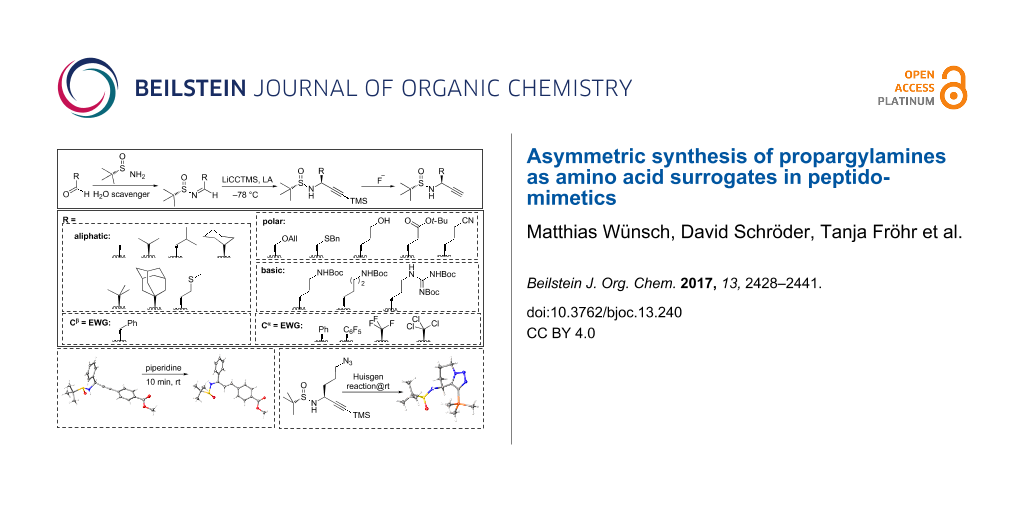
Abstract
The amide moiety of peptides can be replaced for example by a triazole moiety, which is considered to be bioisosteric. Therefore, the carbonyl moiety of an amino acid has to be replaced by an alkyne in order to provide a precursor of such peptidomimetics. As most amino acids have a chiral center at Cα, such amide bond surrogates need a chiral moiety. Here the asymmetric synthesis of a set of 24 N-sulfinyl propargylamines is presented. The condensation of various aldehydes with Ellman’s chiral sulfinamide provides chiral N-sulfinylimines, which were reacted with (trimethylsilyl)ethynyllithium to afford diastereomerically pure N-sulfinyl propargylamines. Diverse functional groups present in the propargylic position resemble the side chain present at the Cα of amino acids. Whereas propargylamines with (cyclo)alkyl substituents can be prepared in a direct manner, residues with polar functional groups require suitable protective groups. The presence of particular functional groups in the side chain in some cases leads to remarkable side reactions of the alkyne moiety. Thus, electron-withdrawing substituents in the Cα-position facilitate a base induced rearrangement to α,β-unsaturated imines, while azide-substituted propargylamines form triazoles under surprisingly mild conditions. A panel of propargylamines bearing fluoro or chloro substituents, polar functional groups, or basic and acidic functional groups is accessible for the use as precursors of peptidomimetics.

Graphical Abstract
Introduction
Terminal alkynes display an intriguing versatility as building blocks in organic and medicinal chemistry, as their reactivity is unique. Their chemistry involves several highly selective reactions, e.g., [3 + 2] cycloadditions with azides and isoelectronic functional groups (among them the copper or ruthenium-catalyzed azide–alkyne cycloaddition, CuAAC and RuAAC), the thiol–yne reaction, Diels–Alder reactions and the Sonogashira cross-coupling. While amino acids with a terminal alkyne in the side chain are well-known, the synthesis of their correlates where the carboxy group is replaced by a terminal alkyne is still tedious. Nevertheless, these propargylamines have been frequently used as precursors for the synthesis of diverse bioactive compounds. Their conversion into triazoles is best investigated, since triazoles as amide bond surrogates are found in several inhibitors of proteases such as cathepsin S [1-6], cysteine proteases [7], cruzain 20 [8,9], caspases [10] and peptidyl aminopeptidases [11]. These protease inhibitors show potential for the treatment of Chagas disease [2,9], Huntington’s disease [10], malaria [11], autoimmune diseases [6] and the imaging of tumor associated macrophages [2]. Whereas the carboxylic acid function of amino acids can be easily converted into amides or esters (Figure 1), propargylamines have been converted into acids, alcohols [12] or olefins in order to obtain natural products like angustureine and cuspareine [13].
![[1860-5397-13-240-1]](/bjoc/content/figures/1860-5397-13-240-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Concept of carboxylic acid or amide bond replacement on the basis of an alkyne moiety.
Figure 1: Concept of carboxylic acid or amide bond replacement on the basis of an alkyne moiety.
Intramolecular Pauson–Khand reaction [14], Diels–Alder reaction [15], gold-catalyzed azetidin-3-one formation [16], as well as various transition metal-mediated additions and cross-coupling reactions [17] represent further important reactions of propargylamines, providing the potential to form innovative peptidomimetics (Figure 2).
![[1860-5397-13-240-2]](/bjoc/content/figures/1860-5397-13-240-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Selection of reactions based on propargylamines as precursors. a) Intramolecular Pauson–Khand reaction, R = (S)-tert-butylsulfinyl, R’ = CH2CH2OTBDPS [14]. b) Diels–Alder reaction, R = pTs, R’ = H, R’’ = Me [15]. c) Gold-catalyzed intramolecular reaction to azetidin-3-ones, R = tert-butylsulfonyl, R’ = aromatics, aliphatics [16]. d) Sonogashira cross-coupling, R = tert-butylsulfinyl, R’ = Me, CHMe2, CH2CHMe2, cyclohexyl [17]. e,f) CuI or RuII-catalyzed [3 + 2] cycloadditions (CuAAC, RuAAC) [18-21]. e) R = Boc, R’ = Bn [19]. f) R = Boc, R’ = Me, CHMe2, CH2CHMe2, R’’ = Me, CHMe2, CH2Ph [21].
Figure 2: Selection of reactions based on propargylamines as precursors. a) Intramolecular Pauson–Khand react...
Our attention has been focused on the synthesis of amino acid analogous propargylamines, furnished with Cα-substituents imitating various amino acid side chains.
The direct conversion of amino acids into propargylamines by the Corey–Fuchs or the Seyferth–Gilbert homologation has been successfully used for the preparation of several natural amino acid analogues [22,23]. However, epimerization in the α-position frequently occurs under the alkaline reaction conditions of the Seyferth–Gilbert and the Corey–Fuchs reaction. In order to access propargylamines with modified side chains, we chose a de novo synthesis strategy, using Ellman’s chiral sulfinamide auxiliary to produce diastereomerically pure amines [7]. Ellman’s chiral sulfinamide can be readily synthesized on a laboratory scale [24]. Moreover, sulfinamides can be cleaved easily under acidic conditions [1,25,26] and the produced sulfinic acid can even be recycled [25,27]. However, the sulfinamide auxiliary has to be treated with care, as it tends to disproportionate quickly in solution at elevated temperature or in chloroform at room temperature [28]. Furthermore, sulfinamides are unstable upon sonication [28]. Sulfinamides are also reported to decompose in the presence of low concentrations of acid under pressure, typical conditions of HPLC analysis. Consequently, the application of the tert-butylsulfinyl as protective group for the amine is restricted to very mild conditions. However, it can be easily cleaved from N-sulfinyl propargylamines by acidic methanolysis and subsequent Boc protection [21]. Here we report on the diastereoselective synthesis of chiral N-sulfinyl propargylamines with amino acid type “side chains” attached to the propargylic position mediated by Ellman’s auxiliary.
Enantiomerically pure amines can be obtained by condensation of aldehydes or ketones with Ellman’s chiral sulfinamide, mediated by KHSO4 [29], Cs2CO3 [30], Ti(OEt)4 [31-33], or CuSO4 [34], followed by either reduction [35-40] or addition of a nucleophile to the imine [41]. In general, there are two options (Figure 3) for the synthesis of enantiomerically pure propargylamines by nucleophilic addition. The synthesis of propargylamines by diastereoselective reductive amination requires alkynyl ketones, which are difficult to prepare and are unstable towards reductive conditions.
![[1860-5397-13-240-3]](/bjoc/content/figures/1860-5397-13-240-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Two different approaches for the stereoselective de novo synthesis of propargylamines using Ellman’s chiral sulfinamide. (a) tert-Butyl sulfinamide, Lewis acid, CH2Cl2. (b) Organometallic compound. (c) (Trimethylsilyl)ethynyllithium.
Figure 3: Two different approaches for the stereoselective de novo synthesis of propargylamines using Ellman’...
In approach I, organometallic nucleophiles are added to N-sulfinyl propargylimines, derived from aldehydes. According to approach II, a metallated terminal alkyne is added to an N-sulfinylaldimine. In approach I, the organometallic nucleophile is transferring the amino acid side chain, in approach II, the amino acid side chain comes from the aldehyde incorporated in the imine.
Results and Discussion
Synthesis of propargylamines, general strategies
To avoid side reactions of the terminal alkyne in approach I, internal alkynes were applied. Benzoate substituents were chosen as they are comparatively inert and convertible to peptidomimetics. At first, iodobenzene derivatives with an ester moiety in p- or m-position were reacted with propargyl alcohol in a Sonogashira reaction to give the phenylpropargyl alcohols 1a and 1b (Figure 4) [42,43], which were transformed in a Swern oxidation to afford the aldehydes 2a and 2b [44]. Condensation of the aldehydes 2a and 2b with (R)-configured tert-butyl sulfinamide led to the enantiomerically pure sulfinylimines 3a and 3b, which were reacted with a variety of organometallic compounds, including iPrMgBr, MeMgBr and BnMgBr.
![[1860-5397-13-240-4]](/bjoc/content/figures/1860-5397-13-240-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Synthesis of propargylamines 4a and 4b by introducing the side chain as nucleophile. (a) HC≡CCH2OH, Cl2Pd(PPh3)2 (1 mol %), CuI (2 mol %), THF/piperidine (3:1), rt, 2 h. (b) (COCl)2, DMSO, NEt3, −78 °C, DCM. (c) (R)-tert-Butyl sulfinamide ((R)-1), CuSO4, DCM, rt, 3 d (see GP-2). (d) iPrMgBr, THF, −38 °C, AlMe3 in n-hexane (4a, 13%, dr 99:1). (e) MeLi in Et2O, toluene, −30 °C, AlMe3 in n-hexane (4b, 4%, dr 52:48). (f) MeMgBr in Et2O, toluene, −35 °C (4b, 10%, dr 51:49).
Figure 4: Synthesis of propargylamines 4a and 4b by introducing the side chain as nucleophile. (a) HC≡CCH2OH,...
The reaction of the enantiomerically pure N-sulfinylimines 3a and 3b with aliphatic organometallic nucleophiles resulted in low yields and diastereoselectivity. The reaction of sulfinylimine 3a with isopropylmagnesium bromide provided the sulfinamide 4a in only 13% yield with a ratio of diastereomers of 99:1. Only 10% of the addition product 4b were obtained by addition of methylmagnesium bromide to the N-sulfinylimine 4b (dr 51:49). In order to prepare phenylalanine analogoues, N-sulfinylimines 3a and 3b were reacted with benzylmagnesium bromide. However, only traces of the desired addition products could be detected by LC–MS analysis of the crude products.
Approach II starts with the condensation of an aldehyde with Ellman’s chiral sulfinamide to provide a sulfinylimine [29-34], which was reacted with (trimethylsilyl)ethynyllithium [1,14,45-48]. The terminal TMS group was cleaved off after the addition reaction [49,50] (Table 1).
Table 1: Preparation of N-sulfinyl propargylamines 7 with aliphatic side chains.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-240-i1.svg?max-width=637&scale=1.0)
|
|||||
| R | (a) | Yield (5) | (b), (c) | Yield (7)b | dr |
| Me | GP-1c | 81% (5a) | GP-3, GP-5 | 47% (7a) | 100:0 |
| iPr | GP-1 | 90% (5b) | GP-3, GP-5 | 61% (7b) | 97:3 |
| CH2-iPr | GP-1 | 92% (5c) | GP-3, GP-5 | 53% (7c) | 97:3 |
| C6H11 | GP-2 | 96% (5d) | GP-3, GP-5 | 59% (7d) | 97:3 |
| t-Bu | GP-2 | 18% (5e) | GP-4, GP-5 | 41% (7e) | 80:20 |
| adamantyl | GP-2 | 42% (5f) | GP-4, GP-5 | 15% (7f) | 100:0 |
| CH2CH2SMe | GP-2c | 89% (5g) | GP-4, GP-6 | 34% (7g)d | 96:4 |
a(a) GP-1: Auxiliary (S)-1, aldehyde, Ti(OiPr)4, 70 °C, 40 min. GP-2: Auxiliary (S)-1, aldehyde, CuSO4, DCM, rt, 3 d. (b) GP-3: (Trimethylsilyl)ethynyllithium, Ti(OiPr)4, THF, −78 °C to rt. GP-4: (Trimethylsilyl)ethynyllithium, AlMe3, toluene, −78 °C to rt. (c) GP-5: TBAF, THF, 0 °C, 3 h. GP-6: KF, 18-crown-6, THF/H2O (98:2), 0 °C. bIsolated yields are given, referring to 5 (over two steps (b) and (c)). cGP-1 had to be modified for the synthesis of 5a: acetaldehyde (5 equiv), MgSO4 (5 equiv), 30 °C, 12 h [51]. d(R)-configured Ellman’s sulfinamide (R)-1 was applied. Hence, the mirror images of 5–7g were obtained.
Several conditions for the condensation of aldehydes with Ellman’s chiral sulfinamide (S)-1 have been described. Catalysis by Brønstedt acids has been reported [29], but was not considered here due to the lability of the tert-butyl sulfinamide moiety towards acids [1,25,26]. Strongly alkaline conditions [30] were also avoided. Liquid aldehydes were readily condensed with tert-butyl sulfinamide (S)-1 in the presence of Ti(OEt)4 as Lewis acid and water scavenger [31-33]. However, the removal of precipitated TiO2 was tedious and time consuming. Therefore, dry CuSO4 as Lewis acid and water scavenger at ambient temperature [34] represented a versatile method, leading to high yields of sulfinylimines 5. (Trimethylsilyl)ethynylmagnesium bromide has already been successfully added to sulfinylimines to produce various N-sulfinyl propargylamines in excellent yields and diastereomeric excesses [13,16]. Several N-sulfinyl propargylamines (analogoues of valine, phenylglycine and tyrosine) have been prepared using a large excess (4 equivalents) of [(trimethylsilyl)ethynyl]dimethylaluminum as the nucleophile [14,48,52]. The reaction of the sulfinylimines 5 with (trimethylsilyl)ethynyllithium provided the crude intermediates 6, which were converted into the N-sulfinyl propargylamines 7 in high yields and satisfactory diastereomeric ratios by cleaving off the TMS protecting group with TBAF. In previous investigations, the choice of solvent and Lewis acid were controversially discussed [13,14,45-50,52-55]. Nonpolar solvents (DCM < THF < Et2O < toluene) have been described to enhance the stability of the transition state, improving the diastereomeric ratio, as well as the reactivity of the nucleophile, resulting in an improved yield [45]. As aldimines have been reported to be rather unreactive electrophiles [53], hard Lewis acids (AlMe3 > AlR3 > Ti(OiPr)4 [14] > BF3 > MgBr2 > ZnCl2 > ZnEt2) have been recommended for activation [45]. However, the Lewis acid BF3 was shown to give products with inverted configuration of the newly formed chiral center [47]. While several authors obtained increased yields upon addition of AlMe3 [1,45,46], others advised against its use as activation agent of aldimines, because side reactions were observed [13,54,55], which will also be further discussed below. We used THF or toluene as solvents and AlMe3 or Ti(OiPr)4 as activation agent. According to Ellman et al., the diastereoselectivity of the addition to sulfinylimines is controlled by the formation of a cyclic, six-membered, chair-like transition state, which is formed by precoordination of the organometallic reagent to aldimine 5 [45]. This cyclic transition state accounts for the preferred re-face attack of the nucleophile at (S)-configured N-sulfinylimines, which leads to amines with the same configuration as the proteinogenic (S)-configured amino acids. This stereoselection was confirmed by various X-ray crystal structures of propargylamines obtained during our investigations like 7a, 7c–e, 7s, 7i–k, 7q and triazole 13w (see Supporting Information File 1). Independent on the substituent, solvent and Lewis acid used, the direction of the nucleophilic attack of (trimethylsilyl)ethynyllithium was always the same, forming the new centers of chirality of all N-sulfinyl propargylamines 7 configured as expected. As already described by Ellman et al. [45], the size of the side chain correlates with the result of the reaction. The diastereomeric excesses increased in a size-dependent manner in the order 6a (alanine) < 6c (leucine) < 6d (cyclohexylglycine) (compare Table 1). The yield of the tert-leucine derivative 6e was reduced due to sterically shielding of the imino moiety by the tert-butyl group.
The cleavage of the TMS groups of the addition products 6 could be accomplished with TBAF in THF [49,50,56]. Basic conditions, like K2CO3 as described in the literature [50,57] did not lead to the desired N-sulfinyl propargylamines 7. We assume that strong bases do not only lead to desilylation, but also induce decomposition of the propargylamine system (see below). Kracker et al. recently demonstrated the substitution of the labile tert-butylsulfinyl group of compounds 7a–c by the more versatile Boc protective group in yields of 56–94% [21].
Synthesis of propargylamines containing electron-withdrawing substituents
Aromatic and carbonyl substituents in the Cβ-position of propargylamines (occurring in analogoues of the amino acids phenylalanine, tyrosine, histidine, tryptophan, aspartic acid and asparagine) increase the acidity of the adjacent protons considerably. In the precursor sulfinylimines 5 of the target propargylamines the acidity of the protons of the methylene moiety is further increased by the electron-withdrawing effect of the adjacent sulfinyl imino moiety. Thus, the nucleophilic addition of (trimethylsilyl)ethynyllithium is competing with deprotonation of the methylene moiety giving rise to the formation of the azaenolate (Figure 5). The resulting anion is reprotonated during aqueous work-up, leading to the starting sulfinylimine 5.
![[1860-5397-13-240-5]](/bjoc/content/figures/1860-5397-13-240-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Reaction of N-sulfinylimine 5h with (trimethylsilyl)ethynyllithium. (a) GP-3 or GP-4. (b) Aqueous work-up, H2O/H+. Deprotonation in benzylic position competes with nucleophilic attack (5h/6h, 7:3).
Figure 5: Reaction of N-sulfinylimine 5h with (trimethylsilyl)ethynyllithium. (a) GP-3 or GP-4. (b) Aqueous w...
Benzyl-substituted N-sulfinyl propargylamine 6h was prepared by the addition of (trimethylsilyl)ethynyllithium to N-sulfinylimine 5h. The starting material 5h and the addition product 6h were isolated in a 7:3 ratio indicating deprotonation to be the predominant reaction. The phenylalanine analogous N-sulfinyl propargylamine 7h was isolated in only 12% yield (over two steps, referring to imine 5h) after desilylation with TBAF (Table 2). As the benzylic proton of sulfinylimine 5h is quite acidic, approach II was not pursued for the synthesis of propargylamines analogous to tyrosine, histidine, tryptophan, and aspartate.
Table 2: Synthesis of propargylamines 7 with electron-withdrawing substituents in the side chain.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-240-i2.svg?max-width=637&scale=1.0)
|
|||||||
| R | (a) | Yield (5) | (b) | Yield (6) | (c) | Yield (7) | dr |
| CH2Ph | GP-2 | 64% (5h) | GP-4 | 34% (6h) | GP-5 | 35% (7h) | 97:3 |
| Ph | GP-1 | 99% (5i) | GP-4 | 54% (6i) | GP-7 | 66% (7i) | 95:5 |
| C6F5 | GP-2 | 88% (5j) | GP-4 | 41% (6j) | GP-6 | 52% (7j) | 100:0 |
| CF3 | GP-2b | n.i. (5k) | GP-4 | 33%c (6k) | GP-6 | 29% (7k) | 93:7 |
| CCl3 | GP-2b | 87% (5l) | GP-4 | 10% (6l) | GP-6 | 57% (7l) | 100:0 |
a(a) GP-1: Auxiliary (S)-1, aldehyde, Ti(OiPr)4, 70 °C, 40 min. GP-2: Auxiliary (S)-1, aldehyde, CuSO4, DCM, rt, 3 d. (b) GP-4: (Trimethylsilyl)ethynyllithium, AlMe3, toluene, −78 °C to rt. (c) GP-5: TBAF, THF, 0 °C, 3 h. GP-6: KF, 18-crown-6, THF/H2O (98:2), 0 °C. GP-7: 1. AgNO3, EtOH, 0 °C; 2. KCN, EtOH, HCl. bFor the synthesis of 5k, procedure GP-2 was modified: The aldehyde was distilled under argon atmosphere, condensed onto a mixture of sulfinamide (S)-1 and molecular sieves 4 Å. Toluene was added and the solution was stirred for 48 h. 5k was directly applied for subsequent reactions and could not be isolated (n.i.). cYield refers to Ellman’s chiral sulfinamide (S)-1.
Proteinogenic amino acids do not contain substituents, which additionally increase the acidity of the α-proton. Nevertheless, glycine derivatives with electron-withdrawing substituents like formylglycine [58,59], phenylglycine [60,61] and fluorinated alanine [62-64] have attracted great attention as peptidomimetics, drugs or in the bioorthogonal functionalization of larger peptides.
The trifluoromethyl moiety has been reported to enhance the activity, stability and selectivity of various pharmacologically active compounds [65,66], e.g., trifluridine [67-70], efavirenz [71-73], fluoxetine [74-76] and fluozolate [77]. As the general structure of fluorinated propargylamines occurs in important drugs, like HIV protease inhibitor DPC 961 and its analogues [78-83], particular effort was put on the synthesis of N-sulfinyl propargylamine 7k. Because of the poor electrophilicity of imines 5, the nucleophilic addition of trifluoromethyl nucleophiles, such as TMS–CF3 [84,85] has been described to be inefficient. Still, the asymmetric synthesis of CF3-substituted propargylamines has been described, using (R)-2-methoxy-1-phenylethan-1-amine as chiral auxiliary [86-88]. However, cleavage of the protective group requires reductive conditions, which also affects the CF3 group and the alkyne [86-88].
Various trifluoromethyl-substituted ketones have been converted into N-sulfinylimines, which were subsequently transformed into sulfinamides attached to a tertiary C-atom by addition of nucleophiles [89-92]. N-Sulfinyl propargylamine 7k was prepared, following approach II under varying conditions (Table 2). Fluoral hydrate was dehydrated with concentrated sulfuric acid to give trifluoroacetaldehyde by distillation [93].
The reaction of trifluoroacetaldehyde with tert-butyl sulfinamide (S)-1 in the presence of molecular sieves 4 Å provided the sulfinylimine 5k, which could not be isolated due to its extremely high electrophilicity. Instead, immediate hydrolysis occurs upon contact with water forming hemiaminal 8k (Figure 6), as already observed by Truong et al. [94]. This frequently observed hemiaminal was isolated by column chromatography and recrystallized from EtOAc.
![[1860-5397-13-240-6]](/bjoc/content/figures/1860-5397-13-240-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Side reactions observed in the course of the conversion of highly electrophilic sulfinylimines 5. (a) Sulfinamide (S)-1, molecular sieves 4 Å, toluene. (b) 1. Dilution with H2O, 2. Extraction with DCM. (c) Addition of Ti(OiPr)4 prior to the conversion with (trimethylslyl)ethynyllithium [94]. (d) Addition of AlMe3 prior to the conversion with (trimethylsilyl)ethynyllithium.
Figure 6: Side reactions observed in the course of the conversion of highly electrophilic sulfinylimines 5. (...
Distillation of imine 5k has been reported to provide a very low yield (22% [94]). The reaction of hemiaminal 8k with (trimethylsilyl)ethynyllithium in the presence of the strong Lewis acid AlMe3 has been proposed by Truong et al. [94], but was reported to give very poor yields and low diastereoselectivity. In contrast to the argumentation of Xiao et al., who strongly recommended hard Lewis acids for the reaction of sulfinylimines with various ethynyllithium reagents [95], the crude imine 5k predominantly reacted with the Lewis acids Ti(OiPr)4 and AlMe3 and gave only low yields of the desired N-sulfinyl propargylamine 6k. The reaction of crude 5k in the presence of the Lewis acid Ti(OiPr)4 afforded the N/O-acetal 9k, whereas the attempt of activation with AlMe3 provided the methylated sulfinamide 10k. Both side products were isolated as colorless crystalline solids. It is assumed, that the undesired side products 9k and 10k were formed by a ligand transfer from the Lewis acids to imine 5k. Nucleophilic substitution of N/O-acetal 9k with two equivalents of (trimethylsilyl)ethynyllithium, in analogy to the conversions reported by Kuduk et al. [96], remained unsuccessful. The (S)-configuration of the newly generated chiral center of amine 10k was determined by X-ray structure analysis (Figure 7), suggesting two possible transition states for the ligand transfer.
![[1860-5397-13-240-7]](/bjoc/content/figures/1860-5397-13-240-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: a) Possible transition states TI and TII for the transfer of the methyl moiety from AlMe3 to the imino moiety. b) X-ray crystal structure analysis of the methyl transfer product 10k.
Figure 7: a) Possible transition states TI and TII for the transfer of the methyl moiety from AlMe3 to the im...
Whereas transition state TII requires only one equivalent of AlMe3, two equivalents of AlMe3 are involved in transition state TI. As in this case almost quantitative conversion of AlMe3 was observed, transition state TII seems to be more probable. Still, more complex coordination geometry (hexagonal or dinuclear complexes) of the Lewis acid are probable as well and hardly depend on the stoichiometry. In the conversion of highly reactive sulfinylimines like 5k, best results were obtained in the absence of a Lewis acid. Analogous investigations with chloral hydrate instead of fluoral hydrate showed that the trichloromethylimine 5l also forms a hemiaminal 8l upon contact with water, but does not undergo comparable side reactions with Ti(OiPr)4 or AlMe3. Sulfinylimine 5l appears to be a weaker electrophile, which is attributed to the lower electronegativity of Cl compared to F and to the larger size of the CCl3 group compared to the CF3 moiety, sterically shielding imine 5 against nucleophilic attack.
Desilylation of N-sulfinyl propargylamine 6i with TBAF (GP-5) resulted in decomposition instead of formation of the free alkyne 7i. In order to obtain the free terminal alkyne 7i, a milder desilylation procedure was required. Therefore, AgNO3 and KCN (GP-7) were used for the desilylation of 6i to afford the free alkyne 7i in 66% yield. N-Sulfinyl propargylamines 6k and 6j, with perfluorinated side chains, required even milder desilylation conditions. KF in the presence of 18-crown-6 (GP-6) provided the free alkynes 7k and 7j in 29% and 52% yield, respectively (results are collected in Table 2).
Desilylation of alkynes 6k and 6j under alkaline conditions with K2CO3, in analogy to the description of Vasella et al. [57] leads to instant decomposition of the starting material in a base-promoted propargyl–allenamide isomerization. According to our hypothesis, basic fluoride leads to deprotonation in the Cα-position of 6i, 6k and 6j, inducing an alkyne rearrangement to form an allene, which rearranges further to provide an α,β-unsaturated imine (Figure 8) [97].
![[1860-5397-13-240-8]](/bjoc/content/figures/1860-5397-13-240-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Base-induced rearrangement of propargylamines bearing electron-withdrawing substituents.
Figure 8: Base-induced rearrangement of propargylamines bearing electron-withdrawing substituents.
One target application of propargylamines 7 is the Sonogashira cross-coupling with halogenated benzoates, forming the scaffold of versatile peptidomimetics 11. According to the proposed rearrangement of alkyne 7 (Figure 9), the choice of base is crucial for such cross-coupling reactions. To get a better understanding of the reactivity of propargylamines, the stability and propensity for base induced rearrangement of 11 were investigated (Figure 9).
![[1860-5397-13-240-9]](/bjoc/content/figures/1860-5397-13-240-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Base-catalyzed rearrangement of propargylamines 11 to α,β-unsaturated imines 12. A) Reaction scheme: (a) methyl 4-iodobenzoate (for the conversion of 7i to 11i, 1.6 equiv) or methyl 3-iodobenzoate (for the conversion of 7k to 11k), DIPEA (6 equiv), THF, Cl2Pd(PPh3)2, CuI, room temperature, 2 h (GP-9). (b) Piperidine/THF (1:3), 0 °C, 30 min (conversion of 11i to 12i). Or LiOH (3 equiv), MeOH/H2O (2:1), 0 °C, 30 min. B) UV–vis spectra of propargylamines 11 and α,β-unsaturated imines 12. C) X-ray crystal structure analysis of propargylamine 11i and α,β-unsaturated imine 12i.
Figure 9: Base-catalyzed rearrangement of propargylamines 11 to α,β-unsaturated imines 12. A) Reaction scheme...
The ester substituted compounds 11i and 11k were obtained by Sonogashira cross-coupling of the terminal alkynes 7i and 7k with methyl 4- and 3-iodobenzoate, respectively. As the tert-butylsulfinyl group can be cleaved off under mild, acidic conditions [1,25,26], it provides a versatile protective group for the amine during the conversion of the alkyne. Additionally, it’s chirality indicates epimerization and, for example in the Pauson–Khand reaction [14], allows the determination of the stereoselectivity of asymmetric conversions by simple 1H NMR experiments. Consequently it was not removed prior to the rearrangement experiments.
Treatment of 11i and 11k with the mild base piperidine led to the formation of α,β-unsaturated sulfinylimines 12i and 12k. The structures of the rearranged products were proven unequivocally by 1H and 13C NMR spectroscopy and X-ray crystal structure analysis.
Due to the extended π-system of the α,β-unsaturated imines 12i and 12k, the progress of the propargylamide–allenylamide rearrangement that eventually leads to the formation of the α,β-unsaturated imines by tautomerism could be easily monitored by UV–vis spectroscopy. The reaction mixture turned brightly yellow when treated with bases like piperidine. The X-ray crystal structures of 11i and 12i confirm unequivocally the structure of the products and thus the postulated two-step rearrangement. Both, the rearrangement of the alkyne to the allene and the subsequent tautomerism to the α,β-unsaturated imine are not reversible. Reversibility of the rearrangement would be fundamental for racemization of propargylamines, which is consequently improbable. However, even in the presence of strong bases like KOt-Bu or LDA, the propargylamide–allenylamide rearrangement could never be observed for propargylamines without an acidifying Cα-substituent.
Synthesis of propargylamines containing polar or acidic functional groups
The synthesis of propargylamines with polar substituents to mimic polar amino acids such as serine (alcohol), cysteine (thiol) or glutamine (carboxamide) requires special protective groups (Table 3).
Table 3: Preparation of N-sulfinyl propargylamines 7 with polar and acidic functional groups in the side chains.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-240-i3.svg?max-width=637&scale=1.0)
|
|||||
| R | (a) | Yield (5) | (b), (c) | Yield (7)b | dr |
| CH2OCPh3 | GP-2 | 47% (5m) | GP-4, GP-6 | 0% (7m)c | n.d. |
| CH2OBn | GP-2 | 81% (5n)d | GP-3, GP-5 | 4% (7n)e | 95/5 |
| CH2OAll | GP-1 | 24% (5o) | GP-4, GP-5 | 31% (7o) | 93/7 |
| CH2SBn | GP-2 | 83% (5p)d | GP-3, GP-5 | 31% (7p)f | 93/7 |
| (CH2)3CN | GP-2 | 80% (5q) | GP-4, GP-6 | 43% (7q) | 95/5 |
| (CH2)2CO2Bn | GP-2 | 79% (5r) | GP-4, GP-5 | 0% (7r)c | n.d. |
| (CH2)2CO2t-Bu | GP-2 | 64% (5s) | GP-4, GP-6 | 47% (7s) | 93/7 |
| (CH2)3OXf | GP-1 | 90% (5t) | GP-3, GP-5 | 27% (7t) | 93/7 |
| (CH2)3Cl | GP-1 | 85% (5u) | GP-4, GP-5 | 0% (7u)c | n.d. |
| (CH2)4N3 | GP-2 | 52% (5v) | f | ||
| (CH2)3N3 | GP-2 | 84% (5w) | f | ||
a(a) GP-1: Auxiliary (S)-1, aldehyde, Ti(OiPr)4, 70 °C, 40 min. GP-2: Auxiliary (S)-1, aldehyde, CuSO4, DCM, rt, 3 d. (b) GP-4: (Trimethylsilyl)ethynyllithium, AlMe3, toluene, −78 °C to rt. (c) GP-5: TBAF, THF, 0 °C, 3 h. GP-6: KF, 18-crown-6, THF/H2O (98:2), 0 °C. bYields of 7 refer to imine 5 (two steps). cCompounds 7m, 7r and 7u could not be isolated and the diastereoselectivity could not be determined (n.d.). dFurther reactions in Table 4. e(R)-Configured Ellman’s sulfinamide (R)-1 was applied. Hence, the mirror images of 5–7n,p were obtained. f5t, X = TBDMS. 7t, X = H.
The cyano moiety was used as precursor of the carboxamide moiety of glutamine, since the cyano group is stable in the presence of nucleophiles and strong bases. The synthesis started with the Kolbe nitrile synthesis of 4-iodobutan-1-ol with NaCN. Performing this transformation in DMSO provided the desired 5-hydroxypentanenitrile and THF in the ratio 5:2 (monitored by 1H NMR spectroscopy, see Supporting Information File 1). Next, 4-cyanobutan-1-ol was oxidized in a Swern oxidation and the resulting aldehyde was condensed with tert-butylsulfinamide (S)-1, according to GP-2. The reaction of sulfinimine 5q with (trimethylsilyl)ethynyllithium led to sulfinamide 6q. In this case, the Lewis acid AlMe3, which could also react with the cyano moiety, was omitted. Finally, cleavage of the TMS group was achieved under mild conditions with KF and 18-crown-6 (GP-6), to yield N-sulfinyl propargylamine 7q as an analogue of the non-proteinogenic amino acid 5-cyano-L-norvaline. Formation of a glutamine analogous side chain by hydrolysis of the nitrile function to an amide remained unsuccessful so far.
Propargylamines with diversely protected alcohol functionalities in the side chain were obtained by approach II. The application of an allyl ether, starting from 2-allyloxyacetaldehyde to form 7o and a benzoate, starting from formylmethyl benzoate to form 7n (Table 3) turned out to be most convenient. In contrast to labile TMS ethers, the sterically more demanding tert-butyl dimethylsilyl ether was successfully applied as protective group to obtain the homologated serine analogous propargylamine 7t similar to the description by Verrier et al. [47,55], starting from TBDMS-protected oxybutanal. Treatment of alkyne 6t with TBAF leads to simultaneous cleavage of both silyl groups. Although sterically shielding protective groups have proven convenient, the trityl group turned out to be inefficient to generate a serine-analogous propargylamine. Trityl-protected imine 5m immediately decomposed, when treated with (trimethylsilyl)ethynyllithium.
The preparation of glutamic acid-analogous propargylamines failed when the acid functionality was protected as a benzyl ester. Fortunately, the sterically more demanding tert-butyl ester was stable and gave high yields of 7s. The synthesis started from the aldehyde tert-butyl-4-oxobutanoate, which could be easily obtained from succinic anhydride [44,98,99]. Selective cleavage of the tert-butyl group was not yet accomplished without affecting the tert-butyl sulfinamide protection group of the amine.
A cysteine-analogous alkyne could be synthesized starting from benzylmercaptan. Unfortunately, only extremely low yields were achieved and N-sulfinyl propargylamine 7p is not stable under the conditions, which are necessary to cleave the thioether [100,101].
Synthesis of propargylamines with basic functional groups in the side chain
Very often, basic amino acids, like lysine or arginine are found in the catalytic center of enzymes. Therefore, propargylamines mimicking these basic amino acids are of particular interest to be incorporated in peptidomimetics. The exchange of basic amino acids has already been performed to develop potent enzyme inhibitors [102-104]. In order to introduce side chains with basic amino moieties into propargylamines, these have to be protected against nucleophiles, bases and deprotonation.
According to the approach of Ye et al., a 3-bromopropyl side chain was used, which was converted to the azide and further transformed into an aminoalkyl group by Staudinger reduction [16]. In order to follow a more convergent approach and to avoid nucleophilic substitution of the halide at a late stage, we decided to start the synthesis with 4-azidobutanal, which was prepared by opening THF with iodine and NaBH4, nucleophilic substitution by sodium azide and Swern oxidation of the alcohol.
4-Azidobutanal was converted with chiral sulfinamide (S)-1 into N-sulfinylimine 5w, which was reacted with (trimethylsilyl)ethynyllithium. However, following the usual procedure GP-4 with warming up the reaction mixture to room temperature before quenching with water led to the formation of triazole 13w, which was isolated in 56% yield (Table 4). The target propargylamine 6wx was obtained in only 19% yield. Cleavage of the TMS group with KF and 18-crown-6 at 0 °C provided azide 7wx in only 22% yield, because triazole 14w was formed in a side reaction. Compound 7wx was converted into triazole 14w even upon standing at room temperature in CDCl3 (monitored by 1H NMR spectroscopy, see Supporting Information File 1). Formation of triazoles 13w and 14w was unexpected, because the uncatalyzed Huisgen reaction usually requires higher temperature or activation by electron-withdrawing substituents at the alkyne or electron-donating substituents at the azide, respectively [105,106]. As none of these requirements are met in the case of 6wx and 7wx, it is assumed, that the preorientation of the azide and the alkyne together with the formation of an energetically favored six-membered ring are the driving forces. As hexose scaffolds similar to 13w and 14w have been successfully applied as inhibitors of β-glucosidases [107] and hexosamidases [108], this intramolecular Huisgen reaction could be exploited to develop novel enzyme inhibitors.
Table 4: Synthesis of lysine, ornithine and arginine-analogous propargylamines 7vy, 7wy and 7x and discovery of an unexpected intramolecular low-temperature Huisgen reaction.
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-240-i4.svg?max-width=637&scale=1.0)
|
||||
| Starting material | Reaction conditions | Product | Yield | dr |
| 5v | (a) (trimethylsilyl)ethynyllithium, Ti(OiPr)4, THF, −78 °C (GP-3) | 6v | 58% | 100:0 |
| 5w | (a) (trimethylsilyl)ethynyllithium, Ti(OiPr)4, THF, −78 °C. (GP-3) | 6w | 14% | n.d. |
| 6v | (b) (trimethylsilyl)ethynyllithium, AlMe3, toluene, −78 °C (GP-4) | 13w | 56% | 100:0 |
| 6v | (c) TBAF (2 equiv), THF, 0 °C, 4 h (GP-5) | 7vx | 58% | 74:26 |
| 6w | (c) TBAF (2 equiv), THF, 0 °C, 4 h (GP-5) | 7wx | 19% | 96:4 |
| 7wx | (d) CDCl3, 7 d, rt. | 14w | 66% | 96:4 |
| 7vx | (e) 1. PPh3, CuSO4, THF; 2. H2O. | 7vy | 0% | n.d. |
| 7wx | (e) 1. PPh3, CuSO4, THF; 2. H2O. | 7wy | 0% | n.d. |
| 6v | (f) 1. NaBH4, CuSO4, THF; 2. H2O. | 7vy | 0% | n.d. |
| 6w | (f) 1. NaBH4, CuSO4, THF; 2. H2O. | 7wy | 0% | n.d. |
| 5v | (g) 1. (trimethylsilyl)ethynyllithium, toluene, −78 °C, 3 h; 2. PPh3 (4 equiv), THF, −78 °C; 3. H2O, rt, 2 h. | 7vy | 68% | 91:9 |
| 5w | (g) 1. (trimethylsilyl)ethynyllithium, toluene, −78 °C, 3 h; 2. PPh3 (4 equiv), THF, −78 °C; 3. H2O, rt, 2 h. | 7wy | 86% | 80:20 |
| 7vy | (h) 1. Boc2O (2 equiv), THF/H2O (1:1), NaHCO3 (3 equiv); 2. imidazole, 4 h. | 7vz | 45% | 91:9 |
| 7wy | (h) 1. Boc2O (2 equiv), THF/H2O (1:1), NaHCO3 (3 equiv); 2. imidazole, 4 h. | 7wz | 63% | 90:10 |
| 7wy | (i) BocHN-C(=NBoc)SMe (1 equiv), DCM, rt, 3 d. | 7x | 79% | 93:7 |
In order to get access to propargylamines with the side chains of lysine, ornithine and arginine, azide 7wx should be reduced. However, all attempts to reduce the isolated azide 7wx with NaBH4 or PPh3 failed, due to the competing triazole formation. Therefore, instant reduction of in situ prepared 6wy was envisaged. The nucleophilic addition of (trimethylsilyl)ethynyllithium to N-sulfinylimine 5w was monitored by analytical HPLC. After complete conversion, PPh3 was added directly to the reaction mixture at −78 °C. After addition of water and stirring for two hours, the primary amine 7wy was isolated in 86% yield. This procedure led to the ornithine analogous propargylamine 7wy, which was reacted with Boc-protected S-methylisothiourea to yield the arginine analogous propargylamine 7x in a yield of 79%. During the decomposition of the azide in the Staudinger reaction, the TMS group at the alkyne was cleaved off simultaneously. Cleavage of TMS groups with PPh3 under similar conditions has not been reported so far. Therefore, an intramolecular mechanism is postulated, in which the iminophosphorane, formed by the reaction of PPh3 with azide 6wy, coordinates to the silyl group. This intramolecular coordination facilitates the fast hydrolytic cleavage of the silyl group during aqueous work-up. This PPh3-induced TMS cleavage could also be successfully applied in the synthesis of the lysine analogous propargylamine 7vy from 5v but could never be reproduced in the formation of other propargylamines, like the tert-butyl-substituted compound 7e, under identical reaction conditions. The amine groups of 7wy and 7vy were protected with a Boc group to give propargylamines 7wz and 7vz.
Conclusion
Ellman’s chiral sulfinamide has been successfully used for the asymmetric synthesis of enantiomerically pure propargylamines. An array of 24 diastereomerically pure N-sulfinyl propargylamines has been prepared, bearing side chains in α-position, which are analogous or similar to amino acid side chains. In general, various aldehydes are condensed with Ellman’s chiral sulfinamide. Diastereoselective re-face addition of (trimethylsilyl)ethynyllithium to the (S)-configured sulfinimines 5 gives the corresponding N-sulfinyl propargylamines 6. Cleavage of the TMS group with TBAF or KF × 18-crown-6 provides N-sulfinyl propargylamines 7 with a terminal alkyne.
Propargylamines with aliphatic side chains were obtained in good yields, depending on the size of the Cα substituent. Various polar and basic substituents in the side chain can be introduced using masked or protected functionalities. Side chains with amino groups were introduced masked as azide. For this purpose, the unprecedented intramolecular Huisgen reaction has to be suppressed. Electron-withdrawing substituents in the Cβ-position could not be used by this approach. Electron-withdrawing substituents in the Cα-position induced an irreversible alkyne–allene-α,β-unsaturated imine rearrangement under mild basic conditions, which makes an alkaline racemization of propargylamines improbable. Altogether, a large set of propargylamines with various amino acid similar substituents are available for application in peptidomimetics and some knowledge on the reactivity of propargylamines has been contributed.
Supporting Information
| Supporting Information File 1: Details about the experiments, methods and materials, the X-ray crystal structures and NMR spectra. | ||
| Format: PDF | Size: 18.8 MB | Download |
References
-
Patterson, A. W.; Ellman, J. A. J. Org. Chem. 2006, 71, 7110–7112. doi:10.1021/jo061160h
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Verdoes, M.; Edgington, L. E.; Scheeren, F. A.; Leyva, M.; Blum, G.; Weiskopf, K.; Bachmann, M. H.; Ellman, J. A.; Bogyo, M. Chem. Biol. 2012, 19, 619–628. doi:10.1016/j.chembiol.2012.03.012
Return to citation in text: [1] [2] [3] -
Patterson, A. W.; Wood, W. J. L.; Hornsby, M.; Lesley, S.; Spraggon, G.; Ellman, J. A. J. Med. Chem. 2006, 49, 6298–6307. doi:10.1021/jm060701s
Return to citation in text: [1] -
Wood, W. J. L.; Patterson, A. W.; Tsuruoka, H.; Jain, R. K.; Ellman, J. A. J. Am. Chem. Soc. 2005, 127, 15521–15527. doi:10.1021/ja0547230
Return to citation in text: [1] -
Inagaki, H.; Tsuruoka, H.; Hornsby, M.; Lesley, S. A.; Spraggon, G.; Ellman, J. A. J. Med. Chem. 2007, 50, 2693–2699. doi:10.1021/jm070111+
Return to citation in text: [1] -
Moss, N.; Xiong, Z.; Burke, M.; Cogan, D.; Gao, D. A.; Haverty, K.; Heim-Riether, A.; Hickey, E. R.; Nagaraja, R.; Netherton, M.; O’Shea, K.; Ramsden, P.; Schwartz, R.; Shih, D.-T.; Ward, Y.; Young, E.; Zhang, Q. Bioorg. Med. Chem. Lett. 2012, 22, 7189–7193. doi:10.1016/j.bmcl.2012.09.054
Return to citation in text: [1] [2] -
Xu, H.-C.; Chowdhury, S.; Ellman, J. A. Nat. Protoc. 2013, 8, 2271–2280. doi:10.1038/nprot.2013.134
Return to citation in text: [1] [2] -
Brak, K.; Doyle, P. S.; McKerrow, J. H.; Ellman, J. A. J. Am. Chem. Soc. 2008, 130, 6404–6410. doi:10.1021/ja710254m
Return to citation in text: [1] -
Brak, K.; Kerr, I. D.; Barrett, K. T.; Fuchi, N.; Debnath, M.; Ang, K.; Engel, J. C.; McKerrow, J. H.; Doyle, P. S.; Brinen, L. S.; Ellman, J. A. J. Med. Chem. 2010, 53, 1763–1773. doi:10.1021/jm901633v
Return to citation in text: [1] [2] -
Leyva, M. J.; DeGiacomo, F.; Kaltenbach, L. S.; Holcomb, J.; Zhang, N.; Gafni, J.; Park, H.; Lo, D. C.; Salvesen, G. S.; Ellerby, L. M.; Ellman, J. A. Chem. Biol. 2010, 17, 1189–1200. doi:10.1016/j.chembiol.2010.08.014
Return to citation in text: [1] [2] -
Deu, E.; Leyva, M. J.; Albrow, V. E.; Rice, M. J.; Ellman, J. A.; Bogyo, M. Chem. Biol. 2010, 17, 808–819. doi:10.1016/j.chembiol.2010.06.007
Return to citation in text: [1] [2] -
Jordan, S.; Starks, S. A.; Whatley, M. F.; Turlington, M. Org. Lett. 2015, 17, 4842–4845. doi:10.1021/acs.orglett.5b02408
Return to citation in text: [1] -
Chen, B.-L.; Wang, B.; Lin, G.-Q. J. Org. Chem. 2010, 75, 941–944. doi:10.1021/jo902424m
Return to citation in text: [1] [2] [3] [4] -
Bauer, R. A.; DiBlasi, C. M.; Tan, D. S. Org. Lett. 2010, 12, 2084–2087. doi:10.1021/ol100574y
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Lee, S. I.; Park, S. Y.; Park, J. H.; Jung, I. G.; Choi, S. Y.; Chung, Y. K.; Lee, B. Y. J. Org. Chem. 2006, 71, 91–96. doi:10.1021/jo051685u
Return to citation in text: [1] [2] -
Ye, L.; He, W.; Zhang, L. Angew. Chem., Int. Ed. 2011, 50, 3236–3239. doi:10.1002/anie.201007624
Return to citation in text: [1] [2] [3] [4] -
Wünsch, M.; Senger, J.; Schultheisz, P.; Schwarzbich, S.; Michalek, C.; Klaß, M.; Goskowitz, S.; Borchert, P.; Jung, M.; Sewald, N. ChemMedChem, in press.
Return to citation in text: [1] [2] -
Nahrwold, M.; Bogner, T.; Eissler, S.; Verma, S.; Sewald, N. Org. Lett. 2010, 12, 1064–1067. doi:10.1021/ol1000473
Return to citation in text: [1] -
Angelo, N. G.; Arora, P. S. J. Am. Chem. Soc. 2005, 127, 17134–17135. doi:10.1021/ja056406z
Return to citation in text: [1] [2] -
Johansson, J. R.; Hermansson, E.; Nordén, B.; Kann, N.; Beke-Somfai, T. Eur. J. Org. Chem. 2014, 2703–2713. doi:10.1002/ejoc.201400018
Return to citation in text: [1] -
Kracker, O.; Góra, J.; Krzciuk-Gula, J.; Marion, A.; Neumann, B.; Stammler, H.-G.; Nieß, A.; Antes, I.; Latajka, R.; Sewald, N. Chem. – Eur. J. submitted.
Return to citation in text: [1] [2] [3] [4] -
Ko, E.; Liu, J.; Perez, L. M.; Lu, G.; Schaefer, A.; Burgess, K. J. Am. Chem. Soc. 2011, 133, 462–477. doi:10.1021/ja1071916
Return to citation in text: [1] -
Temperini, A.; Capperucci, A.; Degl’Innocenti, A.; Terlizzi, R.; Tiecco, M. Tetrahedron Lett. 2010, 51, 4121–4124. doi:10.1016/j.tetlet.2010.05.143
Return to citation in text: [1] -
Weix, D. J.; Ellman, J. A. Org. Synth. 2005, 82, 157–165. doi:10.1002/0471264229.os082.24
Return to citation in text: [1] -
Aggarwal, V. K.; Barbero, N.; McGarrigle, E. M.; Mickle, G.; Navas, R.; Suárez, J. R.; Unthank, M. G.; Yar, M. Tetrahedron Lett. 2009, 50, 3482–3484. doi:10.1016/j.tetlet.2009.03.020
Return to citation in text: [1] [2] [3] [4] -
Ellman, J. A.; Owens, T. D.; Tang, T. P. Acc. Chem. Res. 2002, 35, 984–995. doi:10.1021/ar020066u
Return to citation in text: [1] [2] [3] -
Wakayama, M.; Ellman, J. A. J. Org. Chem. 2009, 74, 2646–2650. doi:10.1021/jo9001883
Return to citation in text: [1] -
Arava, V. R.; Gorentla, L.; Dubey, P. K. Beilstein J. Org. Chem. 2011, 7, 9–12. doi:10.3762/bjoc.7.2
Return to citation in text: [1] [2] -
Huang, Z.; Zhang, M.; Wang, Y.; Qin, Y. Synlett 2005, 1334–1336. doi:10.1055/s-2005-865234
Return to citation in text: [1] [2] [3] -
Higashibayashi, S.; Tohmiya, H.; Mori, T.; Hashimoto, K.; Nakata, M. Synlett 2004, 457–460. doi:10.1055/s-2004-815409
Return to citation in text: [1] [2] [3] -
Liu, G.; Cogan, D. A.; Owens, T. D.; Tang, T. P.; Ellman, J. A. J. Org. Chem. 1999, 64, 1278–1284. doi:10.1021/jo982059i
Return to citation in text: [1] [2] [3] -
Datta, G. K.; Ellman, J. A. J. Org. Chem. 2010, 75, 6283–6285. doi:10.1021/jo1011625
Return to citation in text: [1] [2] [3] -
Collados, J. F.; Toledano, E.; Guijarro, D.; Yus, M. J. Org. Chem. 2012, 77, 5744–5750. doi:10.1021/jo300919x
Return to citation in text: [1] [2] [3] -
Liu, G.; Cogan, D. A.; Ellman, J. A. J. Am. Chem. Soc. 1997, 119, 9913–9914. doi:10.1021/ja972012z
Return to citation in text: [1] [2] [3] -
Foti, M. C.; Johnson, E. R.; Vinqvist, M. R.; Wright, J. S.; Barclay, L. R. C.; Ingold, K. U. J. Org. Chem. 2002, 67, 5190–5196. doi:10.1021/jo020184v
Return to citation in text: [1] -
Peltier, H. M.; McMahon, J. P.; Patterson, A. W.; Ellman, J. A. J. Am. Chem. Soc. 2006, 128, 16018–16019. doi:10.1021/ja067177z
Return to citation in text: [1] -
Patterson, A. W.; Peltier, H. M.; Sasse, F.; Ellman, J. A. Chem. – Eur. J. 2007, 13, 9534–9541. doi:10.1002/chem.200701057
Return to citation in text: [1] -
Patterson, A. W.; Peltier, H. M.; Ellman, J. A. J. Org. Chem. 2008, 73, 4362–4369. doi:10.1021/jo800384x
Return to citation in text: [1] -
Jung, H. H.; Floreancig, P. E. J. Org. Chem. 2007, 72, 7359–7366. doi:10.1021/jo071225w
Return to citation in text: [1] -
González-Gómez, J. C.; Foubelo, F.; Yus, M. Synthesis 2009, 12, 2083–2088. doi:10.1055/s-0029-1216821
Return to citation in text: [1] -
Robak, M. T.; Herbage, M. A.; Ellman, J. A. Chem. Rev. 2010, 110, 3600–3740. doi:10.1021/cr900382t
Return to citation in text: [1] -
Chuang, C. P.; Gallucci, J. C.; Hart, D. J. J. Org. Chem. 1988, 53, 3210–3218. doi:10.1021/jo00249a014
Return to citation in text: [1] -
Yang, X.; Nath, D.; Fleming, F. F. Org. Lett. 2015, 17, 4906–4909. doi:10.1021/acs.orglett.5b02481
Return to citation in text: [1] -
Mancuso, A. J.; Huang, S.-L.; Swern, D. J. Org. Chem. 1978, 43, 2480–2482. doi:10.1021/jo00406a041
Return to citation in text: [1] [2] -
Cogan, D. A.; Liu, G.; Ellman, J. Tetrahedron 1999, 55, 8883–8904. doi:10.1016/S0040-4020(99)00451-2
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Guijarro, D.; Pablo, Ó.; Yus, M. Tetrahedron Lett. 2009, 50, 5386–5388. doi:10.1016/j.tetlet.2009.07.044
Return to citation in text: [1] [2] [3] -
Verrier, C.; Carret, S.; Poisson, J.-F. Org. Biomol. Chem. 2014, 12, 1875–1878. doi:10.1039/c3ob42352b
Return to citation in text: [1] [2] [3] [4] -
Turcaud, S.; Berhal, F.; Royer, J. J. Org. Chem. 2007, 72, 7893–7897. doi:10.1021/jo071139w
Return to citation in text: [1] [2] [3] -
Carreira, E. M.; Du Bois, J. J. Am. Chem. Soc. 1995, 117, 8106–8125. doi:10.1021/ja00136a008
Return to citation in text: [1] [2] [3] -
Nishikawa, T.; Ino, A.; Isobe, M. Tetrahedron 1994, 50, 1449–1468. doi:10.1016/S0040-4020(01)80629-3
Return to citation in text: [1] [2] [3] [4] -
Ferreira, F.; Audouin, M.; Chemla, F. Chem. – Eur. J. 2005, 11, 5269–5278. doi:10.1002/chem.200500268
Return to citation in text: [1] -
Feuvrie, C.; Blanchet, J.; Bonin, M.; Micouin, L. Org. Lett. 2004, 6, 2333–2336. doi:10.1021/ol049346v
Return to citation in text: [1] [2] -
Levin, V. V.; Dilman, A. D.; Belyakov, P. A.; Struchkova, M. I.; Tartakovsky, V. A. Eur. J. Org. Chem. 2008, 5226–5230. doi:10.1002/ejoc.200800820
Return to citation in text: [1] [2] -
Moura-Letts, G.; DiBlasi, C. M.; Bauer, R. A.; Tan, D. S. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6745–6750. doi:10.1073/pnas.1015268108
Return to citation in text: [1] [2] -
Ding, C.-H.; Chen, D.-D.; Luo, Z.-B.; Dai, L.-X.; Hou, X.-L. Synlett 2006, 1272–1274. doi:10.1055/s-2006-939076
Return to citation in text: [1] [2] [3] -
Alzeer, J.; Vasella, A. Helv. Chim. Acta 1995, 78, 177–193. doi:10.1002/hlca.19950780117
Return to citation in text: [1] -
Ernst, A.; Gobbi, L.; Vasella, A. Tetrahedron Lett. 1996, 37, 7959–7962. doi:10.1016/0040-4039(96)01838-2
Return to citation in text: [1] [2] -
Dierks, T.; Lecca, M. R.; Schmidt, B.; von Figura, K. FEBS Lett. 1998, 423, 61–65. doi:10.1016/S0014-5793(98)00065-9
Return to citation in text: [1] -
Rabuka, D.; Rush, J. S.; deHart, G. W.; Wu, P.; Bertozzi, C. R. Nat. Protoc. 2012, 7, 1052–1067. doi:10.1038/nprot.2012.045
Return to citation in text: [1] -
Brewer, G. A., Jr.; Johnson, M. J. Appl. Microbiol. 1953, 1, 163–166.
Return to citation in text: [1] -
Weigel, L. F.; Nitsche, C.; Graf, D.; Bartenschlager, R.; Klein, C. D. J. Med. Chem. 2015, 58, 7719–7733. doi:10.1021/acs.jmedchem.5b00612
Return to citation in text: [1] -
Pollegioni, L.; Harris, C. M.; Molla, G.; Pilone, M. S.; Ghisla, S. FEBS Lett. 2001, 507, 323–326. doi:10.1016/S0014-5793(01)02983-0
Return to citation in text: [1] -
Asimakopoulou, A.; Panopoulos, P.; Chasapis, C. T.; Coletta, C.; Zhou, Z.; Cirino, G.; Giannis, A.; Szabo, C.; Spyroulias, G. A.; Papapetropoulos, A. Br. J. Pharmacol. 2013, 169, 922–932. doi:10.1111/bph.12171
Return to citation in text: [1] -
Shu, M.; Yu, R.; Zhang, Y.; Wang, J.; Yang, L.; Wang, L.; Lin, Z. Med. Chem. (Sharjah, United Arab Emirates) 2013, 9, 32–44. doi:10.2174/157340613804488350
Return to citation in text: [1] -
Shah, P.; Westwell, A. D. J. Enzyme Inhib. Med. Chem. 2007, 22, 527–540. doi:10.1080/14756360701425014
Return to citation in text: [1] -
Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315–8359. doi:10.1021/acs.jmedchem.5b00258
Return to citation in text: [1] -
De Clercq, E. Med. Res. Rev. 2009, 29, 611–645. doi:10.1002/med.20153
Return to citation in text: [1] -
Prober, C. G. Adv. Exp. Med. Biol. 2004, 549, 9–12. doi:10.1007/978-1-4419-8993-2_3
Return to citation in text: [1] -
Heidelberger, C. Ann. N. Y. Acad. Sci. 1975, 255, 317–325. doi:10.1111/j.1749-6632.1975.tb29239.x
Return to citation in text: [1] -
Carmine, A. A.; Brogden, R. N.; Heel, R. C.; Speight, T. M.; Avery, G. S. Drugs 1982, 23, 329–353. doi:10.2165/00003495-198223050-00001
Return to citation in text: [1] -
Corbett, J. W.; Ko, S. S.; Rodgers, J. D.; Gearhart, L. A.; Magnus, N. A.; Bacheler, L. T.; Diamond, S.; Jeffrey, S.; Klabe, R. M.; Cordova, B. C.; Garber, S.; Logue, K.; Trainor, G. L.; Anderson, P. S.; Erickson-Viitanen, S. K. J. Med. Chem. 2000, 43, 2019–2030. doi:10.1021/jm990580e
Return to citation in text: [1] -
Maggiolo, F. J. Antimicrob. Chemother. 2009, 64, 910–928. doi:10.1093/jac/dkp334
Return to citation in text: [1] -
King, J.; Aberg, J. A. AIDS (London, U. K.) 2008, 22, 1709–1717. doi:10.1097/QAD.0b013e32830163ad
Return to citation in text: [1] -
Cipriani, A.; Barbui, C.; Brambilla, P.; Furukawa, T. A.; Hotopf, M.; Geddes, J. R. J. Clin. Psychiatry 2006, 67, 850–864. doi:10.4088/JCP.v67n0601
Return to citation in text: [1] -
Owen, R. T. Drugs Today 2006, 42, 185–192. doi:10.1358/dot.2006.42.3.953589
Return to citation in text: [1] -
Kim, S. S. Ann. Pharmacother. 2003, 37, 890–892. doi:10.1345/aph.1C362
Return to citation in text: [1] -
Foster, R. S.; Jakobi, H.; Harrity, J. P. A. Org. Lett. 2012, 14, 4858–4861. doi:10.1021/ol3021918
Return to citation in text: [1] -
Corbett, J. W.; Ko, S. S.; Rodgers, J. D.; Jeffrey, S.; Bacheler, L. T.; Klabe, R. M.; Diamond, S.; Lai, C. M.; Rabel, S. R.; Saye, J. A.; Adams, S. P.; Trainor, G. L.; Anderson, P. S.; Erickson-Viitanen, S. K. Antimicrob. Agents Chemother. 1999, 43, 2893–2897.
Return to citation in text: [1] -
Jiang, B.; Si, Y.-G. Angew. Chem., Int. Ed. 2004, 43, 216–218. doi:10.1002/anie.200352301
Return to citation in text: [1] -
Magnus, N. A.; Confalone, P. N.; Storace, L. Tetrahedron Lett. 2000, 41, 3015–3019. doi:10.1016/S0040-4039(00)00331-2
Return to citation in text: [1] -
King, R. W.; Klabe, R. M.; Reid, C. D.; Erickson-Viitanen, S. K. Antimicrob. Agents Chemother. 2002, 46, 1640–1646. doi:10.1128/AAC.46.6.1640-1646.2002
Return to citation in text: [1] -
Corbett, J. W. Curr. Med. Chem. - Anti-Infect. Agents 2002, 1, 119–140. doi:10.2174/1568012023354938
Return to citation in text: [1] -
Kauffman, G. S.; Harris, G. D.; Dorow, R. L.; Stone, B. R. P.; Parsons, R. L., Jr.; Pesti, J. A.; Magnus, N. A.; Fortunak, J. M.; Confalone, P. N.; Nugent, W. A. Org. Lett. 2000, 2, 3119–3121. doi:10.1021/ol006321x
Return to citation in text: [1] -
Prakash, G. K. S.; Mandal, M.; Olah, G. A. Angew. Chem., Int. Ed. 2001, 40, 589–590. doi:10.1002/1521-3773(20010202)40:3<589::AID-ANIE589>3.0.CO;2-9
Return to citation in text: [1] -
Prakash, G. K. S.; Mandal, M.; Olah, G. A. Org. Lett. 2001, 3, 2847–2850. doi:10.1021/ol010134x
Return to citation in text: [1] -
Magueur, G.; Crousse, B.; Bonnet-Delpon, D. Tetrahedron Lett. 2005, 46, 2219–2221. doi:10.1016/j.tetlet.2005.02.030
Return to citation in text: [1] [2] -
Magueur, G.; Crousse, B.; Bonnet-Delpon, D. Eur. J. Org. Chem. 2008, 1527–1534. doi:10.1002/ejoc.200701090
Return to citation in text: [1] [2] -
Bégué, J.-P.; Bonnet-Delpon, D.; Crousse, B.; Legros, J. Chem. Soc. Rev. 2005, 34, 562–572. doi:10.1039/b401707m
Return to citation in text: [1] [2] -
Bravo, P.; Crucianelli, M.; Vergani, B.; Zanda, M. Tetrahedron Lett. 1998, 39, 7771–7774. doi:10.1016/S0040-4039(98)01698-0
Return to citation in text: [1] -
Asensio, A.; Bravo, P.; Crucianelli, M.; Farina, A.; Fustero, S.; Soler, J. G.; Meille, S. V.; Panzeri, W.; Viani, F.; Volonterio, A.; Zanda, M. Eur. J. Org. Chem. 2001, 1449–1458. doi:10.1002/1099-0690(200104)2001:8<1449::AID-EJOC1449>3.0.CO;2-2
Return to citation in text: [1] -
Crucianelli, M.; De Angelis, F.; Lazzaro, F.; Malpezzi, L.; Volonterio, A.; Zanda, M. J. Fluorine Chem. 2004, 125, 573–577. doi:10.1016/j.jfluchem.2003.11.034
Return to citation in text: [1] -
Lazzaro, F.; Crucianelli, M.; De Angelis, F.; Frigerio, M.; Malpezzi, L.; Volonterio, A.; Zanda, M. Tetrahedron: Asymmetry 2004, 15, 889–893. doi:10.1016/j.tetasy.2004.01.013
Return to citation in text: [1] -
Greier, G. Synthesis of Novel Trifluormethyl-Substituted Thienothiazines; Vienna University of Technology, 1983.
Return to citation in text: [1] -
Truong, V. L.; Ménard, M. S.; Dion, I. Org. Lett. 2007, 9, 683–685. doi:10.1021/ol063001q
Return to citation in text: [1] [2] [3] [4] -
Xiao, H.; Huang, Y.; Qing, F.-L. Tetrahedron: Asymmetry 2010, 21, 2949–2955. doi:10.1016/j.tetasy.2010.11.028
Return to citation in text: [1] -
Kuduk, S. D.; Di Marco, C. N.; Pitzenberger, S. M.; Tsou, N. Tetrahedron Lett. 2006, 47, 2377–2381. doi:10.1016/j.tetlet.2006.01.154
Return to citation in text: [1] -
Vartanyan, S. A.; Badanyan, S. O. Russ. Chem. Rev. 1967, 36, 670. doi:10.1070/RC1967v036n09ABEH001681
Return to citation in text: [1] -
Srinivasan, R.; Uttamchandani, M.; Yao, S. Q. Org. Lett. 2006, 8, 713–716. doi:10.1021/ol052895w
Return to citation in text: [1] -
Chen, S.; Zhao, X.; Chen, J.; Chen, J.; Kuznetsova, L.; Wong, S. S.; Ojima, I. Bioconjugate Chem. 2010, 21, 979–987. doi:10.1021/bc9005656
Return to citation in text: [1] -
Corrie, J. E. T.; Hlubucek, J. R.; Lowe, G. J. Chem. Soc., Perkin Trans. 1 1977, 1421–1425. doi:10.1039/p19770001421
Return to citation in text: [1] -
Koreeda, M.; Yang, W. Synlett 1994, 3, 201–203. doi:10.1055/s-1994-22794
Return to citation in text: [1] -
Bitonti, A. J.; Casara, P. J.; McCann, P. P.; Bey, P. Biochem. J. 1987, 242, 69–74. doi:10.1042/bj2420069
Return to citation in text: [1] -
Metcalf, B. W.; Jung, M. α-Acetylenic Derivatives of Amines. U.S. Patent US4139563 A, Feb 13, 1979.
Return to citation in text: [1] -
Metcalf, B. W.; Jung, M. α-Acetylenic Derivatives of α-Amino Acids. U.S. Patent US4182891 A, Jan 8, 1980.
Return to citation in text: [1] -
Amantini, D.; Fringuelli, F.; Piermatti, O.; Pizzo, F.; Zunino, E.; Vaccaro, L. J. Org. Chem. 2005, 70, 6526–6529. doi:10.1021/jo0507845
Return to citation in text: [1] -
Huisgen, R. Angew. Chem., Int. Ed. Engl. 1963, 2, 565–598. doi:10.1002/anie.196305651
Return to citation in text: [1] -
Panday, N.; Meyyappan, M.; Vasella, A. Helv. Chim. Acta 2000, 83, 513–538. doi:10.1002/(SICI)1522-2675(20000315)83:3<513::AID-HLCA513>3.0.CO;2-1
Return to citation in text: [1] -
Panday, N.; Vasella, A. Helv. Chim. Acta 2000, 83, 1205–1208. doi:10.1002/1522-2675(20000607)83:6<1205::AID-HLCA1205>3.0.CO;2-L
Return to citation in text: [1]
| 25. | Aggarwal, V. K.; Barbero, N.; McGarrigle, E. M.; Mickle, G.; Navas, R.; Suárez, J. R.; Unthank, M. G.; Yar, M. Tetrahedron Lett. 2009, 50, 3482–3484. doi:10.1016/j.tetlet.2009.03.020 |
| 27. | Wakayama, M.; Ellman, J. A. J. Org. Chem. 2009, 74, 2646–2650. doi:10.1021/jo9001883 |
| 28. | Arava, V. R.; Gorentla, L.; Dubey, P. K. Beilstein J. Org. Chem. 2011, 7, 9–12. doi:10.3762/bjoc.7.2 |
| 84. | Prakash, G. K. S.; Mandal, M.; Olah, G. A. Angew. Chem., Int. Ed. 2001, 40, 589–590. doi:10.1002/1521-3773(20010202)40:3<589::AID-ANIE589>3.0.CO;2-9 |
| 85. | Prakash, G. K. S.; Mandal, M.; Olah, G. A. Org. Lett. 2001, 3, 2847–2850. doi:10.1021/ol010134x |
| 28. | Arava, V. R.; Gorentla, L.; Dubey, P. K. Beilstein J. Org. Chem. 2011, 7, 9–12. doi:10.3762/bjoc.7.2 |
| 86. | Magueur, G.; Crousse, B.; Bonnet-Delpon, D. Tetrahedron Lett. 2005, 46, 2219–2221. doi:10.1016/j.tetlet.2005.02.030 |
| 87. | Magueur, G.; Crousse, B.; Bonnet-Delpon, D. Eur. J. Org. Chem. 2008, 1527–1534. doi:10.1002/ejoc.200701090 |
| 88. | Bégué, J.-P.; Bonnet-Delpon, D.; Crousse, B.; Legros, J. Chem. Soc. Rev. 2005, 34, 562–572. doi:10.1039/b401707m |
| 77. | Foster, R. S.; Jakobi, H.; Harrity, J. P. A. Org. Lett. 2012, 14, 4858–4861. doi:10.1021/ol3021918 |
| 78. | Corbett, J. W.; Ko, S. S.; Rodgers, J. D.; Jeffrey, S.; Bacheler, L. T.; Klabe, R. M.; Diamond, S.; Lai, C. M.; Rabel, S. R.; Saye, J. A.; Adams, S. P.; Trainor, G. L.; Anderson, P. S.; Erickson-Viitanen, S. K. Antimicrob. Agents Chemother. 1999, 43, 2893–2897. |
| 79. | Jiang, B.; Si, Y.-G. Angew. Chem., Int. Ed. 2004, 43, 216–218. doi:10.1002/anie.200352301 |
| 80. | Magnus, N. A.; Confalone, P. N.; Storace, L. Tetrahedron Lett. 2000, 41, 3015–3019. doi:10.1016/S0040-4039(00)00331-2 |
| 81. | King, R. W.; Klabe, R. M.; Reid, C. D.; Erickson-Viitanen, S. K. Antimicrob. Agents Chemother. 2002, 46, 1640–1646. doi:10.1128/AAC.46.6.1640-1646.2002 |
| 82. | Corbett, J. W. Curr. Med. Chem. - Anti-Infect. Agents 2002, 1, 119–140. doi:10.2174/1568012023354938 |
| 83. | Kauffman, G. S.; Harris, G. D.; Dorow, R. L.; Stone, B. R. P.; Parsons, R. L., Jr.; Pesti, J. A.; Magnus, N. A.; Fortunak, J. M.; Confalone, P. N.; Nugent, W. A. Org. Lett. 2000, 2, 3119–3121. doi:10.1021/ol006321x |
| 74. | Cipriani, A.; Barbui, C.; Brambilla, P.; Furukawa, T. A.; Hotopf, M.; Geddes, J. R. J. Clin. Psychiatry 2006, 67, 850–864. doi:10.4088/JCP.v67n0601 |
| 75. | Owen, R. T. Drugs Today 2006, 42, 185–192. doi:10.1358/dot.2006.42.3.953589 |
| 76. | Kim, S. S. Ann. Pharmacother. 2003, 37, 890–892. doi:10.1345/aph.1C362 |
| 41. | Robak, M. T.; Herbage, M. A.; Ellman, J. A. Chem. Rev. 2010, 110, 3600–3740. doi:10.1021/cr900382t |
| 42. | Chuang, C. P.; Gallucci, J. C.; Hart, D. J. J. Org. Chem. 1988, 53, 3210–3218. doi:10.1021/jo00249a014 |
| 43. | Yang, X.; Nath, D.; Fleming, F. F. Org. Lett. 2015, 17, 4906–4909. doi:10.1021/acs.orglett.5b02481 |
| 34. | Liu, G.; Cogan, D. A.; Ellman, J. A. J. Am. Chem. Soc. 1997, 119, 9913–9914. doi:10.1021/ja972012z |
| 94. | Truong, V. L.; Ménard, M. S.; Dion, I. Org. Lett. 2007, 9, 683–685. doi:10.1021/ol063001q |
| 35. | Foti, M. C.; Johnson, E. R.; Vinqvist, M. R.; Wright, J. S.; Barclay, L. R. C.; Ingold, K. U. J. Org. Chem. 2002, 67, 5190–5196. doi:10.1021/jo020184v |
| 36. | Peltier, H. M.; McMahon, J. P.; Patterson, A. W.; Ellman, J. A. J. Am. Chem. Soc. 2006, 128, 16018–16019. doi:10.1021/ja067177z |
| 37. | Patterson, A. W.; Peltier, H. M.; Sasse, F.; Ellman, J. A. Chem. – Eur. J. 2007, 13, 9534–9541. doi:10.1002/chem.200701057 |
| 38. | Patterson, A. W.; Peltier, H. M.; Ellman, J. A. J. Org. Chem. 2008, 73, 4362–4369. doi:10.1021/jo800384x |
| 39. | Jung, H. H.; Floreancig, P. E. J. Org. Chem. 2007, 72, 7359–7366. doi:10.1021/jo071225w |
| 40. | González-Gómez, J. C.; Foubelo, F.; Yus, M. Synthesis 2009, 12, 2083–2088. doi:10.1055/s-0029-1216821 |
| 30. | Higashibayashi, S.; Tohmiya, H.; Mori, T.; Hashimoto, K.; Nakata, M. Synlett 2004, 457–460. doi:10.1055/s-2004-815409 |
| 93. | Greier, G. Synthesis of Novel Trifluormethyl-Substituted Thienothiazines; Vienna University of Technology, 1983. |
| 31. | Liu, G.; Cogan, D. A.; Owens, T. D.; Tang, T. P.; Ellman, J. A. J. Org. Chem. 1999, 64, 1278–1284. doi:10.1021/jo982059i |
| 32. | Datta, G. K.; Ellman, J. A. J. Org. Chem. 2010, 75, 6283–6285. doi:10.1021/jo1011625 |
| 33. | Collados, J. F.; Toledano, E.; Guijarro, D.; Yus, M. J. Org. Chem. 2012, 77, 5744–5750. doi:10.1021/jo300919x |
| 94. | Truong, V. L.; Ménard, M. S.; Dion, I. Org. Lett. 2007, 9, 683–685. doi:10.1021/ol063001q |
| 21. | Kracker, O.; Góra, J.; Krzciuk-Gula, J.; Marion, A.; Neumann, B.; Stammler, H.-G.; Nieß, A.; Antes, I.; Latajka, R.; Sewald, N. Chem. – Eur. J. submitted. |
| 86. | Magueur, G.; Crousse, B.; Bonnet-Delpon, D. Tetrahedron Lett. 2005, 46, 2219–2221. doi:10.1016/j.tetlet.2005.02.030 |
| 87. | Magueur, G.; Crousse, B.; Bonnet-Delpon, D. Eur. J. Org. Chem. 2008, 1527–1534. doi:10.1002/ejoc.200701090 |
| 88. | Bégué, J.-P.; Bonnet-Delpon, D.; Crousse, B.; Legros, J. Chem. Soc. Rev. 2005, 34, 562–572. doi:10.1039/b401707m |
| 29. | Huang, Z.; Zhang, M.; Wang, Y.; Qin, Y. Synlett 2005, 1334–1336. doi:10.1055/s-2005-865234 |
| 89. | Bravo, P.; Crucianelli, M.; Vergani, B.; Zanda, M. Tetrahedron Lett. 1998, 39, 7771–7774. doi:10.1016/S0040-4039(98)01698-0 |
| 90. | Asensio, A.; Bravo, P.; Crucianelli, M.; Farina, A.; Fustero, S.; Soler, J. G.; Meille, S. V.; Panzeri, W.; Viani, F.; Volonterio, A.; Zanda, M. Eur. J. Org. Chem. 2001, 1449–1458. doi:10.1002/1099-0690(200104)2001:8<1449::AID-EJOC1449>3.0.CO;2-2 |
| 91. | Crucianelli, M.; De Angelis, F.; Lazzaro, F.; Malpezzi, L.; Volonterio, A.; Zanda, M. J. Fluorine Chem. 2004, 125, 573–577. doi:10.1016/j.jfluchem.2003.11.034 |
| 92. | Lazzaro, F.; Crucianelli, M.; De Angelis, F.; Frigerio, M.; Malpezzi, L.; Volonterio, A.; Zanda, M. Tetrahedron: Asymmetry 2004, 15, 889–893. doi:10.1016/j.tetasy.2004.01.013 |
| 44. | Mancuso, A. J.; Huang, S.-L.; Swern, D. J. Org. Chem. 1978, 43, 2480–2482. doi:10.1021/jo00406a041 |
| 29. | Huang, Z.; Zhang, M.; Wang, Y.; Qin, Y. Synlett 2005, 1334–1336. doi:10.1055/s-2005-865234 |
| 30. | Higashibayashi, S.; Tohmiya, H.; Mori, T.; Hashimoto, K.; Nakata, M. Synlett 2004, 457–460. doi:10.1055/s-2004-815409 |
| 31. | Liu, G.; Cogan, D. A.; Owens, T. D.; Tang, T. P.; Ellman, J. A. J. Org. Chem. 1999, 64, 1278–1284. doi:10.1021/jo982059i |
| 32. | Datta, G. K.; Ellman, J. A. J. Org. Chem. 2010, 75, 6283–6285. doi:10.1021/jo1011625 |
| 33. | Collados, J. F.; Toledano, E.; Guijarro, D.; Yus, M. J. Org. Chem. 2012, 77, 5744–5750. doi:10.1021/jo300919x |
| 34. | Liu, G.; Cogan, D. A.; Ellman, J. A. J. Am. Chem. Soc. 1997, 119, 9913–9914. doi:10.1021/ja972012z |
| 57. | Ernst, A.; Gobbi, L.; Vasella, A. Tetrahedron Lett. 1996, 37, 7959–7962. doi:10.1016/0040-4039(96)01838-2 |
| 1. | Patterson, A. W.; Ellman, J. A. J. Org. Chem. 2006, 71, 7110–7112. doi:10.1021/jo061160h |
| 14. | Bauer, R. A.; DiBlasi, C. M.; Tan, D. S. Org. Lett. 2010, 12, 2084–2087. doi:10.1021/ol100574y |
| 45. | Cogan, D. A.; Liu, G.; Ellman, J. Tetrahedron 1999, 55, 8883–8904. doi:10.1016/S0040-4020(99)00451-2 |
| 46. | Guijarro, D.; Pablo, Ó.; Yus, M. Tetrahedron Lett. 2009, 50, 5386–5388. doi:10.1016/j.tetlet.2009.07.044 |
| 47. | Verrier, C.; Carret, S.; Poisson, J.-F. Org. Biomol. Chem. 2014, 12, 1875–1878. doi:10.1039/c3ob42352b |
| 48. | Turcaud, S.; Berhal, F.; Royer, J. J. Org. Chem. 2007, 72, 7893–7897. doi:10.1021/jo071139w |
| 97. | Vartanyan, S. A.; Badanyan, S. O. Russ. Chem. Rev. 1967, 36, 670. doi:10.1070/RC1967v036n09ABEH001681 |
| 95. | Xiao, H.; Huang, Y.; Qing, F.-L. Tetrahedron: Asymmetry 2010, 21, 2949–2955. doi:10.1016/j.tetasy.2010.11.028 |
| 96. | Kuduk, S. D.; Di Marco, C. N.; Pitzenberger, S. M.; Tsou, N. Tetrahedron Lett. 2006, 47, 2377–2381. doi:10.1016/j.tetlet.2006.01.154 |
| 94. | Truong, V. L.; Ménard, M. S.; Dion, I. Org. Lett. 2007, 9, 683–685. doi:10.1021/ol063001q |
| 94. | Truong, V. L.; Ménard, M. S.; Dion, I. Org. Lett. 2007, 9, 683–685. doi:10.1021/ol063001q |
| 34. | Liu, G.; Cogan, D. A.; Ellman, J. A. J. Am. Chem. Soc. 1997, 119, 9913–9914. doi:10.1021/ja972012z |
| 13. | Chen, B.-L.; Wang, B.; Lin, G.-Q. J. Org. Chem. 2010, 75, 941–944. doi:10.1021/jo902424m |
| 16. | Ye, L.; He, W.; Zhang, L. Angew. Chem., Int. Ed. 2011, 50, 3236–3239. doi:10.1002/anie.201007624 |
| 30. | Higashibayashi, S.; Tohmiya, H.; Mori, T.; Hashimoto, K.; Nakata, M. Synlett 2004, 457–460. doi:10.1055/s-2004-815409 |
| 31. | Liu, G.; Cogan, D. A.; Owens, T. D.; Tang, T. P.; Ellman, J. A. J. Org. Chem. 1999, 64, 1278–1284. doi:10.1021/jo982059i |
| 32. | Datta, G. K.; Ellman, J. A. J. Org. Chem. 2010, 75, 6283–6285. doi:10.1021/jo1011625 |
| 33. | Collados, J. F.; Toledano, E.; Guijarro, D.; Yus, M. J. Org. Chem. 2012, 77, 5744–5750. doi:10.1021/jo300919x |
| 29. | Huang, Z.; Zhang, M.; Wang, Y.; Qin, Y. Synlett 2005, 1334–1336. doi:10.1055/s-2005-865234 |
| 47. | Verrier, C.; Carret, S.; Poisson, J.-F. Org. Biomol. Chem. 2014, 12, 1875–1878. doi:10.1039/c3ob42352b |
| 55. | Ding, C.-H.; Chen, D.-D.; Luo, Z.-B.; Dai, L.-X.; Hou, X.-L. Synlett 2006, 1272–1274. doi:10.1055/s-2006-939076 |
| 1. | Patterson, A. W.; Ellman, J. A. J. Org. Chem. 2006, 71, 7110–7112. doi:10.1021/jo061160h |
| 25. | Aggarwal, V. K.; Barbero, N.; McGarrigle, E. M.; Mickle, G.; Navas, R.; Suárez, J. R.; Unthank, M. G.; Yar, M. Tetrahedron Lett. 2009, 50, 3482–3484. doi:10.1016/j.tetlet.2009.03.020 |
| 26. | Ellman, J. A.; Owens, T. D.; Tang, T. P. Acc. Chem. Res. 2002, 35, 984–995. doi:10.1021/ar020066u |
| 44. | Mancuso, A. J.; Huang, S.-L.; Swern, D. J. Org. Chem. 1978, 43, 2480–2482. doi:10.1021/jo00406a041 |
| 98. | Srinivasan, R.; Uttamchandani, M.; Yao, S. Q. Org. Lett. 2006, 8, 713–716. doi:10.1021/ol052895w |
| 99. | Chen, S.; Zhao, X.; Chen, J.; Chen, J.; Kuznetsova, L.; Wong, S. S.; Ojima, I. Bioconjugate Chem. 2010, 21, 979–987. doi:10.1021/bc9005656 |
| 49. | Carreira, E. M.; Du Bois, J. J. Am. Chem. Soc. 1995, 117, 8106–8125. doi:10.1021/ja00136a008 |
| 50. | Nishikawa, T.; Ino, A.; Isobe, M. Tetrahedron 1994, 50, 1449–1468. doi:10.1016/S0040-4020(01)80629-3 |
| 1. | Patterson, A. W.; Ellman, J. A. J. Org. Chem. 2006, 71, 7110–7112. doi:10.1021/jo061160h |
| 25. | Aggarwal, V. K.; Barbero, N.; McGarrigle, E. M.; Mickle, G.; Navas, R.; Suárez, J. R.; Unthank, M. G.; Yar, M. Tetrahedron Lett. 2009, 50, 3482–3484. doi:10.1016/j.tetlet.2009.03.020 |
| 26. | Ellman, J. A.; Owens, T. D.; Tang, T. P. Acc. Chem. Res. 2002, 35, 984–995. doi:10.1021/ar020066u |
| 51. | Ferreira, F.; Audouin, M.; Chemla, F. Chem. – Eur. J. 2005, 11, 5269–5278. doi:10.1002/chem.200500268 |
| 14. | Bauer, R. A.; DiBlasi, C. M.; Tan, D. S. Org. Lett. 2010, 12, 2084–2087. doi:10.1021/ol100574y |
| 13. | Chen, B.-L.; Wang, B.; Lin, G.-Q. J. Org. Chem. 2010, 75, 941–944. doi:10.1021/jo902424m |
| 14. | Bauer, R. A.; DiBlasi, C. M.; Tan, D. S. Org. Lett. 2010, 12, 2084–2087. doi:10.1021/ol100574y |
| 45. | Cogan, D. A.; Liu, G.; Ellman, J. Tetrahedron 1999, 55, 8883–8904. doi:10.1016/S0040-4020(99)00451-2 |
| 46. | Guijarro, D.; Pablo, Ó.; Yus, M. Tetrahedron Lett. 2009, 50, 5386–5388. doi:10.1016/j.tetlet.2009.07.044 |
| 47. | Verrier, C.; Carret, S.; Poisson, J.-F. Org. Biomol. Chem. 2014, 12, 1875–1878. doi:10.1039/c3ob42352b |
| 48. | Turcaud, S.; Berhal, F.; Royer, J. J. Org. Chem. 2007, 72, 7893–7897. doi:10.1021/jo071139w |
| 49. | Carreira, E. M.; Du Bois, J. J. Am. Chem. Soc. 1995, 117, 8106–8125. doi:10.1021/ja00136a008 |
| 50. | Nishikawa, T.; Ino, A.; Isobe, M. Tetrahedron 1994, 50, 1449–1468. doi:10.1016/S0040-4020(01)80629-3 |
| 52. | Feuvrie, C.; Blanchet, J.; Bonin, M.; Micouin, L. Org. Lett. 2004, 6, 2333–2336. doi:10.1021/ol049346v |
| 53. | Levin, V. V.; Dilman, A. D.; Belyakov, P. A.; Struchkova, M. I.; Tartakovsky, V. A. Eur. J. Org. Chem. 2008, 5226–5230. doi:10.1002/ejoc.200800820 |
| 54. | Moura-Letts, G.; DiBlasi, C. M.; Bauer, R. A.; Tan, D. S. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6745–6750. doi:10.1073/pnas.1015268108 |
| 55. | Ding, C.-H.; Chen, D.-D.; Luo, Z.-B.; Dai, L.-X.; Hou, X.-L. Synlett 2006, 1272–1274. doi:10.1055/s-2006-939076 |
| 108. | Panday, N.; Vasella, A. Helv. Chim. Acta 2000, 83, 1205–1208. doi:10.1002/1522-2675(20000607)83:6<1205::AID-HLCA1205>3.0.CO;2-L |
| 45. | Cogan, D. A.; Liu, G.; Ellman, J. Tetrahedron 1999, 55, 8883–8904. doi:10.1016/S0040-4020(99)00451-2 |
| 105. | Amantini, D.; Fringuelli, F.; Piermatti, O.; Pizzo, F.; Zunino, E.; Vaccaro, L. J. Org. Chem. 2005, 70, 6526–6529. doi:10.1021/jo0507845 |
| 106. | Huisgen, R. Angew. Chem., Int. Ed. Engl. 1963, 2, 565–598. doi:10.1002/anie.196305651 |
| 14. | Bauer, R. A.; DiBlasi, C. M.; Tan, D. S. Org. Lett. 2010, 12, 2084–2087. doi:10.1021/ol100574y |
| 48. | Turcaud, S.; Berhal, F.; Royer, J. J. Org. Chem. 2007, 72, 7893–7897. doi:10.1021/jo071139w |
| 52. | Feuvrie, C.; Blanchet, J.; Bonin, M.; Micouin, L. Org. Lett. 2004, 6, 2333–2336. doi:10.1021/ol049346v |
| 107. | Panday, N.; Meyyappan, M.; Vasella, A. Helv. Chim. Acta 2000, 83, 513–538. doi:10.1002/(SICI)1522-2675(20000315)83:3<513::AID-HLCA513>3.0.CO;2-1 |
| 1. | Patterson, A. W.; Ellman, J. A. J. Org. Chem. 2006, 71, 7110–7112. doi:10.1021/jo061160h |
| 2. | Verdoes, M.; Edgington, L. E.; Scheeren, F. A.; Leyva, M.; Blum, G.; Weiskopf, K.; Bachmann, M. H.; Ellman, J. A.; Bogyo, M. Chem. Biol. 2012, 19, 619–628. doi:10.1016/j.chembiol.2012.03.012 |
| 3. | Patterson, A. W.; Wood, W. J. L.; Hornsby, M.; Lesley, S.; Spraggon, G.; Ellman, J. A. J. Med. Chem. 2006, 49, 6298–6307. doi:10.1021/jm060701s |
| 4. | Wood, W. J. L.; Patterson, A. W.; Tsuruoka, H.; Jain, R. K.; Ellman, J. A. J. Am. Chem. Soc. 2005, 127, 15521–15527. doi:10.1021/ja0547230 |
| 5. | Inagaki, H.; Tsuruoka, H.; Hornsby, M.; Lesley, S. A.; Spraggon, G.; Ellman, J. A. J. Med. Chem. 2007, 50, 2693–2699. doi:10.1021/jm070111+ |
| 6. | Moss, N.; Xiong, Z.; Burke, M.; Cogan, D.; Gao, D. A.; Haverty, K.; Heim-Riether, A.; Hickey, E. R.; Nagaraja, R.; Netherton, M.; O’Shea, K.; Ramsden, P.; Schwartz, R.; Shih, D.-T.; Ward, Y.; Young, E.; Zhang, Q. Bioorg. Med. Chem. Lett. 2012, 22, 7189–7193. doi:10.1016/j.bmcl.2012.09.054 |
| 102. | Bitonti, A. J.; Casara, P. J.; McCann, P. P.; Bey, P. Biochem. J. 1987, 242, 69–74. doi:10.1042/bj2420069 |
| 103. | Metcalf, B. W.; Jung, M. α-Acetylenic Derivatives of Amines. U.S. Patent US4139563 A, Feb 13, 1979. |
| 104. | Metcalf, B. W.; Jung, M. α-Acetylenic Derivatives of α-Amino Acids. U.S. Patent US4182891 A, Jan 8, 1980. |
| 16. | Ye, L.; He, W.; Zhang, L. Angew. Chem., Int. Ed. 2011, 50, 3236–3239. doi:10.1002/anie.201007624 |
| 100. | Corrie, J. E. T.; Hlubucek, J. R.; Lowe, G. J. Chem. Soc., Perkin Trans. 1 1977, 1421–1425. doi:10.1039/p19770001421 |
| 101. | Koreeda, M.; Yang, W. Synlett 1994, 3, 201–203. doi:10.1055/s-1994-22794 |
| 11. | Deu, E.; Leyva, M. J.; Albrow, V. E.; Rice, M. J.; Ellman, J. A.; Bogyo, M. Chem. Biol. 2010, 17, 808–819. doi:10.1016/j.chembiol.2010.06.007 |
| 16. | Ye, L.; He, W.; Zhang, L. Angew. Chem., Int. Ed. 2011, 50, 3236–3239. doi:10.1002/anie.201007624 |
| 45. | Cogan, D. A.; Liu, G.; Ellman, J. Tetrahedron 1999, 55, 8883–8904. doi:10.1016/S0040-4020(99)00451-2 |
| 10. | Leyva, M. J.; DeGiacomo, F.; Kaltenbach, L. S.; Holcomb, J.; Zhang, N.; Gafni, J.; Park, H.; Lo, D. C.; Salvesen, G. S.; Ellerby, L. M.; Ellman, J. A. Chem. Biol. 2010, 17, 1189–1200. doi:10.1016/j.chembiol.2010.08.014 |
| 17. | Wünsch, M.; Senger, J.; Schultheisz, P.; Schwarzbich, S.; Michalek, C.; Klaß, M.; Goskowitz, S.; Borchert, P.; Jung, M.; Sewald, N. ChemMedChem, in press. |
| 8. | Brak, K.; Doyle, P. S.; McKerrow, J. H.; Ellman, J. A. J. Am. Chem. Soc. 2008, 130, 6404–6410. doi:10.1021/ja710254m |
| 9. | Brak, K.; Kerr, I. D.; Barrett, K. T.; Fuchi, N.; Debnath, M.; Ang, K.; Engel, J. C.; McKerrow, J. H.; Doyle, P. S.; Brinen, L. S.; Ellman, J. A. J. Med. Chem. 2010, 53, 1763–1773. doi:10.1021/jm901633v |
| 14. | Bauer, R. A.; DiBlasi, C. M.; Tan, D. S. Org. Lett. 2010, 12, 2084–2087. doi:10.1021/ol100574y |
| 1. | Patterson, A. W.; Ellman, J. A. J. Org. Chem. 2006, 71, 7110–7112. doi:10.1021/jo061160h |
| 45. | Cogan, D. A.; Liu, G.; Ellman, J. Tetrahedron 1999, 55, 8883–8904. doi:10.1016/S0040-4020(99)00451-2 |
| 46. | Guijarro, D.; Pablo, Ó.; Yus, M. Tetrahedron Lett. 2009, 50, 5386–5388. doi:10.1016/j.tetlet.2009.07.044 |
| 7. | Xu, H.-C.; Chowdhury, S.; Ellman, J. A. Nat. Protoc. 2013, 8, 2271–2280. doi:10.1038/nprot.2013.134 |
| 15. | Lee, S. I.; Park, S. Y.; Park, J. H.; Jung, I. G.; Choi, S. Y.; Chung, Y. K.; Lee, B. Y. J. Org. Chem. 2006, 71, 91–96. doi:10.1021/jo051685u |
| 13. | Chen, B.-L.; Wang, B.; Lin, G.-Q. J. Org. Chem. 2010, 75, 941–944. doi:10.1021/jo902424m |
| 54. | Moura-Letts, G.; DiBlasi, C. M.; Bauer, R. A.; Tan, D. S. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6745–6750. doi:10.1073/pnas.1015268108 |
| 55. | Ding, C.-H.; Chen, D.-D.; Luo, Z.-B.; Dai, L.-X.; Hou, X.-L. Synlett 2006, 1272–1274. doi:10.1055/s-2006-939076 |
| 6. | Moss, N.; Xiong, Z.; Burke, M.; Cogan, D.; Gao, D. A.; Haverty, K.; Heim-Riether, A.; Hickey, E. R.; Nagaraja, R.; Netherton, M.; O’Shea, K.; Ramsden, P.; Schwartz, R.; Shih, D.-T.; Ward, Y.; Young, E.; Zhang, Q. Bioorg. Med. Chem. Lett. 2012, 22, 7189–7193. doi:10.1016/j.bmcl.2012.09.054 |
| 12. | Jordan, S.; Starks, S. A.; Whatley, M. F.; Turlington, M. Org. Lett. 2015, 17, 4842–4845. doi:10.1021/acs.orglett.5b02408 |
| 45. | Cogan, D. A.; Liu, G.; Ellman, J. Tetrahedron 1999, 55, 8883–8904. doi:10.1016/S0040-4020(99)00451-2 |
| 11. | Deu, E.; Leyva, M. J.; Albrow, V. E.; Rice, M. J.; Ellman, J. A.; Bogyo, M. Chem. Biol. 2010, 17, 808–819. doi:10.1016/j.chembiol.2010.06.007 |
| 13. | Chen, B.-L.; Wang, B.; Lin, G.-Q. J. Org. Chem. 2010, 75, 941–944. doi:10.1021/jo902424m |
| 47. | Verrier, C.; Carret, S.; Poisson, J.-F. Org. Biomol. Chem. 2014, 12, 1875–1878. doi:10.1039/c3ob42352b |
| 10. | Leyva, M. J.; DeGiacomo, F.; Kaltenbach, L. S.; Holcomb, J.; Zhang, N.; Gafni, J.; Park, H.; Lo, D. C.; Salvesen, G. S.; Ellerby, L. M.; Ellman, J. A. Chem. Biol. 2010, 17, 1189–1200. doi:10.1016/j.chembiol.2010.08.014 |
| 53. | Levin, V. V.; Dilman, A. D.; Belyakov, P. A.; Struchkova, M. I.; Tartakovsky, V. A. Eur. J. Org. Chem. 2008, 5226–5230. doi:10.1002/ejoc.200800820 |
| 2. | Verdoes, M.; Edgington, L. E.; Scheeren, F. A.; Leyva, M.; Blum, G.; Weiskopf, K.; Bachmann, M. H.; Ellman, J. A.; Bogyo, M. Chem. Biol. 2012, 19, 619–628. doi:10.1016/j.chembiol.2012.03.012 |
| 9. | Brak, K.; Kerr, I. D.; Barrett, K. T.; Fuchi, N.; Debnath, M.; Ang, K.; Engel, J. C.; McKerrow, J. H.; Doyle, P. S.; Brinen, L. S.; Ellman, J. A. J. Med. Chem. 2010, 53, 1763–1773. doi:10.1021/jm901633v |
| 2. | Verdoes, M.; Edgington, L. E.; Scheeren, F. A.; Leyva, M.; Blum, G.; Weiskopf, K.; Bachmann, M. H.; Ellman, J. A.; Bogyo, M. Chem. Biol. 2012, 19, 619–628. doi:10.1016/j.chembiol.2012.03.012 |
| 14. | Bauer, R. A.; DiBlasi, C. M.; Tan, D. S. Org. Lett. 2010, 12, 2084–2087. doi:10.1021/ol100574y |
| 16. | Ye, L.; He, W.; Zhang, L. Angew. Chem., Int. Ed. 2011, 50, 3236–3239. doi:10.1002/anie.201007624 |
| 14. | Bauer, R. A.; DiBlasi, C. M.; Tan, D. S. Org. Lett. 2010, 12, 2084–2087. doi:10.1021/ol100574y |
| 15. | Lee, S. I.; Park, S. Y.; Park, J. H.; Jung, I. G.; Choi, S. Y.; Chung, Y. K.; Lee, B. Y. J. Org. Chem. 2006, 71, 91–96. doi:10.1021/jo051685u |
| 50. | Nishikawa, T.; Ino, A.; Isobe, M. Tetrahedron 1994, 50, 1449–1468. doi:10.1016/S0040-4020(01)80629-3 |
| 57. | Ernst, A.; Gobbi, L.; Vasella, A. Tetrahedron Lett. 1996, 37, 7959–7962. doi:10.1016/0040-4039(96)01838-2 |
| 21. | Kracker, O.; Góra, J.; Krzciuk-Gula, J.; Marion, A.; Neumann, B.; Stammler, H.-G.; Nieß, A.; Antes, I.; Latajka, R.; Sewald, N. Chem. – Eur. J. submitted. |
| 45. | Cogan, D. A.; Liu, G.; Ellman, J. Tetrahedron 1999, 55, 8883–8904. doi:10.1016/S0040-4020(99)00451-2 |
| 49. | Carreira, E. M.; Du Bois, J. J. Am. Chem. Soc. 1995, 117, 8106–8125. doi:10.1021/ja00136a008 |
| 50. | Nishikawa, T.; Ino, A.; Isobe, M. Tetrahedron 1994, 50, 1449–1468. doi:10.1016/S0040-4020(01)80629-3 |
| 56. | Alzeer, J.; Vasella, A. Helv. Chim. Acta 1995, 78, 177–193. doi:10.1002/hlca.19950780117 |
| 24. | Weix, D. J.; Ellman, J. A. Org. Synth. 2005, 82, 157–165. doi:10.1002/0471264229.os082.24 |
| 1. | Patterson, A. W.; Ellman, J. A. J. Org. Chem. 2006, 71, 7110–7112. doi:10.1021/jo061160h |
| 25. | Aggarwal, V. K.; Barbero, N.; McGarrigle, E. M.; Mickle, G.; Navas, R.; Suárez, J. R.; Unthank, M. G.; Yar, M. Tetrahedron Lett. 2009, 50, 3482–3484. doi:10.1016/j.tetlet.2009.03.020 |
| 26. | Ellman, J. A.; Owens, T. D.; Tang, T. P. Acc. Chem. Res. 2002, 35, 984–995. doi:10.1021/ar020066u |
| 22. | Ko, E.; Liu, J.; Perez, L. M.; Lu, G.; Schaefer, A.; Burgess, K. J. Am. Chem. Soc. 2011, 133, 462–477. doi:10.1021/ja1071916 |
| 23. | Temperini, A.; Capperucci, A.; Degl’Innocenti, A.; Terlizzi, R.; Tiecco, M. Tetrahedron Lett. 2010, 51, 4121–4124. doi:10.1016/j.tetlet.2010.05.143 |
| 67. | De Clercq, E. Med. Res. Rev. 2009, 29, 611–645. doi:10.1002/med.20153 |
| 68. | Prober, C. G. Adv. Exp. Med. Biol. 2004, 549, 9–12. doi:10.1007/978-1-4419-8993-2_3 |
| 69. | Heidelberger, C. Ann. N. Y. Acad. Sci. 1975, 255, 317–325. doi:10.1111/j.1749-6632.1975.tb29239.x |
| 70. | Carmine, A. A.; Brogden, R. N.; Heel, R. C.; Speight, T. M.; Avery, G. S. Drugs 1982, 23, 329–353. doi:10.2165/00003495-198223050-00001 |
| 7. | Xu, H.-C.; Chowdhury, S.; Ellman, J. A. Nat. Protoc. 2013, 8, 2271–2280. doi:10.1038/nprot.2013.134 |
| 71. | Corbett, J. W.; Ko, S. S.; Rodgers, J. D.; Gearhart, L. A.; Magnus, N. A.; Bacheler, L. T.; Diamond, S.; Jeffrey, S.; Klabe, R. M.; Cordova, B. C.; Garber, S.; Logue, K.; Trainor, G. L.; Anderson, P. S.; Erickson-Viitanen, S. K. J. Med. Chem. 2000, 43, 2019–2030. doi:10.1021/jm990580e |
| 72. | Maggiolo, F. J. Antimicrob. Chemother. 2009, 64, 910–928. doi:10.1093/jac/dkp334 |
| 73. | King, J.; Aberg, J. A. AIDS (London, U. K.) 2008, 22, 1709–1717. doi:10.1097/QAD.0b013e32830163ad |
| 19. | Angelo, N. G.; Arora, P. S. J. Am. Chem. Soc. 2005, 127, 17134–17135. doi:10.1021/ja056406z |
| 62. | Pollegioni, L.; Harris, C. M.; Molla, G.; Pilone, M. S.; Ghisla, S. FEBS Lett. 2001, 507, 323–326. doi:10.1016/S0014-5793(01)02983-0 |
| 63. | Asimakopoulou, A.; Panopoulos, P.; Chasapis, C. T.; Coletta, C.; Zhou, Z.; Cirino, G.; Giannis, A.; Szabo, C.; Spyroulias, G. A.; Papapetropoulos, A. Br. J. Pharmacol. 2013, 169, 922–932. doi:10.1111/bph.12171 |
| 64. | Shu, M.; Yu, R.; Zhang, Y.; Wang, J.; Yang, L.; Wang, L.; Lin, Z. Med. Chem. (Sharjah, United Arab Emirates) 2013, 9, 32–44. doi:10.2174/157340613804488350 |
| 21. | Kracker, O.; Góra, J.; Krzciuk-Gula, J.; Marion, A.; Neumann, B.; Stammler, H.-G.; Nieß, A.; Antes, I.; Latajka, R.; Sewald, N. Chem. – Eur. J. submitted. |
| 65. | Shah, P.; Westwell, A. D. J. Enzyme Inhib. Med. Chem. 2007, 22, 527–540. doi:10.1080/14756360701425014 |
| 66. | Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315–8359. doi:10.1021/acs.jmedchem.5b00258 |
| 17. | Wünsch, M.; Senger, J.; Schultheisz, P.; Schwarzbich, S.; Michalek, C.; Klaß, M.; Goskowitz, S.; Borchert, P.; Jung, M.; Sewald, N. ChemMedChem, in press. |
| 58. | Dierks, T.; Lecca, M. R.; Schmidt, B.; von Figura, K. FEBS Lett. 1998, 423, 61–65. doi:10.1016/S0014-5793(98)00065-9 |
| 59. | Rabuka, D.; Rush, J. S.; deHart, G. W.; Wu, P.; Bertozzi, C. R. Nat. Protoc. 2012, 7, 1052–1067. doi:10.1038/nprot.2012.045 |
| 18. | Nahrwold, M.; Bogner, T.; Eissler, S.; Verma, S.; Sewald, N. Org. Lett. 2010, 12, 1064–1067. doi:10.1021/ol1000473 |
| 19. | Angelo, N. G.; Arora, P. S. J. Am. Chem. Soc. 2005, 127, 17134–17135. doi:10.1021/ja056406z |
| 20. | Johansson, J. R.; Hermansson, E.; Nordén, B.; Kann, N.; Beke-Somfai, T. Eur. J. Org. Chem. 2014, 2703–2713. doi:10.1002/ejoc.201400018 |
| 21. | Kracker, O.; Góra, J.; Krzciuk-Gula, J.; Marion, A.; Neumann, B.; Stammler, H.-G.; Nieß, A.; Antes, I.; Latajka, R.; Sewald, N. Chem. – Eur. J. submitted. |
| 60. | Brewer, G. A., Jr.; Johnson, M. J. Appl. Microbiol. 1953, 1, 163–166. |
| 61. | Weigel, L. F.; Nitsche, C.; Graf, D.; Bartenschlager, R.; Klein, C. D. J. Med. Chem. 2015, 58, 7719–7733. doi:10.1021/acs.jmedchem.5b00612 |
© 2017 Wünsch et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)