume, A.; Maegawa, T.; Monguchi, Y. Chem. – Eur. J. 2008, 14, 5109–5111. doi:10.1002/chem.200800535 |
| 28. | Hauwert, P.; Maestri, G.; Sprengers, J. W.; Catellani, M.; Elsevier, C. J. Angew. Chem., Int. Ed. 2008, 47, 3223–3226. doi:10.1002/anie.200705638 |
| 23. | Frémont, L. Life Sci. 2000, 66, 663–673. doi:10.1016/S0024-3205(99)00410-5 |
| 24. | Dong, Z. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2003, 523–524, 145–150. doi:10.1016/S0027-5107(02)00330-5 |
| 25. | Hauser, F. M.; Ganguly, D. J. Org. Chem. 2000, 65, 1842–1849. doi:10.1021/jo991483w |
| 26. | You, Z.-W.; Jiang, Z.-X.; Wang, B.-L.; Qing, F.-L. J. Org. Chem. 2006, 71, 7261–7267. doi:10.1021/jo061012r |
| 36. | Acetylene chemistry: chemistry, biology and material science; Diederich, F.; Stang, P. J.; Tykwinski, R. R., Eds.; Wiley-VCH: Weinheim, 2005; pp 51 ff. |
| 37. | McGallion, G. D. Curr. Org. Chem. 1999, 3, 67–76. |
| 12. | Bacsa, I.; Jójárt, R.; Schneider, G.; Wölfling, J.; Maróti, P.; Herman, B. E.; Szécsi, M.; Mernyák, E. Steroids 2015, 104, 230–236. doi:10.1016/j.steroids.2015.10.008 |
| 13. | Herman, B. E.; Szabó, J.; Bacsa, I.; Wölfling, J.; Schneider, G.; Bálint, M.; Hetényi, C.; Mernyák, E.; Szécsi, M. J. Enzyme Inhib. Med. Chem. 2016, 31, 61–69. doi:10.1080/14756366.2016.1204610 |
| 32. | Zhang, W.; Go, M. L. Eur. J. Med. Chem. 2007, 42, 841–850. doi:10.1016/j.ejmech.2006.12.012 |
| 33. | Jagt, D. L. V.; Deck, L. M.; Royer, R. E.; Heidrich, J. E.; Sillerud, L. O. Substituted cis- and trans-stilbenes as therapeutic agents. U. S. Pat. Appl. US 20130178536 A1, July 11, 2013. |
| 34. | Arcadi, A.; Marinelli, F.; Cacchi, S. Synthesis 1986, 749–751. doi:10.1055/s-1986-31764 |
| 35. | Omelchuk, O. A.; Tikhomirov, A. S.; Shchekotikhin, A. E. Russ. Chem. Rev. 2016, 85, 817–835. doi:10.1070/RCR4613 |
| 31. | Parihar, S.; Kumar, A.; Chaturvedi, A. K.; Sachan, N. K.; Luqman, S.; Changkija, B.; Manohar, M.; Prakash, O.; Chanda, D.; Khan, F.; Chanotiya, C. S.; Shanker, K.; Dwivedi, A.; Konwar, R.; Negi, A. S. J. Steroid Biochem. Mol. Biol. 2013, 137, 332–344. doi:10.1016/j.jsbmb.2013.02.009 |
| 32. | Zhang, W.; Go, M. L. Eur. J. Med. Chem. 2007, 42, 841–850. doi:10.1016/j.ejmech.2006.12.012 |
| 33. | Jagt, D. L. V.; Deck, L. M.; Royer, R. E.; Heidrich, J. E.; Sillerud, L. O. Substituted cis- and trans-stilbenes as therapeutic agents. U. S. Pat. Appl. US 20130178536 A1, July 11, 2013. |
| 29. | Corey, E. J.; Pasto, D. J.; Mock, W. L. J. Am. Chem. Soc. 1961, 83, 2957–2958. doi:10.1021/ja01474a043 |
| 30. | Li, J.; Hua, R.; Liu, T. J. Org. Chem. 2010, 75, 2966–2970. doi:10.1021/jo100247a |
© 2017 Bacsa et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)
