
Abstract
Merging of photo- and mechanochemical activation permitted studying the role of eosin Y in the borylation of aryldiazonium salts in a ball mill. Simultaneous neat grinding/irradiation of the reactants and the photocatalyst led to the formation of boronates in a molten state. On the other hand, the catalyst-free liquid-assisted grinding/irradiation reaction also led to product formation, featuring a direct photolysis pathway facilitated by substrate–solvent charge-transfer complex formation.
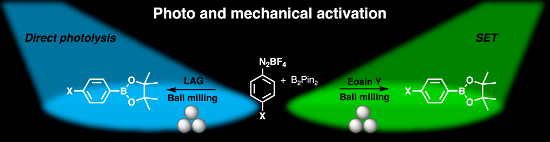
Graphical Abstract
Introduction
The use of mechanical force to process materials or to induce chemical transformations is perhaps as old as the history of mankind itself [1]. Similarly, from time immemorial light has also been present on earth, being perhaps photosynthesis and visions the most fundamental connections between light and living organisms [2]. However, combining synergistically photo- and mechanical activations in organic synthesis is still challenging despite the enormous potential of having both activation modes acting simultaneously.
In recent years, mechanochemistry, which encompasses the use of mechanical means by milling, grinding, shearing, cavitation or pulling to induce chemical transformations [3] has become fundamental for discovering new chemical reactivity [4,5] and to develop more sustainable syntheses. Typically, mechanochemical reactions by milling are conducted in non-translucent containers (e.g., agate, ceramics, steels, and tungsten carbide). While this diversity of milling media materials enables controlling, for example, the energy input during the mechanical process, it becomes an obstacle for in situ characterization of mechanochemical reactions, or to facilitate synergistic activation types involving, for example, light and mechanical energy. Recently, however, the in situ study of mechanochemical transformations has been accomplished by combining translucent milling vessels made of poly(methyl methacrylate) (PMMA) with powder X-ray diffraction (PXRD) [6,7], Raman spectroscopy [8], or a combination of both techniques [9]. On the other hand, attempts to combine photo- and mechanical activation to favor chemical processes have been mainly explored in the photodimerizations of olefins by manual grinding of the reactants followed by long UV-light exposure [10], or by vortex grinding [11]. However, until now, studies of photocatalyzed mechanochemical reactions involving, for example, metal complexes [12] or organic photocatalysts (PC) [13] has been underexplored [14], despite photocatalysis could clearly benefit from the excellent mixing under neat or liquid-assisted grinding (LAG) [15] conditions. Additionally, in contrast to solution-based methods, reactions by milling do not suffer from solubility restrictions due to the possibility to bring reactants and catalysts of very different solubility, into close proximity for achieving chemical reactivity. This last aspect is foreseen as highly valuable in transformations using low-soluble PCs (e.g., porphyrins) [16] or during the photochemical synthesis or modification of polymers [17].
The aforementioned context makes one wonder about the potential for conducting chemical reactions under simultaneous photo- and mechanical activation. To test this idea, the photocatalyzed borylation of aryldiazonium salts, first reported in solution by Yan and co-workers was selected as a model reaction [18]. In the original study, irradiation for 18 h of a MeCN solution of aryldiazonium salts, bis(pinacolato)diboron (B2pin2, 2) and eosin Y with a 25 W visible light lamp led to the corresponding arylboronates in moderate to good yields [18].
Results and Discussion
To commence, a PMMA milling jar was designed to enable external light irradiation of the reaction mixture while having simultaneously the high-speed ball milling acting on the mixture of reactants and PC (Figure 1; for details, see Supporting Information File 1).
![[1860-5397-13-144-1]](/bjoc/content/figures/1860-5397-13-144-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: (a) Cartoon representing the merging of light and mechanical energy. (b) 25 mL transparent PMMA milling jar. (c) Experimental setup for simultaneous photo- and mechanical-activation with an external light source.
Figure 1: (a) Cartoon representing the merging of light and mechanical energy. (b) 25 mL transparent PMMA mil...
Subsequently, with the aim to determine the role of the light, PC and the mechanical milling in the borylation of the aryldiazonium salts, and especially to exclude a potential background borylation reaction triggered by either thermal, mechanical or light-induced heterolytic cleavage of aryldiazonium salts, various control reactions were conducted. First, an equimolar mixture of the diazonium salt 1a and B2pin2 (2) was milled for 2 h at 25 Hz in a mixer mill, using a Teflon milling jar and ZrO2 ball bearings. The safe use of diazonium salt under ball milling conditions has been previously reported in the literature [19]. The analysis of the reaction mixture by 1H NMR spectroscopy revealed just the presence of both reactants, both in the presence or absence of the organic photocatalyst eosin Y (5.0 mol %). Ruling out a sole mechanochemical activation pathway (Table 1, entries 1 and 2).
Table 1: Screening of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-144-i3.svg?max-width=637&scale=1.0)
|
||||
| Entry | Eosin Y (mol %) | Time (h) | Light | 1a:3a (%)b |
|---|---|---|---|---|
| 1c | – | 2 | – | 100:0 |
| 2c | (5) | 2 | – | 100:0 |
| 3 | (5) | 2 | ambient | 100:0 |
| 4 | – | 2 | blue LEDs | 100:0 |
| 5d | (5) | 2 | blue LEDs | 94:6 |
| 6 | (5) | 0.5 | blue LEDs | 83:17 |
| 7 | (5) | 1 | blue LEDs | 54:46 |
| 8 | (5) | 1.5 | blue LEDs | 27:73 |
| 9 | (5) | 2 | blue LEDs | 15:85 |
| 10e | (5) | 2 | blue LEDs | 59:41 |
| 11f | (5) | 2 | blue LEDs | 51:49 |
| 12 | (5) | 1 | green LEDs | 6:94 |
| 13 | (0.5) | 1.5 | green LEDs | 63:37 |
aReaction conditions: a mixture of 1a (0.369 mmol), 2 (0.369 mmol) and eosin Y was mixed in a 25 mL PMMA milling jar with 15 ZrO2 balls of 5 mm in diameter at 25 Hz. bDetermined by 1H NMR spectroscopy. cA 25 mL Teflon milling jar was used. d1a, 2 and the PC were mixed for 30 s in the PMMA jar, then the mixing was stopped and the milling jar was exposed to the light irradiation for 2 h. eThe irradiation was stopped after 1 h of reaction. fThe milling was stopped after 1 h of reaction.
Repeating the reaction in the presence of the PC in the transparent PPMA milling jar yielded the same negative result proving that ambient light did not mediate the photoredox catalytic borylation reaction under mechanochemical conditions (Table 1, entry 3). Furthermore, neat grinding of a catalyst-free mixture of 1a and 2 under blue LEDs (light-emitting diodes) light did not afford the borylated product 3a. In addition to the 1H NMR analysis in solution, this result was confirmed by immediate ex situ analysis using IR spectroscopy of the solid reaction mixture, which revealed only the presence of both starting materials. Thereby, excluding a direct thermal [20,21] or photolysis pathway operating under solventless conditions (Table 1, entry 4). Then, a premilled mixture of 1a, B2pin2, and eosin Y was subjected to irradiation with blue LEDs for 2 h in the absence of milling (Table 1, entry 5). After the irradiation was halted, the reaction mixture was immediately analyzed by 1H NMR spectroscopy. This time trace quantities of product 3a were detected (Table 1, entry 5). This interesting result under solvent-free conditions encouraged performing the light irradiation accompanied by milling to improve mixing and to increase the surface exposure of the reaction mixture. In a following set of experiments, milling of the reactants and PC was carried out for a time in the range of 15 min to 2 h. The analysis of the composition of the reaction mixture showed significant formation of the product after 30 min of milling/irradiation (Table 1, entry 6). Monitoring the progress of a mixture of the reactants and PC in CD3CN at room temperature by 1H NMR spectroscopy over a period of time of 20 h showed a composition (98:2; 1a:3a), ruling out the formation of the 3a during the standard analysis time. Furthermore, the presence of 3a in the mixture coincided with the observation of an initial molten state of the mixture inside the milling jar [22]. This more homogeneous mixture could have increased the mobility of the reactants favoring the SET process. Reaching the molten state clearly required having both activation modes acting simultaneously, since only milling of 1a (mp 138–141 °C), 2 (mp 139–140 °C) and eosin Y (mp 305–307 °C), or just irradiation of the mixture did not lead to an observable melting of the solids (Table 1, entries 2–4 and 5). Besides, propagation of this molten state could have been favored by the gradual rise in concentration of the lower-melting product 3a (mp 69–70 °C) in the mixture. Indeed, milling the product 3a under the standard milling conditions using the LEDs led to its melt. Similarly, milling a mixture of 1a, 2 and 3a for 1 h under light irradiation reached a eutectic melt phase. The need for simultaneous light and mechanical milling was also confirmed after conducting experiments for 2 h where either the milling or the irradiation was stopped after the first hour. In both cases the outcome of the reaction gave similar results compared to having both energy sources acting together for 1 h (Table 1, entries 7, 10, and 11).
Next, further tuning of the reaction conditions revealed green LEDs to be a more efficient light source for the reaction with eosin Y (for details, see Table S1 in Supporting Information File 1). This change permitted the transformation to take place after 1 h of milling/irradiation. Under these conditions, the ratio 1a:3a in the reaction mixture reached 6:94 (entry 12 in Table 1). Alternatively, longer reaction times allowed reducing the amount of the organic photocatalyst to 1.0 mol % and 0.5 mol % (entry 13 in Table 1; for more details, see Table S1 in Supporting Information File 1). Then, using the green LEDs an experiment in the presence of 1,1-diphenylethene (4) as a radical inhibitor was conducted. After the standard 1 h of milling, the formation of 3a was slowed down and the analysis of the reaction mixture by gas chromatography–mass spectrometry showed the presence of the phenyl radical trapping adduct 5 (Scheme 1).
![[1860-5397-13-144-i1]](/bjoc/content/inline/1860-5397-13-144-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Borylation of 1a in the presence of 1,1-diphenylethene (4).
Scheme 1: Borylation of 1a in the presence of 1,1-diphenylethene (4).
With the optimized reaction conditions in hand, we explored the photomechanochemical borylation of the halogenated aryldiazonium salts 1a–d (Table 2).
Table 2: Borylation of aryl diazonium salts 1 with 2.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-144-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Aryldiazonium salt | Product | Time (min) | Yield (%)b |
|---|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-144-i5.svg?max-width=637&scale=1.0)
1b |
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-144-i6.svg?max-width=637&scale=1.0)
3b |
90 | 60 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-13-144-i7.svg?max-width=637&scale=1.0)
1c |
![[Graphic 6]](/bjoc/content/inline/1860-5397-13-144-i8.svg?max-width=637&scale=1.0)
3c |
45 | 55 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-13-144-i9.svg?max-width=637&scale=1.0)
1a |
![[Graphic 8]](/bjoc/content/inline/1860-5397-13-144-i10.svg?max-width=637&scale=1.0)
3a |
60 | 68 |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-13-144-i11.svg?max-width=637&scale=1.0)
1d |
![[Graphic 10]](/bjoc/content/inline/1860-5397-13-144-i12.svg?max-width=637&scale=1.0)
3d |
120 | 41 |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-13-144-i13.svg?max-width=637&scale=1.0)
1e |
![[Graphic 12]](/bjoc/content/inline/1860-5397-13-144-i14.svg?max-width=637&scale=1.0)
3e |
120 | 49 |
aReaction conditions: a mixture of 1 (0.369 mmol), 2 (140.6 mg; 0.554 mmol) and photocatalyst (5 mol %) was mixed in a 25 mL PMMA milling jar with 15 ZrO2 balls of 5 mm in diameter at 25 Hz. bAfter column chromatography.
Analogously to the case of 1a, the fluoro and chloro substituted aryldiazonium salts 1b and 1c did react affording the boronates 3b and 3c (Table 2, entries 1 and 2). It was noticed, however, that the milling/irradiation time required for these substrates to react varied in comparison with the reaction of 1a. Furthermore, attempts to react the 4-iodobenzenediazonium tetrafluoroborate (1d) with 2 were made with low success even after 2 h of reaction time (Table 2, entry 4). As indicated above, the development of a molten state upon irradiation and milling appears to be a key prerequisite for the photomechanochemical borylation to occur [14,22]. Reaching that molten state in the reaction with the iodo derivative 1d proved challenging, only a part of the reaction mixture seemed homogeneous. Differential scanning calorimetry (DSC) analysis of the aryldiazonium salts 1a–d revealed that 1a–c melt followed by decomposition of the samples. However, the DSC profile of the iodobenzenediazonium salt 1d showed a direct thermal decomposition upon heating. NMR analysis of the molten 1d revealed the presence of 1-fluoro-4-iodobenzene (for details, see Supporting Information File 1). Therefore, the difference in melting point temperatures of the substrates and their thermal stability could have a direct correlation with the observed reactivity in the ball mill (Table 2). Control experiments by stirring/heating 1a–d and 2 in an oil bath until the melting of the mixture was reached, showed predominantly thermal decomposition of the aryldiazonium salts 1a–d into the 1-halo-4-fluorobenzene derivatives [23] and only in some cases trace quantities of 3a–d were detected. Meaning that the external light contributes to both, heating the reaction mixture to its eutectic and it also initiated the photoredox process. Similarly, a photocatalyzed reaction between 2 and 4-nitrobenzenediazonium tetrafluoroborate (1e), a salt found also to undergo decomposition at 423 K, turned out difficult (for the DSC traces of 1a–e see Supporting Information File 1).
After 2 h of milling/irradiation the corresponding product 3e was obtained in moderate yield. In general, after the milling/irradiation experiments no aryldiazonium salt was observed in the reaction mixture, however, the moderate yields for the products 3a–e, even in the presence of 1.5 equiv of 2, could have been the result of background reactions undergone by 1a–e under the reaction conditions, especially due to the rise in temperature observed upon light irradiation.
As mentioned above, LAG, an alternative to the standard neat grinding, has become an useful parameter in mechanochemistry to control chemical selectivity and product composition by having catalytic volumes of organic solvent during the milling [24]. Here, a change in the milling approach from neat to LAG was anticipated to have the potential for switching the activation mode from a SET process to a direct heterolytic photolysis. Jacobi von Wangelin et al. noticed that the borylation product 3a could also occurred in the absence of eosin Y, upon irradiation of a MeCN solution of the reactants with white LEDs [23,25]. Pleasingly, LAG (MeCN or DMSO η = 0.25) of 1a and 2 under blue LEDs, and in the absence of eosin Y did also generate the product 3a. In contrast, LAG experiments using n-heptane failed at producing 3a (Scheme 2; for details, see Table S2 in Supporting Information File 1).
![[1860-5397-13-144-i2]](/bjoc/content/inline/1860-5397-13-144-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Light-mediated LAG borylation of 1a. aDetermined by 1H NMR spectroscopy using internal standard. bAfter column chromatography.
Scheme 2: Light-mediated LAG borylation of 1a. aDetermined by 1H NMR spectroscopy using internal standard. bA...
Similarly, control LAG/irradiation experiments conducted in a Teflon milling jar, only formed trace quantities of the product 3a, ruling out a sole thermal activation of the system by the light source. These results not only illustrates the versatility of mechanochemistry to control the chemical reaction pathway operating in the process, but also sheds light on the role of the photocatalyst in the borylation of the aryldiazonium salts.
Under non-catalyzed LAG/irradiation conditions, charge-transfer complexes between 1a and appropriate organic solvents could be responsible for the fast generation of the aryl cations in the ball mill, leading to the direct formation of 3a [26,27]. This observation is in agreement with the findings by Jacobi von Wangelin et al., who described that upon irradiation of a solution of 1a in MeCN direct heterolytic cleavage of the aryldiazonium salt occurred [23,25]. However, the formation of 3a under solvent-free milling conditions (vide supra) could have been indeed the result of a photoredox transformation where the organic photocatalyst eosin Y played a key role in triggering the SET process.
Conclusion
In summary, simultaneous activation of an organic system by light and ball milling techniques has been successfully accomplished for the first time. The utilization of translucent milling vessels permitted the study of the photoborylation of aryldiazonium salts in the presence and in the absence of the organic photocatalyst eosin Y. The results of this proof-of-concept demonstration revealed that under neat grinding conditions the PC does play a role in initiating a SET borylation. Furthermore, the implementation of a LAG/irradiation approach allowed the borylation reaction to occur under catalyst-free conditions. This observation is supported by the tendency of the electrophilic aryldiazonium salts to undergo direct heterolytic photolysis facilitated by organic solvents, upon exposure to near-UV or blue light. In addition to this, the contribution from the increase in temperature experienced during the light exposure and mechanical milling was observed to be vital for the neat grinding, facilitating the formation of molten reaction mixtures.
Despite the still existing technical challenges for merging light and mechanical energy, the positive cooperative synergism between light and mechanical activation reported here, will certainly stimulate the design of more innovative experimental setups [28] and, more important, the exploration of new photomechanochemical organic reactions, where solubility constrains caused by working with low-soluble photocatalysts, substrates or products can be bypassed by mechanochemical means.
Supporting Information
| Supporting Information File 1: Experimental procedures, experimental set-ups and characterization data, NMR spectra, and DSC traces. | ||
| Format: PDF | Size: 2.1 MB | Download |
Acknowledgements
This research was possible thanks to the financial support from the RWTH Aachen University through the 1) RWTH Start-up grant StUpPD_221-16 funded by the Excellence Initiative of the German federal and state governments and 2) the Distinguished Professorship Program. Dr. J. Langanke, Dr. C. Rosorius, M.Sc. A. Ernst (CAT Catalytic Center Aachen) are acknowledge for the access to DSC and the technical assistance. Prof. Dr. Carsten Bolm is gratefully acknowledged for generous support and valuable advice.
References
-
Takacs, L. Chem. Soc. Rev. 2013, 42, 7649–7659. doi:10.1039/C2CS35442J
Return to citation in text: [1] -
Valentini, A.; Rivero, D.; Zapata, F.; García-Iriepa, C.; Marazzi, M.; Palmeiro, R.; Galván, I. F.; Sampedro, D.; Olivucci, M.; Frutos, L. M. Angew. Chem., Int. Ed. 2017, 56, 3842–3846. doi:10.1002/anie.201611265
Return to citation in text: [1] -
James, S. L.; Adams, C. J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K. D. M.; Hyett, G.; Jones, W.; Krebs, A.; Mack, J.; Maini, L.; Orpen, A. G.; Parkin, I. P.; Shearouse, W. C.; Steed, J. W.; Waddell, D. C. Chem. Soc. Rev. 2012, 41, 413–447. doi:10.1039/C1CS15171A
Return to citation in text: [1] -
Hernández, J. G.; Bolm, C. J. Org. Chem. 2017, 82, 4007–4019. doi:10.1021/acs.joc.6b02887
Return to citation in text: [1] -
Do, J.-L.; Friščić, T. ACS Cent. Sci. 2017, 3, 13–19. doi:10.1021/acscentsci.6b00277
Return to citation in text: [1] -
Friščić, T.; Halasz, I.; Beldon, P. J.; Belenguer, A. M.; Adams, F.; Kimber, S. A. J.; Honkimäki, V.; Dinnebier, R. E. Nat. Chem. 2013, 5, 66–73. doi:10.1038/nchem.1505
Return to citation in text: [1] -
Užarević, K.; Halasz, I.; Friščić, T. J. Phys. Chem. Lett. 2015, 6, 4129–4140. doi:10.1021/acs.jpclett.5b01837
Return to citation in text: [1] -
Gracin, D.; Štrukil, V.; Friščić, T.; Halasz, I.; Užarević, K. Angew. Chem., Int. Ed. 2014, 53, 6193–6197. doi:10.1002/anie.201402334
Return to citation in text: [1] -
Batzdorf, L.; Fischer, F.; Wilke, M.; Wenzel, K.-J.; Emmerling, F. Angew. Chem., Int. Ed. 2015, 54, 1799–1802. doi:10.1002/anie.201409834
Return to citation in text: [1] -
Sokolov, A. N.; Bučar, D.-K.; Baltrusaitis, J.; Gu, S. X.; MacGillivray, L. R. Angew. Chem., Int. Ed. 2010, 49, 4273–4277. doi:10.1002/anie.201000874
Return to citation in text: [1] -
Stojaković, J.; Farris, B. S.; MacGillivray, L. R. Faraday Discuss. 2014, 170, 35–40. doi:10.1039/C4FD00006D
Return to citation in text: [1] -
Teegardin, K.; Day, J. I.; Chan, J.; Weaver, J. Org. Process Res. Dev. 2016, 20, 1156–1163. doi:10.1021/acs.oprd.6b00101
Return to citation in text: [1] -
Hari, D. P.; König, B. Chem. Commun. 2014, 50, 6688–6699. doi:10.1039/C4CC00751D
Return to citation in text: [1] -
Obst, M.; König, B. Beilstein J. Org. Chem. 2016, 12, 2358–2363. doi:10.3762/bjoc.12.229
Return to citation in text: [1] [2] -
Friščić, T.; Childs, S. L.; Rizvi, S. A. A.; Jones, W. CrystEngComm 2009, 11, 418–426. doi:10.1039/B815174A
Return to citation in text: [1] -
Rybicka-Jasińska, K.; König, B.; Gryko, D. Eur. J. Org. Chem. 2017, 2104–2107. doi:10.1002/ejoc.201601518
Return to citation in text: [1] -
Corrigan, N.; Shanmugam, S.; Xu, J.; Boyer, C. Chem. Soc. Rev. 2016, 45, 6165–6212. doi:10.1039/C6CS00185H
Return to citation in text: [1] -
Yu, J.; Zhang, L.; Yan, G. Adv. Synth. Catal. 2012, 354, 2625–2628. doi:10.1002/adsc.201200416
Return to citation in text: [1] [2] -
Mukherjee, N.; Chatterjee, T.; Ranu, B. C. J. Org. Chem. 2013, 78, 11110–11114. doi:10.1021/jo402071b
Return to citation in text: [1] -
Kulla, H.; Wilke, M.; Fischer, F.; Rölling, M.; Maierhofer, C.; Emmerling, F. Chem. Commun. 2017, 53, 1664–1667. doi:10.1039/C6CC08950J
Return to citation in text: [1] -
Zhu, C.; Yamane, M. Org. Lett. 2012, 14, 4560–4563. doi:10.1021/ol302024m
Return to citation in text: [1] -
Rothenberg, G.; Downie, A. P.; Raston, C. L.; Scott, J. L. J. Am. Chem. Soc. 2001, 123, 8701–8708. doi:10.1021/ja0034388
Return to citation in text: [1] [2] -
Majek, M.; Jacobi von Wangelin, A. Acc. Chem. Res. 2016, 49, 2316–2327. doi:10.1021/acs.accounts.6b00293
Return to citation in text: [1] [2] [3] -
Hasa, D.; Miniussi, E.; Jones, W. Cryst. Growth Des. 2016, 16, 4582–4588. doi:10.1021/acs.cgd.6b00682
Return to citation in text: [1] -
Majek, M.; Filace, F.; Jacobi von Wangelin, A. Beilstein J. Org. Chem. 2014, 10, 981–989. doi:10.3762/bjoc.10.97
Return to citation in text: [1] [2] -
Hirose, Y.; Wahl, G. H., Jr.; Zollinger, H. Helv. Chim. Acta 1976, 59, 1427–1437. doi:10.1002/hlca.19760590504
Return to citation in text: [1] -
Szele, I.; Zollinger, H. Helv. Chim. Acta 1978, 61, 1721–1729. doi:10.1002/hlca.19780610520
Return to citation in text: [1] -
Obst, M.; Shaikh, R. S.; König, B. React. Chem. Eng. 2017. doi:10.1039/C6RE00220J
Return to citation in text: [1]
| 6. | Friščić, T.; Halasz, I.; Beldon, P. J.; Belenguer, A. M.; Adams, F.; Kimber, S. A. J.; Honkimäki, V.; Dinnebier, R. E. Nat. Chem. 2013, 5, 66–73. doi:10.1038/nchem.1505 |
| 7. | Užarević, K.; Halasz, I.; Friščić, T. J. Phys. Chem. Lett. 2015, 6, 4129–4140. doi:10.1021/acs.jpclett.5b01837 |
| 17. | Corrigan, N.; Shanmugam, S.; Xu, J.; Boyer, C. Chem. Soc. Rev. 2016, 45, 6165–6212. doi:10.1039/C6CS00185H |
| 4. | Hernández, J. G.; Bolm, C. J. Org. Chem. 2017, 82, 4007–4019. doi:10.1021/acs.joc.6b02887 |
| 5. | Do, J.-L.; Friščić, T. ACS Cent. Sci. 2017, 3, 13–19. doi:10.1021/acscentsci.6b00277 |
| 18. | Yu, J.; Zhang, L.; Yan, G. Adv. Synth. Catal. 2012, 354, 2625–2628. doi:10.1002/adsc.201200416 |
| 3. | James, S. L.; Adams, C. J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K. D. M.; Hyett, G.; Jones, W.; Krebs, A.; Mack, J.; Maini, L.; Orpen, A. G.; Parkin, I. P.; Shearouse, W. C.; Steed, J. W.; Waddell, D. C. Chem. Soc. Rev. 2012, 41, 413–447. doi:10.1039/C1CS15171A |
| 15. | Friščić, T.; Childs, S. L.; Rizvi, S. A. A.; Jones, W. CrystEngComm 2009, 11, 418–426. doi:10.1039/B815174A |
| 2. | Valentini, A.; Rivero, D.; Zapata, F.; García-Iriepa, C.; Marazzi, M.; Palmeiro, R.; Galván, I. F.; Sampedro, D.; Olivucci, M.; Frutos, L. M. Angew. Chem., Int. Ed. 2017, 56, 3842–3846. doi:10.1002/anie.201611265 |
| 16. | Rybicka-Jasińska, K.; König, B.; Gryko, D. Eur. J. Org. Chem. 2017, 2104–2107. doi:10.1002/ejoc.201601518 |
| 11. | Stojaković, J.; Farris, B. S.; MacGillivray, L. R. Faraday Discuss. 2014, 170, 35–40. doi:10.1039/C4FD00006D |
| 13. | Hari, D. P.; König, B. Chem. Commun. 2014, 50, 6688–6699. doi:10.1039/C4CC00751D |
| 10. | Sokolov, A. N.; Bučar, D.-K.; Baltrusaitis, J.; Gu, S. X.; MacGillivray, L. R. Angew. Chem., Int. Ed. 2010, 49, 4273–4277. doi:10.1002/anie.201000874 |
| 14. | Obst, M.; König, B. Beilstein J. Org. Chem. 2016, 12, 2358–2363. doi:10.3762/bjoc.12.229 |
| 9. | Batzdorf, L.; Fischer, F.; Wilke, M.; Wenzel, K.-J.; Emmerling, F. Angew. Chem., Int. Ed. 2015, 54, 1799–1802. doi:10.1002/anie.201409834 |
| 8. | Gracin, D.; Štrukil, V.; Friščić, T.; Halasz, I.; Užarević, K. Angew. Chem., Int. Ed. 2014, 53, 6193–6197. doi:10.1002/anie.201402334 |
| 12. | Teegardin, K.; Day, J. I.; Chan, J.; Weaver, J. Org. Process Res. Dev. 2016, 20, 1156–1163. doi:10.1021/acs.oprd.6b00101 |
| 20. | Kulla, H.; Wilke, M.; Fischer, F.; Rölling, M.; Maierhofer, C.; Emmerling, F. Chem. Commun. 2017, 53, 1664–1667. doi:10.1039/C6CC08950J |
| 21. | Zhu, C.; Yamane, M. Org. Lett. 2012, 14, 4560–4563. doi:10.1021/ol302024m |
| 18. | Yu, J.; Zhang, L.; Yan, G. Adv. Synth. Catal. 2012, 354, 2625–2628. doi:10.1002/adsc.201200416 |
| 19. | Mukherjee, N.; Chatterjee, T.; Ranu, B. C. J. Org. Chem. 2013, 78, 11110–11114. doi:10.1021/jo402071b |
| 23. | Majek, M.; Jacobi von Wangelin, A. Acc. Chem. Res. 2016, 49, 2316–2327. doi:10.1021/acs.accounts.6b00293 |
| 25. | Majek, M.; Filace, F.; Jacobi von Wangelin, A. Beilstein J. Org. Chem. 2014, 10, 981–989. doi:10.3762/bjoc.10.97 |
| 28. | Obst, M.; Shaikh, R. S.; König, B. React. Chem. Eng. 2017. doi:10.1039/C6RE00220J |
| 23. | Majek, M.; Jacobi von Wangelin, A. Acc. Chem. Res. 2016, 49, 2316–2327. doi:10.1021/acs.accounts.6b00293 |
| 25. | Majek, M.; Filace, F.; Jacobi von Wangelin, A. Beilstein J. Org. Chem. 2014, 10, 981–989. doi:10.3762/bjoc.10.97 |
| 26. | Hirose, Y.; Wahl, G. H., Jr.; Zollinger, H. Helv. Chim. Acta 1976, 59, 1427–1437. doi:10.1002/hlca.19760590504 |
| 27. | Szele, I.; Zollinger, H. Helv. Chim. Acta 1978, 61, 1721–1729. doi:10.1002/hlca.19780610520 |
| 23. | Majek, M.; Jacobi von Wangelin, A. Acc. Chem. Res. 2016, 49, 2316–2327. doi:10.1021/acs.accounts.6b00293 |
| 24. | Hasa, D.; Miniussi, E.; Jones, W. Cryst. Growth Des. 2016, 16, 4582–4588. doi:10.1021/acs.cgd.6b00682 |
| 22. | Rothenberg, G.; Downie, A. P.; Raston, C. L.; Scott, J. L. J. Am. Chem. Soc. 2001, 123, 8701–8708. doi:10.1021/ja0034388 |
| 14. | Obst, M.; König, B. Beilstein J. Org. Chem. 2016, 12, 2358–2363. doi:10.3762/bjoc.12.229 |
| 22. | Rothenberg, G.; Downie, A. P.; Raston, C. L.; Scott, J. L. J. Am. Chem. Soc. 2001, 123, 8701–8708. doi:10.1021/ja0034388 |
© 2017 Hernández; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)
