Abstract
Bifunctional organocatalysts bearing amino and urea functional groups in a chiral molecular skeleton were applied to the enantioselective synthesis of axially chiral benzamides via aromatic electrophilic bromination. The results demonstrate the versatility of bifunctional organocatalysts for the enantioselective construction of axially chiral compounds. Moderate to good enantioselectivities were afforded with a range of benzamide substrates. Mechanistic investigations were also carried out.

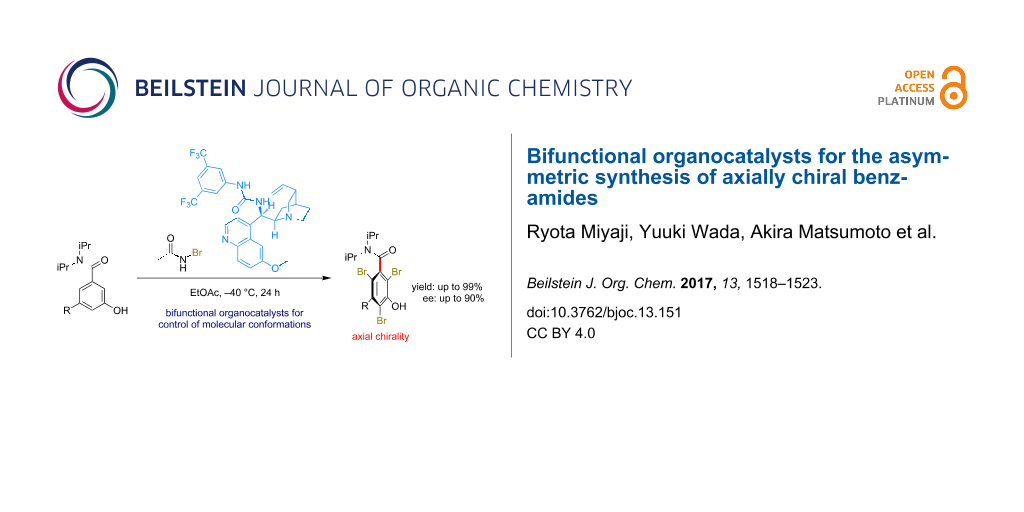
Graphical Abstract
Introduction
Bifunctional organocatalysts have significantly contributed to the field of asymmetric synthesis [1-6]. In these catalysts, (thio)urea and tertiary amino functional groups cooperatively activate a nucleophile and an electrophile simultaneously, in a suitable spatial configuration. Thus, they enable various stereoselective addition reactions to occur. Organocatalysts have also been employed in several asymmetric cyclization reactions via intramolecular hetero-Michael addition [7-16]. In these reactions, multipoint recognition by the catalysts favors the specific conformations of the substrates in the transition state. Several successful results and a recent trend in organocatalytic atroposelective reactions, including enantioselective formation of chiral axes [17-24], dynamic kinetic resolution [25-41], kinetic resolution [42-47], desymmetrization [48-54], de novo annulation [55-61], and point-to-axial chirality transfer [58,59] (for reviews, see references [31,62,63]), motivated us to expand on the utility of this class of small-molecule catalysts. We have recently demonstrated that bifunctional organocatalysts can also be applied to the asymmetric synthesis of axially chiral compounds (biaryls bearing isoquinoline N-oxides or quinolines and phenolic moieties) by translating a specific conformation, recognized by bifunctional organocatalysts, into axial chirality [36,37]. Thus, we assumed that this method could be further applied to the enantioselective synthesis of a range of axially chiral compounds. In this study, we present the enantioselective synthesis of 3-hydroxybenzamides via aromatic electrophilic bromination [28,29]. The 3-hydroxybenzamide substrates comprise both amide and phenolic moieties. These can interact with a hydrogen-bond donor and a hydrogen-bond acceptor, respectively. Such interactions are expected to recognize a specific conformation of the substrate molecule to realize the enantioselective construction of axially chiral benzamides [36,37].
Results and Discussion
We initiated our investigations using 3-hydroxy-N,N-diisopropylbenzamide (1a) and N-bromoacetamide (NBA, 4a) as a brominating reagent, with 10 mol % quinidine-derived bifunctional catalyst 3a, in toluene, at −40 °C. As expected, the tribrominated product 2a was formed enantioselectively (Table 1, entry 1). Although a lower temperature did not improve the enantioselectivity (Table 1, entry 2), lowering the concentration of the reaction mixture was effective (Table 1, entry 3). The screening of solvents identified ethyl acetate as the most suitable solvent (Table 1, entries 4–7). Other brominating reagents (Figure 1) were also investigated; however, NBA (4a) still afforded the best enantioselective results (Table 1, entries 8–10). In addition, other bifunctional organocatalysts derived from easily available cinchona alkaloids exhibited similarly good enantioselectivities; 3c and 3d afforded the opposite enantiomer of the product (Table 1, entries 11–13, results of further catalyst screening are described in the Supporting Information File 1).
Table 1: Optimization of conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-151-i7.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Catalyst | Brominating reagent | Solvent | Yield (%)b | ee (%) |
|---|---|---|---|---|---|
| 1c | 3a | NBA (4a) | toluene | 88 | 78 |
| 2c,d | 3a | NBA (4a) | toluene | 48 | 78 |
| 3 | 3a | NBA (4a) | toluene | 58 | 84 |
| 4 | 3a | NBA (4a) | CHCl3 | 73 | 84 |
| 5 | 3a | NBA (4a) | Et2O | 66 | 42 |
| 6 | 3a | NBA (4a) | THF | 69 | 82 |
| 7 | 3a | NBA (4a) | EtOAc | 84 | 87 |
| 8e | 3a | DBH (4b) | EtOAc | 99 | 77 |
| 9 | 3a | NBS (4c) | EtOAc | 99 | 51 |
| 10 | 3a | NBP (4d) | EtOAc | 99 | 72 |
| 11 | 3b | NBA (4a) | EtOAc | 56 | 84 |
| 12 | 3c | NBA (4a) | EtOAc | 89 | −81 |
| 13 | 3d | NBA (4a) | EtOAc | 76 | −80 |
aReactions were run using 1a (0.1 mmol), the catalyst (0.01 mmol), and the brominating reagent (0.3 mmol) in the solvent (10 mL). bIsolated yields. cReactions were run in 0.5 mL of toluene. dReaction was run at −45 °C. e1.5 equiv of 4b was used for the reaction.
![[1860-5397-13-151-1]](/bjoc/content/figures/1860-5397-13-151-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
We then investigated substrates bearing other substituents on the amino group (Scheme 1). Dimethyl- and diisobutylamide groups resulted in much lower enantioselectivities (2b and 2c). Substrates bearing cyclohexyl groups or a piperidinyl moiety provided the corresponding products in high yields; however, the enantioselectivities were not as high as that of 2a. The absolute configuration of 2d was determined by X-ray analysis (see the Supporting Information File 1 for details), and the configurations of all other examples were assigned analogously.
![[1860-5397-13-151-i1]](/bjoc/content/inline/1860-5397-13-151-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Optimization of the substituents of the amide group. Reactions were run using 1 (0.1 mmol), 3a (0.01 mmol), and 4a (0.3 mmol) in EtOAc (10 mL). Yields represent material isolated after silica gel column chromatography.
Scheme 1: Optimization of the substituents of the amide group. Reactions were run using 1 (0.1 mmol), 3a (0.0...
Once the optimal conditions for the transformation were established, we next proceeded to explore the substrate scope (Scheme 2). The substrate bearing a phenyl group yielded the product with the highest enantioselectivity (Scheme 2, 2f). However, a decrease in enantioselectivity was observed when the phenyl group was replaced by substituted phenyl groups (Scheme 2, 2g and 2h). The substrate bearing a naphthyl group afforded the corresponding product in moderate enantioselectivity (Scheme 2, 2i). In addition, a benzamide with a cyclopropyl group also provided the product in good enantioselectivity (Scheme 2, 2j). Furthermore, when the reaction was carried out using 1k and 1l with 2 equiv of NBA (4a), dibromination proceeded in high yields and moderate enantioselectivities (Scheme 3); both 1k and 1l comprise a substituent ortho to the hydroxy group. These brominated axially chiral benzamides can further be derivatized for the synthesis of functional molecules [64].
![[1860-5397-13-151-i2]](/bjoc/content/inline/1860-5397-13-151-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope. Reactions were run using 1 (0.1 mmol), 3a (0.01 mmol), and 4a (0.3 mmol) in EtOAc (10 mL). Yields represent material isolated after silica gel column chromatography.
Scheme 2: Substrate scope. Reactions were run using 1 (0.1 mmol), 3a (0.01 mmol), and 4a (0.3 mmol) in EtOAc ...
![[1860-5397-13-151-i3]](/bjoc/content/inline/1860-5397-13-151-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reactions of substrates with substituted phenols.
Scheme 3: Reactions of substrates with substituted phenols.
To gain insight into the reaction mechanism, the reactions were performed using substrates 1m and 1n, previously monobrominated at the ortho-positions of the rotational axis. Much lower enantioselectivities than that afforded by 1a were observed in both reactions (Scheme 4). In addition, the reaction was also carried out with 1 equiv of NBA (4a). The sole product afforded was 1m and most of the starting material was recovered (see Supporting Information File 1 for details). These results imply that the first bromination, occurring at the ortho-position of the axis (probably at the 2-position), is the enantiodetermining step of the reaction. Moreover, once one of the ortho-positions is brominated, racemization through bond rotation is negligible during further brominations [65]. Indeed, the rotational barrier of substrate 1a, calculated at the B3YLP/6-31G(d) level of theory, is only 7.6 kcal/mol; on the other hand, that of the monobrominated intermediate 1m is 19.0 kcal/mol (Scheme 5). However, this latter value is not high enough to inhibit bond rotation at room temperature. This explains why the reactions must be carried out at such a low temperature (−40 °C) to afford high enantioselectivities. Compound 1o, with both ortho-positions brominated, has a rotational barrier that is high enough to enable the isolation of the optically active form, even at room temperature. Furthermore, it is also important to employ substrates bearing bulky substituents on the nitrogen atom. Such substrates limit the bond rotation about the chiral axis to realize high enantioselectivity (Scheme 1). The rotational barriers of monobrominated compounds 1p and 1q (bearing methyl and isobutyl groups, respectively, on the amide moiety) are lower than that of 1m. Although racemization of 2b, the rotational barrier of which is 22.9 kcal/mol, was observed after a lot of months, it is enough slow to enable the immediate analysis of the reaction selectivity (the decrease of the enantiomeric purity of 2b was negligible after a day).
![[1860-5397-13-151-i4]](/bjoc/content/inline/1860-5397-13-151-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reactions of monobrominated substrates.
Scheme 4: Reactions of monobrominated substrates.
![[1860-5397-13-151-i5]](/bjoc/content/inline/1860-5397-13-151-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Rotational barriers of substrates and intermediates calculated at the B3YLP/6-31G(d) level of theory.
Scheme 5: Rotational barriers of substrates and intermediates calculated at the B3YLP/6-31G(d) level of theor...
Furthermore, the reaction of benzamide 5, bearing a protected phenol, was carried out (Scheme 6). It failed to give the corresponding product 6, indicating the significance of multipoint activation involving the phenolic hydroxy group.
![[1860-5397-13-151-i6]](/bjoc/content/inline/1860-5397-13-151-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reaction of substrate with protected phenol.
Scheme 6: Reaction of substrate with protected phenol.
Conclusion
In summary, we demonstrated a novel enantioselective synthesis of axially chiral benzamides, using bifunctional organocatalysts, via aromatic electrophilic halogenation. Moderate to good enantioselectiveties were accomplished with various benzamide substrates. These results, along with ones reported in our previous work and other literature [35-38,58,59], verify the utility of bifunctional organocatalysts for application in the synthesis of various axially chiral compounds. Further studies regarding the detailed clarification of the reaction mechanism and application of this method to the construction of other axially chiral structures are currently underway and will be reported in due course.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, copies of the 1H, 13C NMR spectra, HPLC chromatogram profiles, and the ORTEP drawing. | ||
| Format: PDF | Size: 3.3 MB | Download |
| Supporting Information File 2: Crystallographic information file of compound 2d. | ||
| Format: CIF | Size: 30.9 KB | Download |
Acknowledgements
We thank Professor Takuya Kurahashi (Kyoto University) for X-ray crystallographic analysis. This work was supported financially by the Japanese Ministry of Education, Culture, Sports, Science and Technology (15H05845 and 16K13994). K.A. also acknowledges the Asahi Glass Foundation, Toyota Physical and Chemical Research Institute, Tokyo Institute of Technology Foundation, the Naito Foundation, Research Institute for Production Development, the Tokyo Biochemical Research Foundation, the Uehara Memorial Foundation, and the Kyoto University Foundation. R.M. and A.M. also acknowledge the Japan Society for the Promotion of Science for Young Scientists for the fellowship support.
References
-
Okino, T.; Hoashi, Y.; Takemoto, Y. J. Am. Chem. Soc. 2003, 125, 12672. doi:10.1021/ja036972z
Return to citation in text: [1] -
Okino, T.; Hoashi, Y.; Furukawa, T.; Xu, X.; Takemoto, Y. J. Am. Chem. Soc. 2005, 127, 119. doi:10.1021/ja044370p
Return to citation in text: [1] -
Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967. doi:10.1021/ol050431s
Return to citation in text: [1] -
Hamza, A.; Schubert, G.; Soós, T.; Pápai, I. J. Am. Chem. Soc. 2006, 128, 13151. doi:10.1021/ja063201x
Return to citation in text: [1] -
Connon, S. J. Chem. – Eur. J. 2006, 12, 5418. doi:10.1002/chem.200501076
Return to citation in text: [1] -
Zhu, J.-L.; Zhang, Y.; Liu, C.; Zheng, A.-M.; Wang, W. J. Org. Chem. 2012, 77, 9813. doi:10.1021/jo302133n
Return to citation in text: [1] -
Asano, K.; Matsubara, S. J. Am. Chem. Soc. 2011, 133, 16711. doi:10.1021/ja207322d
Return to citation in text: [1] -
Asano, K.; Matsubara, S. Org. Lett. 2012, 14, 1620. doi:10.1021/ol3003755
Return to citation in text: [1] -
Okamura, T.; Asano, K.; Matsubara, S. Chem. Commun. 2012, 48, 5076. doi:10.1039/c2cc31602a
Return to citation in text: [1] -
Fukata, Y.; Asano, K.; Matsubara, S. Chem. Lett. 2013, 42, 355. doi:10.1246/cl.121245
Return to citation in text: [1] -
Fukata, Y.; Miyaji, R.; Okamura, T.; Asano, K.; Matsubara, S. Synthesis 2013, 45, 1627. doi:10.1055/s-0032-1316920
Return to citation in text: [1] -
Miyaji, R.; Asano, K.; Matsubara, S. Org. Lett. 2013, 15, 3658. doi:10.1021/ol401538b
Return to citation in text: [1] -
Fukata, Y.; Asano, K.; Matsubara, S. J. Am. Chem. Soc. 2013, 135, 12160. doi:10.1021/ja407027e
Return to citation in text: [1] -
Miyaji, R.; Asano, K.; Matsubara, S. Org. Biomol. Chem. 2014, 12, 119. doi:10.1039/c3ob41938j
Return to citation in text: [1] -
Yoneda, N.; Hotta, A.; Asano, K.; Matsubara, S. Org. Lett. 2014, 16, 6264. doi:10.1021/ol503104b
Return to citation in text: [1] -
Yoneda, N.; Fukata, Y.; Asano, K.; Matsubara, S. Angew. Chem., Int. Ed. 2015, 54, 15497. doi:10.1002/anie.201508405
Return to citation in text: [1] -
Brandes, S.; Bella, M.; Kjærsgaard, A.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2006, 45, 1147. doi:10.1002/anie.200503042
Return to citation in text: [1] -
Brandes, S.; Niess, B.; Bella, M.; Prieto, A.; Overgaard, J.; Jørgensen, K. A. Chem. – Eur. J. 2006, 12, 6039. doi:10.1002/chem.200600495
Return to citation in text: [1] -
Li, G.-Q.; Gao, H.; Keene, C.; Devonas, M.; Ess, D. H.; Kürti, L. J. Am. Chem. Soc. 2013, 135, 7414. doi:10.1021/ja401709k
Return to citation in text: [1] -
Wang, J.-Z.; Zhou, J.; Xu, C.; Sun, H.; Kürti, L.; Xu, Q.-L. J. Am. Chem. Soc. 2016, 138, 5202. doi:10.1021/jacs.6b01458
Return to citation in text: [1] -
De, C. K.; Pesciaioli, F.; List, B. Angew. Chem., Int. Ed. 2013, 52, 9293. doi:10.1002/anie.201304039
Return to citation in text: [1] -
Chen, Y.-H.; Cheng, D.-J.; Zhang, J.; Wang, Y.; Liu, X.-Y.; Tan, B. J. Am. Chem. Soc. 2015, 137, 15062. doi:10.1021/jacs.5b10152
Return to citation in text: [1] -
Moliterno, M.; Cari, R.; Puglisi, A.; Antenucci, A.; Sperandio, C.; Moretti, E.; Di Sabato, A.; Salvio, R.; Bella, M. Angew. Chem., Int. Ed. 2016, 55, 6525. doi:10.1002/anie.201601660
Return to citation in text: [1] -
Zhang, H.-H.; Wang, C.-S.; Li, C.; Mei, G.-J.; Li, Y.; Shi, F. Angew. Chem., Int. Ed. 2017, 56, 116. doi:10.1002/anie.201608150
Return to citation in text: [1] -
Chan, V.; Kim, J. G.; Jimeno, C.; Carroll, P. J.; Walsh, P. J. Org. Lett. 2004, 6, 2051. doi:10.1021/ol0492952
Return to citation in text: [1] -
Gustafson, J. L.; Lim, D.; Miller, S. J. Science 2010, 328, 1251. doi:10.1126/science.1188403
Return to citation in text: [1] -
Pathak, T. P.; Miller, S. J. J. Am. Chem. Soc. 2012, 134, 6120. doi:10.1021/ja301566t
Return to citation in text: [1] -
Barrett, K. T.; Miller, S. J. J. Am. Chem. Soc. 2013, 135, 2963. doi:10.1021/ja400082x
Return to citation in text: [1] [2] -
Barrett, K. T.; Metrano, A. J.; Rablen, P. R.; Miller, S. J. Nature 2014, 509, 71. doi:10.1038/nature13189
Return to citation in text: [1] [2] -
Diener, M. E.; Metrano, A. J.; Kusano, S.; Miller, S. J. J. Am. Chem. Soc. 2015, 137, 12369. doi:10.1021/jacs.5b07726
Return to citation in text: [1] -
Cozzi, P. G.; Emer, E.; Gualandi, A. Angew. Chem., Int. Ed. 2011, 50, 3847. doi:10.1002/anie.201008031
Return to citation in text: [1] [2] -
Shirakawa, S.; Liu, K.; Maruoka, K. J. Am. Chem. Soc. 2012, 134, 916. doi:10.1021/ja211069f
Return to citation in text: [1] -
Liu, K.; Wu, X.; Kan, S. B. J.; Shirakawa, S.; Maruoka, K. Chem. – Asian J. 2013, 8, 3214. doi:10.1002/asia.201301036
Return to citation in text: [1] -
Nushiro, K.; Kikuchi, S.; Yamada, T. Chem. Lett. 2013, 42, 165. doi:10.1246/cl.2013.165
Return to citation in text: [1] -
Zhao, P.; Beaudry, C. M. Angew. Chem., Int. Ed. 2014, 53, 10500. doi:10.1002/anie.201406621
Return to citation in text: [1] [2] -
Miyaji, R.; Asano, K.; Matsubara, S. J. Am. Chem. Soc. 2015, 137, 6766. doi:10.1021/jacs.5b04151
Return to citation in text: [1] [2] [3] [4] -
Miyaji, R.; Asano, K.; Matsubara, S. Chem. – Eur. J. 2017, 23, 9996. doi:10.1002/chem.201701707
Return to citation in text: [1] [2] [3] [4] -
Yu, C.; Huang, H.; Li, X.; Zhang, Y.; Wang, W. J. Am. Chem. Soc. 2016, 138, 6956. doi:10.1021/jacs.6b03609
Return to citation in text: [1] [2] -
Mori, K.; Itakura, T.; Akiyama, T. Angew. Chem., Int. Ed. 2016, 55, 11642. doi:10.1002/anie.201606063
Return to citation in text: [1] -
Staniland, S.; Adams, R. W.; McDouall, J. J. W.; Maffucci, I.; Contini, A.; Grainger, D. M.; Turner, N. J.; Clayden, J. Angew. Chem., Int. Ed. 2016, 55, 10755. doi:10.1002/anie.201605486
Return to citation in text: [1] -
Jolliffe, J. D.; Armstrong, R. J.; Smith, M. D. Nat. Chem. 2017, 9, 558. doi:10.1038/nchem.2710
Return to citation in text: [1] -
Shirakawa, S.; Wu, X.; Maruoka, K. Angew. Chem., Int. Ed. 2013, 52, 14200. doi:10.1002/anie.201308237
Return to citation in text: [1] -
Shirakawa, S.; Wu, X.; Liu, S.; Maruoka, K. Tetrahedron 2016, 72, 5163. doi:10.1016/j.tet.2015.10.074
Return to citation in text: [1] -
Cheng, D.-J.; Yan, L.; Tian, S.-K.; Wu, M.-Y.; Wang, L.-X.; Fan, Z.-L.; Zheng, S.-C.; Liu, X.-Y.; Tan, B. Angew. Chem., Int. Ed. 2014, 53, 3684. doi:10.1002/anie.201310562
Return to citation in text: [1] -
Lu, S.; Poh, S. B.; Zhao, Y. Angew. Chem., Int. Ed. 2014, 53, 11041. doi:10.1002/anie.201406192
Return to citation in text: [1] -
Ma, G.; Deng, J.; Sibi, M. P. Angew. Chem., Int. Ed. 2014, 53, 11818. doi:10.1002/anie.201406684
Return to citation in text: [1] -
Wang, J.; Chen, M.-W.; Ji, Y.; Hu, S.-B.; Zhou, Y.-G. J. Am. Chem. Soc. 2016, 138, 10413. doi:10.1021/jacs.6b06009
Return to citation in text: [1] -
Matsumoto, T.; Konegawa, T.; Nakamura, T.; Suzuki, K. Synlett 2002, 122. doi:10.1055/s-2002-19349
Return to citation in text: [1] -
Okuyama, K.; Shingubara, K.; Tsujiyama, S.; Suzuki, K.; Matsumoto, T. Synlett 2009, 941. doi:10.1055/s-0028-1088215
Return to citation in text: [1] -
Mori, K.; Ichikawa, Y.; Kobayashi, M.; Shibata, Y.; Yamanaka, M.; Akiyama, T. J. Am. Chem. Soc. 2013, 135, 3964. doi:10.1021/ja311902f
Return to citation in text: [1] -
Mori, K.; Ichikawa, Y.; Kobayashi, M.; Shibata, Y.; Yamanaka, M.; Akiyama, T. Chem. Sci. 2013, 4, 4235. doi:10.1039/c3sc52142g
Return to citation in text: [1] -
Mori, K.; Kobayashi, M.; Itakura, T.; Akiyama, T. Adv. Synth. Catal. 2015, 357, 35. doi:10.1002/adsc.201400611
Return to citation in text: [1] -
Armstrong, R. J.; Smith, M. D. Angew. Chem., Int. Ed. 2014, 53, 12822. doi:10.1002/anie.201408205
Return to citation in text: [1] -
Zhang, J.-W.; Xu, J.-H.; Cheng, D.-J.; Shi, C.; Liu, X.-Y.; Tan, B. Nat. Commun. 2016, 7, No. 10677. doi:10.1038/ncomms10677
Return to citation in text: [1] -
Link, A.; Sparr, C. Angew. Chem., Int. Ed. 2014, 53, 5458. doi:10.1002/anie.201402441
Return to citation in text: [1] -
Lotter, D.; Neuburger, M.; Rickhaus, M.; Häussinger, D.; Sparr, C. Angew. Chem., Int. Ed. 2016, 55, 2920. doi:10.1002/anie.201510259
Return to citation in text: [1] -
Fäseke, V. C.; Sparr, C. Angew. Chem., Int. Ed. 2016, 55, 7261. doi:10.1002/anie.201602689
Return to citation in text: [1] -
Quinonero, O.; Jean, M.; Vanthuyne, N.; Roussel, C.; Bonne, D.; Constantieux, T.; Bressy, C.; Bugaut, X.; Rodriguez, J. Angew. Chem., Int. Ed. 2016, 55, 1401. doi:10.1002/anie.201509967
Return to citation in text: [1] [2] [3] -
Raut, V. S.; Jean, M.; Vanthuyne, N.; Roussel, C.; Constantieux, T.; Bressy, C.; Bugaut, X.; Bonne, D.; Rodriguez, J. J. Am. Chem. Soc. 2017, 139, 2140. doi:10.1021/jacs.6b11079
Return to citation in text: [1] [2] [3] -
Li, S.; Zhang, J.-W.; Li, X.-L.; Cheng, D.-J.; Tan, B. J. Am. Chem. Soc. 2016, 138, 16561. doi:10.1021/jacs.6b11435
Return to citation in text: [1] -
Zhang, L.; Zhang, J.; Ma, J.; Cheng, D.-J.; Tan, B. J. Am. Chem. Soc. 2017, 139, 1714. doi:10.1021/jacs.6b09634
Return to citation in text: [1] -
Shirakawa, S.; Liu, S.; Kaneko, S. Chem. – Asian J. 2016, 11, 330. doi:10.1002/asia.201500951
Return to citation in text: [1] -
Wencel-Delord, J.; Panossian, A.; Leroux, F. R.; Colobert, F. Chem. Soc. Rev. 2015, 44, 3418. doi:10.1039/c5cs00012b
Return to citation in text: [1] -
Barrett, K. T.; Miller, S. J. Org. Lett. 2015, 17, 580. doi:10.1021/ol503593y
Return to citation in text: [1] -
Clayden, J.; Pink, J. H. Angew. Chem., Int. Ed. 1998, 37, 1937. doi:10.1002/(SICI)1521-3773(19980803)37:13/14<1937::AID-ANIE1937>3.0.CO;2-4
Return to citation in text: [1]
| 1. | Okino, T.; Hoashi, Y.; Takemoto, Y. J. Am. Chem. Soc. 2003, 125, 12672. doi:10.1021/ja036972z |
| 2. | Okino, T.; Hoashi, Y.; Furukawa, T.; Xu, X.; Takemoto, Y. J. Am. Chem. Soc. 2005, 127, 119. doi:10.1021/ja044370p |
| 3. | Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967. doi:10.1021/ol050431s |
| 4. | Hamza, A.; Schubert, G.; Soós, T.; Pápai, I. J. Am. Chem. Soc. 2006, 128, 13151. doi:10.1021/ja063201x |
| 5. | Connon, S. J. Chem. – Eur. J. 2006, 12, 5418. doi:10.1002/chem.200501076 |
| 6. | Zhu, J.-L.; Zhang, Y.; Liu, C.; Zheng, A.-M.; Wang, W. J. Org. Chem. 2012, 77, 9813. doi:10.1021/jo302133n |
| 42. | Shirakawa, S.; Wu, X.; Maruoka, K. Angew. Chem., Int. Ed. 2013, 52, 14200. doi:10.1002/anie.201308237 |
| 43. | Shirakawa, S.; Wu, X.; Liu, S.; Maruoka, K. Tetrahedron 2016, 72, 5163. doi:10.1016/j.tet.2015.10.074 |
| 44. | Cheng, D.-J.; Yan, L.; Tian, S.-K.; Wu, M.-Y.; Wang, L.-X.; Fan, Z.-L.; Zheng, S.-C.; Liu, X.-Y.; Tan, B. Angew. Chem., Int. Ed. 2014, 53, 3684. doi:10.1002/anie.201310562 |
| 45. | Lu, S.; Poh, S. B.; Zhao, Y. Angew. Chem., Int. Ed. 2014, 53, 11041. doi:10.1002/anie.201406192 |
| 46. | Ma, G.; Deng, J.; Sibi, M. P. Angew. Chem., Int. Ed. 2014, 53, 11818. doi:10.1002/anie.201406684 |
| 47. | Wang, J.; Chen, M.-W.; Ji, Y.; Hu, S.-B.; Zhou, Y.-G. J. Am. Chem. Soc. 2016, 138, 10413. doi:10.1021/jacs.6b06009 |
| 35. | Zhao, P.; Beaudry, C. M. Angew. Chem., Int. Ed. 2014, 53, 10500. doi:10.1002/anie.201406621 |
| 36. | Miyaji, R.; Asano, K.; Matsubara, S. J. Am. Chem. Soc. 2015, 137, 6766. doi:10.1021/jacs.5b04151 |
| 37. | Miyaji, R.; Asano, K.; Matsubara, S. Chem. – Eur. J. 2017, 23, 9996. doi:10.1002/chem.201701707 |
| 38. | Yu, C.; Huang, H.; Li, X.; Zhang, Y.; Wang, W. J. Am. Chem. Soc. 2016, 138, 6956. doi:10.1021/jacs.6b03609 |
| 58. | Quinonero, O.; Jean, M.; Vanthuyne, N.; Roussel, C.; Bonne, D.; Constantieux, T.; Bressy, C.; Bugaut, X.; Rodriguez, J. Angew. Chem., Int. Ed. 2016, 55, 1401. doi:10.1002/anie.201509967 |
| 59. | Raut, V. S.; Jean, M.; Vanthuyne, N.; Roussel, C.; Constantieux, T.; Bressy, C.; Bugaut, X.; Bonne, D.; Rodriguez, J. J. Am. Chem. Soc. 2017, 139, 2140. doi:10.1021/jacs.6b11079 |
| 25. | Chan, V.; Kim, J. G.; Jimeno, C.; Carroll, P. J.; Walsh, P. J. Org. Lett. 2004, 6, 2051. doi:10.1021/ol0492952 |
| 26. | Gustafson, J. L.; Lim, D.; Miller, S. J. Science 2010, 328, 1251. doi:10.1126/science.1188403 |
| 27. | Pathak, T. P.; Miller, S. J. J. Am. Chem. Soc. 2012, 134, 6120. doi:10.1021/ja301566t |
| 28. | Barrett, K. T.; Miller, S. J. J. Am. Chem. Soc. 2013, 135, 2963. doi:10.1021/ja400082x |
| 29. | Barrett, K. T.; Metrano, A. J.; Rablen, P. R.; Miller, S. J. Nature 2014, 509, 71. doi:10.1038/nature13189 |
| 30. | Diener, M. E.; Metrano, A. J.; Kusano, S.; Miller, S. J. J. Am. Chem. Soc. 2015, 137, 12369. doi:10.1021/jacs.5b07726 |
| 31. | Cozzi, P. G.; Emer, E.; Gualandi, A. Angew. Chem., Int. Ed. 2011, 50, 3847. doi:10.1002/anie.201008031 |
| 32. | Shirakawa, S.; Liu, K.; Maruoka, K. J. Am. Chem. Soc. 2012, 134, 916. doi:10.1021/ja211069f |
| 33. | Liu, K.; Wu, X.; Kan, S. B. J.; Shirakawa, S.; Maruoka, K. Chem. – Asian J. 2013, 8, 3214. doi:10.1002/asia.201301036 |
| 34. | Nushiro, K.; Kikuchi, S.; Yamada, T. Chem. Lett. 2013, 42, 165. doi:10.1246/cl.2013.165 |
| 35. | Zhao, P.; Beaudry, C. M. Angew. Chem., Int. Ed. 2014, 53, 10500. doi:10.1002/anie.201406621 |
| 36. | Miyaji, R.; Asano, K.; Matsubara, S. J. Am. Chem. Soc. 2015, 137, 6766. doi:10.1021/jacs.5b04151 |
| 37. | Miyaji, R.; Asano, K.; Matsubara, S. Chem. – Eur. J. 2017, 23, 9996. doi:10.1002/chem.201701707 |
| 38. | Yu, C.; Huang, H.; Li, X.; Zhang, Y.; Wang, W. J. Am. Chem. Soc. 2016, 138, 6956. doi:10.1021/jacs.6b03609 |
| 39. | Mori, K.; Itakura, T.; Akiyama, T. Angew. Chem., Int. Ed. 2016, 55, 11642. doi:10.1002/anie.201606063 |
| 40. | Staniland, S.; Adams, R. W.; McDouall, J. J. W.; Maffucci, I.; Contini, A.; Grainger, D. M.; Turner, N. J.; Clayden, J. Angew. Chem., Int. Ed. 2016, 55, 10755. doi:10.1002/anie.201605486 |
| 41. | Jolliffe, J. D.; Armstrong, R. J.; Smith, M. D. Nat. Chem. 2017, 9, 558. doi:10.1038/nchem.2710 |
| 17. | Brandes, S.; Bella, M.; Kjærsgaard, A.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2006, 45, 1147. doi:10.1002/anie.200503042 |
| 18. | Brandes, S.; Niess, B.; Bella, M.; Prieto, A.; Overgaard, J.; Jørgensen, K. A. Chem. – Eur. J. 2006, 12, 6039. doi:10.1002/chem.200600495 |
| 19. | Li, G.-Q.; Gao, H.; Keene, C.; Devonas, M.; Ess, D. H.; Kürti, L. J. Am. Chem. Soc. 2013, 135, 7414. doi:10.1021/ja401709k |
| 20. | Wang, J.-Z.; Zhou, J.; Xu, C.; Sun, H.; Kürti, L.; Xu, Q.-L. J. Am. Chem. Soc. 2016, 138, 5202. doi:10.1021/jacs.6b01458 |
| 21. | De, C. K.; Pesciaioli, F.; List, B. Angew. Chem., Int. Ed. 2013, 52, 9293. doi:10.1002/anie.201304039 |
| 22. | Chen, Y.-H.; Cheng, D.-J.; Zhang, J.; Wang, Y.; Liu, X.-Y.; Tan, B. J. Am. Chem. Soc. 2015, 137, 15062. doi:10.1021/jacs.5b10152 |
| 23. | Moliterno, M.; Cari, R.; Puglisi, A.; Antenucci, A.; Sperandio, C.; Moretti, E.; Di Sabato, A.; Salvio, R.; Bella, M. Angew. Chem., Int. Ed. 2016, 55, 6525. doi:10.1002/anie.201601660 |
| 24. | Zhang, H.-H.; Wang, C.-S.; Li, C.; Mei, G.-J.; Li, Y.; Shi, F. Angew. Chem., Int. Ed. 2017, 56, 116. doi:10.1002/anie.201608150 |
| 64. | Barrett, K. T.; Miller, S. J. Org. Lett. 2015, 17, 580. doi:10.1021/ol503593y |
| 7. | Asano, K.; Matsubara, S. J. Am. Chem. Soc. 2011, 133, 16711. doi:10.1021/ja207322d |
| 8. | Asano, K.; Matsubara, S. Org. Lett. 2012, 14, 1620. doi:10.1021/ol3003755 |
| 9. | Okamura, T.; Asano, K.; Matsubara, S. Chem. Commun. 2012, 48, 5076. doi:10.1039/c2cc31602a |
| 10. | Fukata, Y.; Asano, K.; Matsubara, S. Chem. Lett. 2013, 42, 355. doi:10.1246/cl.121245 |
| 11. | Fukata, Y.; Miyaji, R.; Okamura, T.; Asano, K.; Matsubara, S. Synthesis 2013, 45, 1627. doi:10.1055/s-0032-1316920 |
| 12. | Miyaji, R.; Asano, K.; Matsubara, S. Org. Lett. 2013, 15, 3658. doi:10.1021/ol401538b |
| 13. | Fukata, Y.; Asano, K.; Matsubara, S. J. Am. Chem. Soc. 2013, 135, 12160. doi:10.1021/ja407027e |
| 14. | Miyaji, R.; Asano, K.; Matsubara, S. Org. Biomol. Chem. 2014, 12, 119. doi:10.1039/c3ob41938j |
| 15. | Yoneda, N.; Hotta, A.; Asano, K.; Matsubara, S. Org. Lett. 2014, 16, 6264. doi:10.1021/ol503104b |
| 16. | Yoneda, N.; Fukata, Y.; Asano, K.; Matsubara, S. Angew. Chem., Int. Ed. 2015, 54, 15497. doi:10.1002/anie.201508405 |
| 65. | Clayden, J.; Pink, J. H. Angew. Chem., Int. Ed. 1998, 37, 1937. doi:10.1002/(SICI)1521-3773(19980803)37:13/14<1937::AID-ANIE1937>3.0.CO;2-4 |
| 31. | Cozzi, P. G.; Emer, E.; Gualandi, A. Angew. Chem., Int. Ed. 2011, 50, 3847. doi:10.1002/anie.201008031 |
| 62. | Shirakawa, S.; Liu, S.; Kaneko, S. Chem. – Asian J. 2016, 11, 330. doi:10.1002/asia.201500951 |
| 63. | Wencel-Delord, J.; Panossian, A.; Leroux, F. R.; Colobert, F. Chem. Soc. Rev. 2015, 44, 3418. doi:10.1039/c5cs00012b |
| 28. | Barrett, K. T.; Miller, S. J. J. Am. Chem. Soc. 2013, 135, 2963. doi:10.1021/ja400082x |
| 29. | Barrett, K. T.; Metrano, A. J.; Rablen, P. R.; Miller, S. J. Nature 2014, 509, 71. doi:10.1038/nature13189 |
| 58. | Quinonero, O.; Jean, M.; Vanthuyne, N.; Roussel, C.; Bonne, D.; Constantieux, T.; Bressy, C.; Bugaut, X.; Rodriguez, J. Angew. Chem., Int. Ed. 2016, 55, 1401. doi:10.1002/anie.201509967 |
| 59. | Raut, V. S.; Jean, M.; Vanthuyne, N.; Roussel, C.; Constantieux, T.; Bressy, C.; Bugaut, X.; Bonne, D.; Rodriguez, J. J. Am. Chem. Soc. 2017, 139, 2140. doi:10.1021/jacs.6b11079 |
| 36. | Miyaji, R.; Asano, K.; Matsubara, S. J. Am. Chem. Soc. 2015, 137, 6766. doi:10.1021/jacs.5b04151 |
| 37. | Miyaji, R.; Asano, K.; Matsubara, S. Chem. – Eur. J. 2017, 23, 9996. doi:10.1002/chem.201701707 |
| 55. | Link, A.; Sparr, C. Angew. Chem., Int. Ed. 2014, 53, 5458. doi:10.1002/anie.201402441 |
| 56. | Lotter, D.; Neuburger, M.; Rickhaus, M.; Häussinger, D.; Sparr, C. Angew. Chem., Int. Ed. 2016, 55, 2920. doi:10.1002/anie.201510259 |
| 57. | Fäseke, V. C.; Sparr, C. Angew. Chem., Int. Ed. 2016, 55, 7261. doi:10.1002/anie.201602689 |
| 58. | Quinonero, O.; Jean, M.; Vanthuyne, N.; Roussel, C.; Bonne, D.; Constantieux, T.; Bressy, C.; Bugaut, X.; Rodriguez, J. Angew. Chem., Int. Ed. 2016, 55, 1401. doi:10.1002/anie.201509967 |
| 59. | Raut, V. S.; Jean, M.; Vanthuyne, N.; Roussel, C.; Constantieux, T.; Bressy, C.; Bugaut, X.; Bonne, D.; Rodriguez, J. J. Am. Chem. Soc. 2017, 139, 2140. doi:10.1021/jacs.6b11079 |
| 60. | Li, S.; Zhang, J.-W.; Li, X.-L.; Cheng, D.-J.; Tan, B. J. Am. Chem. Soc. 2016, 138, 16561. doi:10.1021/jacs.6b11435 |
| 61. | Zhang, L.; Zhang, J.; Ma, J.; Cheng, D.-J.; Tan, B. J. Am. Chem. Soc. 2017, 139, 1714. doi:10.1021/jacs.6b09634 |
| 48. | Matsumoto, T.; Konegawa, T.; Nakamura, T.; Suzuki, K. Synlett 2002, 122. doi:10.1055/s-2002-19349 |
| 49. | Okuyama, K.; Shingubara, K.; Tsujiyama, S.; Suzuki, K.; Matsumoto, T. Synlett 2009, 941. doi:10.1055/s-0028-1088215 |
| 50. | Mori, K.; Ichikawa, Y.; Kobayashi, M.; Shibata, Y.; Yamanaka, M.; Akiyama, T. J. Am. Chem. Soc. 2013, 135, 3964. doi:10.1021/ja311902f |
| 51. | Mori, K.; Ichikawa, Y.; Kobayashi, M.; Shibata, Y.; Yamanaka, M.; Akiyama, T. Chem. Sci. 2013, 4, 4235. doi:10.1039/c3sc52142g |
| 52. | Mori, K.; Kobayashi, M.; Itakura, T.; Akiyama, T. Adv. Synth. Catal. 2015, 357, 35. doi:10.1002/adsc.201400611 |
| 53. | Armstrong, R. J.; Smith, M. D. Angew. Chem., Int. Ed. 2014, 53, 12822. doi:10.1002/anie.201408205 |
| 54. | Zhang, J.-W.; Xu, J.-H.; Cheng, D.-J.; Shi, C.; Liu, X.-Y.; Tan, B. Nat. Commun. 2016, 7, No. 10677. doi:10.1038/ncomms10677 |
| 36. | Miyaji, R.; Asano, K.; Matsubara, S. J. Am. Chem. Soc. 2015, 137, 6766. doi:10.1021/jacs.5b04151 |
| 37. | Miyaji, R.; Asano, K.; Matsubara, S. Chem. – Eur. J. 2017, 23, 9996. doi:10.1002/chem.201701707 |
© 2017 Miyaji et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)