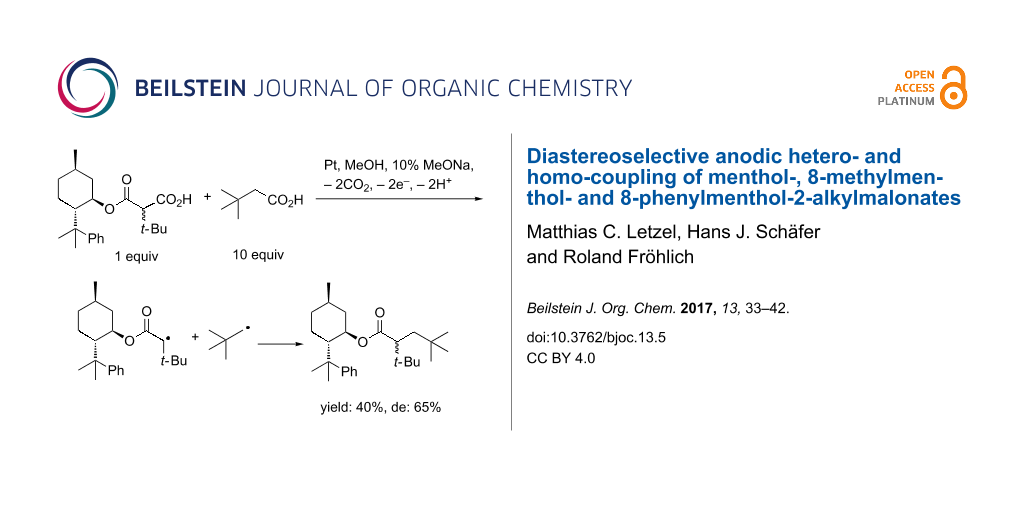
Abstract
Diastereoselective radical coupling was achieved with chiral auxiliaries. The radicals were generated by anodic decarboxylation of five malonic acid derivatives. These were prepared from benzyl malonates and four menthol auxiliaries. Coelectrolyses with 3,3-dimethylbutanoic acid in methanol at platinum electrodes in an undivided cell afforded hetero-coupling products in 22–69% yield with a diastereoselectivity ranging from 5 to 65% de. Electrolyses without a coacid led to diastereomeric homo-coupling products in 21–50% yield with ratios of diastereomers being 1.17:2.00:0.81 to 7.03:2.00. The stereochemistry of the new stereogenic centers was confirmed by X-ray structure analysis and 13C NMR data.

Graphical Abstract
Introduction
Intermolecular radical additions with high diastereoselectivity have been described for a number of cases [1-9]. There are much fewer reports on intermolecular diastereoselective radical coupling reactions [10-16]. There are some examples of high diastereoselectivity in the coupling of radicals generated from azo compounds [10,11], in intramolecular coupling of radicals, that are obtained by photochemical activation [12-14], and in intermolecular coupling of radicals generated by anodic decarboxylation of carboxylic acids [15,16].
At the anode radicals can be generated by anodic decarboxylation of carboxylic acids in molar quantities, in a simple procedure, unaffected by cage effects and in large diversity. For that reason, we were interested in exploring the structural influence of chiral auxiliaries (Figure 1) and the carboxylic acid on the diastereoselectivity in the anodic coupling of carboxylic acids. We report here on the diastereoselectivity found in the anodic hetero- and homo-coupling of menthol- (1)-, 8-methylmenthol- (2)-, 8-phenylmenthol- (3)-, and 8-p-anisylmenthol- (4)-2-alkylmalonates (Figure 1).
![[1860-5397-13-5-1]](/bjoc/content/figures/1860-5397-13-5-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Menthol auxiliaries 1–4 used in the following anodic coupling reactions.
Figure 1: Menthol auxiliaries 1–4 used in the following anodic coupling reactions.
Results and Discussion
Anodic hetero- and homo-coupling of carboxylates
The carboxylic acids 13a/b–18a/b for the Kolbe electrolyses were synthesized according to Scheme 1. The chiral auxiliary 1–4 is acylated with a benzylmalonyl chloride prepared from 5 and 6 and the benzyl group is subsequently removed by hydrogenation to afford the free carboxylic acids 13a/b–18a/b for the electrolyses.
![[1860-5397-13-5-i1]](/bjoc/content/inline/1860-5397-13-5-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of carboxylic acids 13a/b–18a/b.
Scheme 1: Synthesis of carboxylic acids 13a/b–18a/b.
Benzyl 2-isopropylmalonate (5) was prepared in high yield, using a method of Strating et al. [17], by deprotonation of the benzyl ester 19 with lithium diisopropylamide and quenching the enolate with carbon dioxide (Scheme 2a). Benzyl 2-tert-butylmalonate (6) was prepared in good yield using a method of Krapcho et al. [18], by double deprotonation of 3,3-dimethylbutyric acid (20) with LDA and quenching the dianion with benzyl chloroformate (Scheme 2b).
![[1860-5397-13-5-i2]](/bjoc/content/inline/1860-5397-13-5-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: (a) Preparation of benzyl 2-isopropylmalonate (5) and (b) preparation of benzyl 2-tert-butylmalonate (6).
Scheme 2: (a) Preparation of benzyl 2-isopropylmalonate (5) and (b) preparation of benzyl 2-tert-butylmalonat...
The benzyl malonates 5 and 6 are converted to the corresponding benzyl malonyl chlorides with thionyl chloride. To these at −40 °C the auxiliary menthol (1), 8-p-anisylmenthol (4) [19], 8-methylmenthol (2) [20] or 8-phenylmenthol (3) [21] was added in the presence of triethylamine, whereby the menthyl esters 7–12 were obtained in good yield after flash chromatography as a mixture of two diastereomers (Scheme 1).
The benzyl menthyl malonates 7–12 were hydrogenated under atmospheric pressure to afford the free carboxylic acids 13a/b–18a/b in high yield and purity (Scheme 1).
Anodic hetero-coupling of carboxylates
Carboxylates of aliphatic carboxylic acids are oxidized to carbonyloxy radicals at a platinum electrode at potentials being generally higher than 2.0 V (vs NHE). The carbonyloxy radicals undergo fast decarboxylation to alkyl radicals at or near the electrode surface [22]. The generation of radicals in a thin reaction layer at the electrode surface leads to high radical concentrations that favor bimolecular radical coupling. Electrolysis of equal carboxylic acids leads to a symmetrical dimer by homo-coupling of the radicals. Coelectrolysis of two different carboxylic acids affords unsymmetrical coupling products by hetero-coupling. In the latter case three different coupling products are obtained: two symmetrical dimers and one unsymmetrical coupling product, whose ratio is in most cases determined by the statistical coupling of the intermediate radicals. An excess of one coacid leads to mainly two products: the unsymmetrical coupling product and the dimer of the coacid used in excess.
The coelectrolysis (hetero-coupling) of carboxylic acids 13–17 with ten equivalents of 3,3-dimethylbutyric acid (20) afforded the hetero-coupling products 21–25 in 22–69% yield of two diastereomers, whereby one of the two diastereomers was formed in 5–65% diastereomeric excess (Scheme 3).
![[1860-5397-13-5-i3]](/bjoc/content/inline/1860-5397-13-5-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Coelectrolysis (hetero-coupling) of carboxylic acids 13–17 with 3,3-dimethylbutyric acid (20).
Scheme 3: Coelectrolysis (hetero-coupling) of carboxylic acids 13–17 with 3,3-dimethylbutyric acid (20).
Because of the low diastereomeric excess for compounds 21 and 22, the configuration of the major diastereomer was not determined. The configurations for the major diastereomers of 23–25 were obtained from crystal structure analyses and the correlation of the 13C NMR data.
According to an X-ray analysis the minor diastereomer 23b has the configuration (S) at the new formed stereogenic center (Figure 2) [23].
![[1860-5397-13-5-2]](/bjoc/content/figures/1860-5397-13-5-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Crystal structure of the minor diastereomer 23b.
Figure 2: Crystal structure of the minor diastereomer 23b.
The configuration of the new stereogenic centers in compounds 24 and 25 were assigned on the basis of a characteristic down field shift in the 13C NMR of the carbonyl and the methine carbon atom in the major diastereomer (Table 1).
The higher diastereomeric excess in compounds 24 and 25 obtained with 8-phenylmenthol (3) as auxiliary is attributed to an electronic interaction between the carbonyl and the phenyl group [24]. In order to enhance this interaction, the phenyl group was replaced for the more electron-rich anisyl group by applying 8-anisylmenthol (4) as chiral auxiliary. However, with acetic acid or 3,3-dimethylbutyric acid (20) as coacid no radical hetero-coupling product of carboxylic acid 18 was found, but polar compounds were obtained, whose structures were not determined.
To explain this failure, it is assumed that under the electrolysis conditions the electron-rich aromatic ring system is preferentially oxidized. Cyclic voltammetry of the malonates 15a/b, 16a/b and 18a/b supports this assumption. The cyclic voltammogram of 18a/b shows an oxidation peak at 1.75 V to 1.85 V (Figure 3).
![[1860-5397-13-5-3]](/bjoc/content/figures/1860-5397-13-5-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Cyclic voltammograms of the malonic derivatives 15a/b, 16a/b and 18a/b (scan rate: 500 mA/s, solvent: acetonitrile, supporting electrolyte: tetrabutylammonium perchlorate (TBAClO4), reference electrode: Ag/AgCl).
Figure 3: Cyclic voltammograms of the malonic derivatives 15a/b, 16a/b and 18a/b (scan rate: 500 mA/s, solven...
In the cyclic voltammogram of malonic acid derivative 16a/b a peak appears at a potential of 2.4 V (Figure 3). The malonic acid derivative 15a/b does not show any oxidation peak below 3 V (Figure 3). In this case the increase in current at 3 V is caused by the oxidation of the solvent. These results clearly attribute the peak at 1.75 to 1.85 V in carboxylic acid 18a/b to the oxidation of the anisyl group. This is also in accord with oxidation potentials from the literature [25], where the oxidation potential for anisole and toluene was determined to be 1.15 V and 1.35 V (vs Ag/Ag+), respectively.
In the Kolbe electrolysis a critical potential of 1.9 to 2.2 V (vs Ag/AgCl) has to be exceeded. At this potential the coverage of the electrode with carboxylate ions increases sharply, the oxygen evolution is inhibited, solvent oxidation is retarded and the Kolbe electrolysis is promoted [26].
If in the carboxylate an additional electrophore with a lower oxidation potential than the critical potential is present it is oxidized instead of the carboxylate group. This is the case for 18a/b. Even in the case of 16a/b and 17a/b a significant portion of polar products is found, indicating a partial oxidation of the phenyl group.
Anodic homo-coupling of carboxylates
The carboxylic acids 13a/b–16a/b have also been subjected to homo-coupling and the diastereomers 26a/b/c–29a/b/c were afforded in 50–21% yield (Scheme 4).
![[1860-5397-13-5-i4]](/bjoc/content/inline/1860-5397-13-5-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Homo-coupling of carboxylic acids 13a/b–16a/b to diesters 26a/b/c–29a/b/c (n.d.: not determined).
Scheme 4: Homo-coupling of carboxylic acids 13a/b–16a/b to diesters 26a/b/c–29a/b/c (n.d.: not determined).
In the homo-coupling reaction two identical radicals combine to form two stereogenic centers. Three diastereomers can be formed by re,re-, re,si- and si,si-coupling of the prostereogenic radical centers. Without facial selectivity they will be formed in a statistical ratio of 1:2:1. Facial selectivity is indicated by deviation from this statistical ratio. The selectivity for re,re-coupling vs re,si-coupling is given by the ratio 2 (a:b), the selectivity for re,si- vs si,si-coupling by b:2c (Table 2).
The absolute configuration of the major diastereomers 28a and 29a was determined by X-ray analysis (Figure 4 [23] and Figure 5 [23], hydrogen atoms are left out, except those formed at the new stereogenic centers). The new stereogenic centers in both cases have (2’R,3’R)-configuration. For the diastereomers 26b, 27b, 28b and 29b a set of 1H and 13C NMR signals can be observed for the protons and the carbon atoms of each half of the molecule. Based on this observation a (2’R,3’S)-configuration was assigned to 26b, 27b, 28b and 29b. For the diastereomers 26a/c, 27a/c, 28a and 29a only a single set of signals is obtained, because both halves of the molecule are chemically equivalent due to its C2-symmetry.
![[1860-5397-13-5-4]](/bjoc/content/figures/1860-5397-13-5-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Crystal structure of major diastereomer 28a.
Figure 4: Crystal structure of major diastereomer 28a.
![[1860-5397-13-5-5]](/bjoc/content/figures/1860-5397-13-5-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Crystal structure of major diastereomer 29a.
Figure 5: Crystal structure of major diastereomer 29a.
In Figure 4 and Figure 5 the C2-symmetry can be easily seen. From Figure 2, Figure 4 and Figure 5 the conformation of the radicals that undergo the coupling reaction can be deduced. Assuming a homolytic cleavage of the bond between C2’–C3’ in compound 23b (Figure 2) and converting the sp3 hybridized carbon atoms C2’ and C3’ into sp2 hybrids shows that the minor stereoisomer is formed by the shielding of the re-face through the tert-butyl group. Hence the major diastereomer 23a is formed by an attack via the less shielded si-face.
The same consideration for compound 28a (Figure 4) and compound 29a (Figure 5) with a cleavage of the C2’–C3’ bond in the succinic acid derivatives shows that the major diastereomer is formed via the si-face, which is less shielded by the tert-butyl or cumyl group.
Discussion
8-Phenylmenthol has been successfully used as a chiral auxiliary [27] to control the stereochemistry in Diels–Alder reactions [28], [2 + 2]- and [3 + 2]-cycloadditions and asymmetric ene reactions [29]; furthermore, it has been applied in 1,4-cuprate additions [30], Grignard additions [31], in supramolecular chemistry [32] and in alkylation of malonic acid derivatives [33]. In the literature, there are only a few examples where menthol auxiliaries have been used to control the stereochemistry in radical reactions [34].
In first experiments using (−)-menthol (1) as a chiral auxiliary we observed a low but nevertheless promising preference for one diastereomer, depending on the substituent R (Scheme 3). With the tert-butyl group in 22a/b a higher selectivity was observed than with the isopropyl group in 21a/b.
Still higher discrimination between both diastereomeric faces had been expected using a modified menthol with a phenyl group in 8-position, which is 8-phenylmenthol (Figure 6).
![[1860-5397-13-5-6]](/bjoc/content/figures/1860-5397-13-5-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Discrimination of diastereomeric faces in the menthol substituted radical A and in the 8-phenylmenthol substituted radical B.
Figure 6: Discrimination of diastereomeric faces in the menthol substituted radical A and in the 8-phenylment...
This hypothesis was proven to be accurate, obtaining an astonishingly high diastereoselectivity of 65% de for 25a/b with R = tert-butyl and 8-phenylmenthol (3) (Scheme 3). In the same coupling reaction using 2,5-dimethylpyrrolidine as auxiliary the analogous coupling product was obtained with a selectivity of 65% de as well [15]. The selectivity decreases to 38% de for 24a/b with R = isopropyl. When 8-methylmenthol (2) was used, 23a/b was obtained with a moderate selectivity of 27% de, but a good yield of 69%.
The facial selectivity for a single radical center can be calculated from the diastereomeric ratio obtained in the homo-coupling reactions, assuming that there is no simple diastereoselectivity favoring either the (2R,3R)/(2S,3S)- or the (2R,3S)-diastereomers (Table 3). In coupling reactions without auxiliary control, no significant preference for either d, l or meso had been observed [35], proving this precondition to be legitimate.
Table 3: Comparison of the diastereoselectivity in hetero- and homo-coupling reactions.
| Electrolysis | Homo-coupling | Hetero-couplinga |
|---|---|---|
| of compound |
facial diastereoselectivity
re:si |
diastereomeric ratio
R:S |
| 13a/b | 54.5:45.5b | 52.5:47.5b |
| 14a/b | 59.0:41.0b | 55.0:45.0b |
| 15a/b | 83.5:16.8 | 63.5:36.5 |
| 16a/b | 87.5:12.5 | 82.5:17.5 |
a3,3-Dimethylbutanoic acid was used as coacid in all cases. bThe absolute configuration of the major diastereomer has not been determined. Therefore, the facial stereoselectivity is assumed to have the same preference as in the cases where the configuration is known.
Comparing the stereoselectivity in the homo- and hetero-coupling reaction (Table 3), a slightly better selectivity is observed for the homo-coupling reaction. This is probably due to the bulkier coradical in the homo-coupling, compared with the 2,2-dimethylpropyl radical in the hetero-coupling reaction.
With auxiliaries like 2,5-dimethylpyrrolidine, 2,5-dimethoxymethylpyrrolidine or camphorsultam side products resulting from decarboxylation, disproportionation and further oxidation of the intermediate radical have been observed [35-37]. In contrast, hardly any side products of that kind were obtained using menthol related auxiliaries in anodic radical coupling reactions.
Products analogous to these of the radical homo-coupling above can be obtained by auxiliary controlled oxidative enolate coupling in high yield with good stereocontrol [38-40]. With the auxiliary controlled alkylation of enolates products being analogous to radical hetero-coupling can be synthesized [41,42]. However, both methods require costlier reaction conditions as inert gas, dry solvents and LDA as reagent, and are probably harder to perform in a large scale as the Kolbe electrolysis.
There is strong evidence that in the Kolbe electrolysis free radicals are involved in the coupling reaction. Coupling of absorbed radicals is ruled out by Eberson [43-45]. It was shown that the product ratio of cyanoalkyl radicals, which can yield C–C or C–N coupling products, does not differ in the Kolbe electrolysis from the ratio obtained by the coupling of photochemically produced cyanoalkyl radicals. Optically active carboxylates, which have the asymmetric center at C2 racemize in mixed Kolbe electrolyses [46,47]. This finding points to a free radical or its reversible adsorption at the electrode. However, the latter pathway was ruled out by experiments of Utley et al. [48,49], when comparing the coupling of unsaturated and saturated carboxylic acids and the effect of adsorption.
Ester 30a has been cleaved with lithium aluminium hydride in THF to 3 and 31a in 90% (Scheme 5) and with 30b (Scheme 5) and DIBAL in toluene 92% of 31b were obtained [50]. 30c could be cleaved with lithium aluminium hydride in THF to 83% 3 and 90% 31c [51]. However, to our great disappointment the successful cleavage with 30c could not be transferred to the structurally similar compounds 23a/b and 25a/b. Using the same reaction conditions as with 30a both with lithium aluminium hydride in THF and DIBAL in toluene with 23a/b and 25a/b the starting material was completely reisolated. Apparently the carbonyl group is so much shielded by the tert-butyl group and the auxiliary that the reducing agent is not able to reach the carbonyl group.
![[1860-5397-13-5-i5]](/bjoc/content/inline/1860-5397-13-5-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reductive cleavage of 30a–c to 8-phenylmenthol (3) and 31a–c.
Scheme 5: Reductive cleavage of 30a–c to 8-phenylmenthol (3) and 31a–c.
However, the phenyl group at C2 of the ester 30c causes lower yields (11%) of anodic coupling product, because it facilitates the oxidation of the intermediate radical and also lowers the diastereoselectivity significantly (de 39%) compared to the bulkier tert-butyl group with 65% de in 25a/b (Scheme 3) [51]. Therefore, the tert-butyl group appears to be important for a higher product yield and the diastereoselectivity of the coupling. On the other side this bulky group prohibits the cleavage with the so far successful reagents LiAlH4 and DIBAL. Therefore, in the future bulky chiral auxiliaries must be developed that can be partially disassembled before their cleavage and slim cleavage reagents have to be found and explored.
Conclusion
We developed an efficient procedure for the synthesis of menthyl 2-tert-butyl and 2-isopropyl-4,4-dimethylpentanoates (21a/b–25a/b) with a chiral centre at C2 of the acid component. For that purpose, malonic half-esters with substituted menthols as chiral auxiliaries were prepared. The half-esters were electrochemically decarboxylated to alkyl radicals. In the presence of an excess of 3,3-dimethylbutylbutanoic acid that decarboxylated to a 2,2-dimethylpropyl radical preferentially the hetero-coupling products 21a/b–25a/b were obtained in up to 69% yield and diastereomeric ratios of up to 82.5:17.5 induced by the menthyl auxiliaries. However, the removal of the auxiliary in the esters is not yet resolved. Without a coacid succinic acid derivatives 26a/b/c–29a/b/c were obtained by dimerization of the half-ester radicals with yields of up to 50% and a facial diastereoselectivity of up to 87.5:12.5.
Products that are similar to these of the anodic coupling can be obtained by auxiliary controlled oxidative enolate coupling. However, this method often requires more expensive reagents, functional groups are frequently more sensible to the used reagents and an up-scale is more difficult to perform.
On the other side anodic coupling of sodium carboxylates is easy and simply to do. Methanol can be mostly used as solvent, the alkali salt of the carboxylic acid serves at the same time as substrate and supporting electrolyte. The required equipment is simple, inexpensive and readily available. An undivided beaker-type cell is sufficient; electrolysis can be performed current-controlled with a cheap power supply and an up-scale of the reaction is mostly easy.
Experimental
General remarks
Optical rotations: Perkin-Elmer polarimeter 241, specific rotations in grad·cm3·dm−1·g−1, c in g/100 mL. FTIR: Bruker IFS 28. 1H, 13C NMR: Bruker WM 300. X-ray structures: Data sets were collected with an Enraf Nonius CAD4 diffractometer. Programs used: data reduction MolEN, structure solution SHELXS-86, structure refinement SHELXL-93, graphics SCHAKAL-92. MS: Finnigan MAT, Varian Saturn II (ion trap) with capillary column HP 5 (25 m, 0.2 mm i.d., 0.33 μm film); DCI with ammonia as reactant gas. GLC: Hewlett-Packard HP 5890 Series II, Shimadzu GC-14A, Carlo Erba 4200; quartz capillary columns: Macherey and Nagel FS-HP1 (25 m, 0.32 mm i.d., 0.25 μm film), AT 35 (2.5 m, 0.32 mm i.d., 0.25 μm film). Preparative scale electrolyses: Undivided beaker-type glass cell, double walled for constant temperature control, capacity: 3 mL (micro beaker-type cell). Platinum foils serving as anode and cathode materials (platinum foils: 1 × 1 cm). Convection was achieved by magnetic stirring. Current source: Heinzinger galvanostat – potentiostat HN 600–600 and TNs 300–1500.
General procedure for the Kolbe electrolysis
Hetero-coupling reaction: 0.4 mmol of the auxiliary modified carboxylic acid and 4 mmol of the coacid were dissolved in 2.2 mL of a 0.1 N methanolic solution of sodium methanolate (5% neutralisation) and transferred into the micro-beaker-type cell. Dry methanol was added to a total volume of 3 mL, which was electrolyzed maintaining a current density of 400 mA/cm2 and the temperature of the solution in the range of 5–10 °C with a coolant of −25 °C. When the pH of the solution reached 8–9 the electrolysis was stopped, the mixture was poured into 30 mL of water and extracted with ether (5 × 20 mL). The combined organic layers were washed with saturated sodium hydrogen carbonate (2 × 20 mL) and dried over MgSO4. After evaporation of the solvent in the vacuum the product was purified by flash chromatography.
Homo-coupling reaction: 1.6 mmol of the auxiliary modified carboxylic acid was dissolved in 1.6 mL of a 0.1 N methanol solution of sodium methoxide (5% neutralization) and transferred into the micro-beaker-type cell. Dry methanol was added to a total volume of 3 mL, which was electrolyzed maintaining a current density of 400 mA/cm2 and the temperature of the solution in the range of 5–10 °C with a coolant of −25 °C. The further work-up follows the procedure given for the hetero-coupling reaction.
General procedure for the preparation of the menthol esters: To a solution of 4.3 mmol of monobenzyl malonate 5 or 6 in 20 mL dry methylene chloride 0.8 mL thionyl chloride (1.3 g, 10.8 mmol) were added and the mixture was refluxed for 3 h. After evaporation of the solvent and the unreacted thionyl chloride in vacuum, the remaining yellow oil was dissolved in 20 mL methylene chloride without further purification and cooled to −40 °C. Under an argon atmosphere 2.81 mmol auxiliary, dissolved in 10 mL dry methylene chloride and 10 mL dry triethylamine, were added slowly to the carboxylic acid chloride at −40 °C. After the complete addition, the mixture was allowed to warm up to rt and stirred for 20 h. Then 20 mL of water were added and the mixture was extracted with methylene chloride (4 × 20 mL). The combined organic layers were washed successively with 2 N HCl (3 × 20 mL), saturated sodium hydrogen carbonate (2 × 20 mL) and brine (20 mL) and dried (MgSO4). After evaporation of the solvent the product was purified by flash chromatography.
General procedure for the hydrogenation of the benzyl esters: To a solution of 6.4 mmol benzyl ester in 100 mL of methanol 800 mg of the palladium catalyst (10% Pd on carbon) were added. Hydrogenation was performed at atmospheric pressure until no further hydrogen was consumed. After filtration through celite the solvent was evaporated in vacuum. The residue was dissolved in a small amount of diethyl ether, filtrated through celite and dried with MgSO4. After evaporation of the solvent the free carboxylic acid was obtained with high purity as a sticky oil.
The preparation of compounds 5–29, their spectroscopic data and their elemental analyses are reported in Supporting Information File 1.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 360.0 KB | Download |
References
-
Porter, N. A.; Giese, B.; Curran, D. P. Acc. Chem. Res. 1991, 24, 296–304. doi:10.1021/ar00010a003
Return to citation in text: [1] -
Smadja, W. Synlett 1994, 1–26. doi:10.1055/s-1994-22728
Return to citation in text: [1] -
Curran, D. P.; Porter, N. A.; Giese, B. Stereochemistry of Radical Reactions; VCH: Weinheim, 1996.
Return to citation in text: [1] -
Sibi, M. P.; Porter, N. A. Acc. Chem. Res. 1999, 32, 163–171. doi:10.1021/ar9600547
Return to citation in text: [1] -
Sibi, M. P.; Manyem, S.; Zimmerman, J. Chem. Rev. 2003, 103, 3263–3296. doi:10.1021/cr020044l
Return to citation in text: [1] -
Srikanth, G. S. C.; Castle, S. L. Tetrahedron 2005, 61, 10377–10441. doi:10.1016/j.tet.2005.07.077
Return to citation in text: [1] -
Subramanian, H.; Landais, Y.; Sibi, M. P. Radical Addition Reactions. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P.; Molander, G. A., Eds.; Elsevier, 2014; Vol. 4, pp 699–741.
Return to citation in text: [1] -
Yang, Y.-H.; Sibi, M. P. Stereoselective Radical Reactions. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu, C.; Studer, A., Eds.; John Wiley and Sons, 2012; Vol. 2, pp 655–691.
Return to citation in text: [1] -
Guindon, Y.; Godin, F.; Mochirian, P. A.; Prevost, M. Selected diastereoselective reactions: free radical additions and cyclizations. In Comprehensive Chirality; Carreira, E. M.; Yamamoto, H., Eds.; Elsevier, 2012; Vol. 2, pp 472–503. doi:10.1016/B978-0-08-095167-6.00215-9
Return to citation in text: [1] -
Eichin, K.-H.; Beckhaus, H.-D.; Hellmann, S.; Fritz, H.; Peters, E.-M.; Peters, K.; von Schnering, H.-G.; Rüchardt, C. Chem. Ber. 1983, 116, 1787–1821. doi:10.1002/cber.19831160509
Return to citation in text: [1] [2] -
Peyman, A.; Beckhaus, H.-D.; Kramer, D.; Peters, K.; von Schnering, H.-G.; Rüchardt, C. Chem. Ber. 1991, 124, 1989–1996. doi:10.1002/cber.19911240918
Return to citation in text: [1] [2] -
Wessig, P.; Wettstein, P.; Giese, B.; Neuburger, M.; Zehnder, M. Helv. Chim. Acta 1994, 77, 829–837. doi:10.1002/hlca.19940770322
Return to citation in text: [1] [2] -
Wessig, P.; Lindemann, U.; Neuburger, M.; Zehnder, M. J. Inf. Rec. 1996, 23, 19–21.
Return to citation in text: [1] [2] -
Lomölder, R.; Schäfer, H. J. Angew. Chem. 1987, 99, 1282–1283. doi:10.1002/ange.19870991211
Angew. Chem., Int. Ed. Engl. 1987, 26, 1253–1254. doi:10.1002/anie.198712531
Return to citation in text: [1] [2] -
Klotz-Berendes, B.; Schäfer, H. J.; Grehl, M.; Fröhlich, R. Angew. Chem. 1995, 107, 218–220. doi:10.1002/ange.19951070217
Angew. Chem., Int. Ed. Engl. 1995, 34, 189–191. doi:10.1002/anie.199501891
Return to citation in text: [1] [2] [3] -
Schäfer, H. J. In Electroorganic Chemistry, 5th ed.; Hammerich, O.; Speiser, B., Eds.; CRC Press, 2016; pp 705–773.
Return to citation in text: [1] [2] -
Reiffers, S.; Wynberg, H.; Strating, J. Tetrahedron Lett. 1971, 32, 3001–3004. doi:10.1016/S0040-4039(01)97075-3
Return to citation in text: [1] -
Krapcho, A. P.; Jahngen, E. G. E., Jr.; Kashdan, D. S. Tetrahedron Lett. 1974, 15, 2721–2723. doi:10.1016/S0040-4039(01)91723-X
Return to citation in text: [1] -
8-p-Anisylmenthol (4) was synthesized following parts of the procedures given in [20,21]. The diastereomeric menthols were separated by HPLC.
Return to citation in text: [1] -
Buschmann, H.; Scharf, H. D. Synthesis 1988, 827–830. doi:10.1055/s-1988-27724
Return to citation in text: [1] [2] -
Červinka, O.; Svatoš, A.; Masojídková, M. Collect. Czech. Chem. Commun. 1990, 55, 491–498. doi:10.1135/cccc19900491
Return to citation in text: [1] [2] -
Schäfer, H. J. Top. Curr. Chem. 1990, 152, 92–151.
Return to citation in text: [1] -
Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC-101327. Copies of the data can be obtained free of charge on application to The Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: int. code +44(1223)336-033, e-mail: deposit@ccdc.cam.ac.uk].
Return to citation in text: [1] [2] [3] -
Johnes, G. B.; Chapman, B. J. Synthesis 1995, 475–497. doi:10.1055/s-1995-3957
Return to citation in text: [1] -
Effenberger, F.; Kottmann, K. Tetrahedron 1985, 41, 4171–4182. doi:10.1016/S0040-4020(01)97192-3
Return to citation in text: [1] -
Dickinson, T.; Wynne-Jones, W. F. K. Trans. Faraday Soc. 1962, 58, 382–387. doi:10.1039/TF9625800382
Return to citation in text: [1] -
Kipphardt, H.; Enders, D. Kontakte (Darmstadt) 1985, 2, 37–45.
Return to citation in text: [1] -
Bednarski, M.; Danishefsky, S. J. Am. Chem. Soc. 1986, 108, 7060–7067. doi:10.1021/ja00282a035
Return to citation in text: [1] -
Whitesell, J. K.; Bhattacharya, A.; Buchanan, C. M.; Chen, H. H.; Deyo, D.; James, D.; Liu, C.-L.; Minton, M. A. Tetrahedron 1986, 42, 2993–3001. doi:10.1016/S0040-4020(01)90590-3
Return to citation in text: [1] -
Oppolzer, W.; Löhner, H. J. Helv. Chim. Acta 1981, 64, 2808–2811. doi:10.1002/hlca.19810640842
Return to citation in text: [1] -
Comins, D. L.; Guerra-Weltzien, L. Tetrahedron Lett. 1996, 37, 3807–3810. doi:10.1016/0040-4039(96)00709-5
Return to citation in text: [1] -
Siering, C.; Grimme, S.; Waldvogel, S. R. Chem. – Eur. J. 2005, 11, 1877–1888. doi:10.1002/chem.200401002
Return to citation in text: [1] -
Ihara, M.; Takahashi, M.; Taniguchi, N.; Yagui, K.; Niitsuma, H.; Fukumoto, K. J. Chem. Soc., Perkin Trans. 1 1991, 525–535. doi:10.1039/P19910000525
Return to citation in text: [1] -
Zhang, A.; Mohan, R. M.; Cook, L.; Kazanis, S.; Peisach, D.; Foxman, B. M.; Snider, B. B. J. Org. Chem. 1993, 58, 7640–7651. doi:10.1021/jo00079a006
Return to citation in text: [1] -
Klotz-Berendes, B. Studien zur diastereoselektiven Kupplung anodisch erzeugter Radikale. Ph.D. Thesis, Universität Münster, 1994.
Return to citation in text: [1] [2] -
Letzel, M., Auxiliarkontrollierte dimeriserung von 2-tert-butylmalonsäureamid zu 2,3-di-tert-butylbernsteinsäurederivaten durch Kolbe elektrolyse. Master Thesis, Universität Münster, 1994.
Return to citation in text: [1] -
Letzel, M. Studien zur diastereoselektiven Kupplung anodisch erzeugter Radikale in Hetero- und Homokupplungen. Ph.D. Thesis, Universität Münster, 1997.
Return to citation in text: [1] -
Porter, N. A.; Su, Q.; Harp, J. J.; Rosenstein, I. J.; McPhail, A. T. Tetrahedron Lett. 1993, 34, 4457–4460. doi:10.1016/0040-4039(93)88058-Q
Return to citation in text: [1] -
Langer, T.; Illich, M.; Helmchen, G. Synlett 1996, 1137–1139. doi:10.1055/s-1996-5668
Return to citation in text: [1] -
Langer, T.; Illich, M.; Helmchen, G. Tetrahedron Lett. 1995, 36, 4409–4412. doi:10.1016/0040-4039(95)00786-C
Return to citation in text: [1] -
Evans, D. A.; Takacs, J. M. Tetrahedron Lett. 1980, 21, 4233–4236. doi:10.1016/S0040-4039(00)92870-3
Return to citation in text: [1] -
Evans, D. A.; Ennis, M. D.; Mathre, D. J. J. Am. Chem. Soc. 1982, 104, 1737–1739. doi:10.1021/ja00370a050
Return to citation in text: [1] -
Eberson, L. Electrochim. Acta 1967, 12, 1473–1478. doi:10.1016/0013-4686(67)80061-6
Return to citation in text: [1] -
Eberson, L.; Gränse, S.; Olofsson, B. Acta Chem. Scand. 1968, 22, 2462–2470. doi:10.3891/acta.chem.scand.22-2462
Return to citation in text: [1] -
Eberson, L. Acta Chem. Scand. 1963, 17, 2004–2018. doi:10.3891/acta.chem.scand.17-2004
Return to citation in text: [1] -
Eberson, L.; Ryde-Petterson, G. Acta Chem. Scand. 1973, 27, 1159–1161. doi:10.3891/acta.chem.scand.27-1159
Return to citation in text: [1] -
Eberson, L.; Nyberg, K.; Servin, R. Acta Chem. Scand., Ser. B 1976, 30, 906–907. doi:10.3891/acta.chem.scand.30b-0906
Return to citation in text: [1] -
Hawkes, G. E.; Utley, J. H. P.; Yates, G. B. J. Chem. Soc., Perkin Trans. 2 1976, 1709–1716. doi:10.1039/p29760001709
Return to citation in text: [1] -
Utley, J. H. P.; Yates, G. B. J. Chem. Soc., Perkin Trans. 2 1978, 395–400. doi:10.1039/p29780000395
Return to citation in text: [1] -
Solladié-Cavallo, A.; Csaky, A. G.; Gantz, I.; Suffert, J. J. Org. Chem. 1994, 59, 5343–5346. doi:10.1021/jo00097a041
Return to citation in text: [1] -
Nguyen, P. Q., Master Thesis, University of Münster, 1998, p. 39. The experimental procedure for the conversion of 30c into 31c and 3 is given in Supporting Information File 1.
Return to citation in text: [1] [2]
| 35. | Klotz-Berendes, B. Studien zur diastereoselektiven Kupplung anodisch erzeugter Radikale. Ph.D. Thesis, Universität Münster, 1994. |
| 36. | Letzel, M., Auxiliarkontrollierte dimeriserung von 2-tert-butylmalonsäureamid zu 2,3-di-tert-butylbernsteinsäurederivaten durch Kolbe elektrolyse. Master Thesis, Universität Münster, 1994. |
| 37. | Letzel, M. Studien zur diastereoselektiven Kupplung anodisch erzeugter Radikale in Hetero- und Homokupplungen. Ph.D. Thesis, Universität Münster, 1997. |
| 38. | Porter, N. A.; Su, Q.; Harp, J. J.; Rosenstein, I. J.; McPhail, A. T. Tetrahedron Lett. 1993, 34, 4457–4460. doi:10.1016/0040-4039(93)88058-Q |
| 39. | Langer, T.; Illich, M.; Helmchen, G. Synlett 1996, 1137–1139. doi:10.1055/s-1996-5668 |
| 40. | Langer, T.; Illich, M.; Helmchen, G. Tetrahedron Lett. 1995, 36, 4409–4412. doi:10.1016/0040-4039(95)00786-C |
| 41. | Evans, D. A.; Takacs, J. M. Tetrahedron Lett. 1980, 21, 4233–4236. doi:10.1016/S0040-4039(00)92870-3 |
| 42. | Evans, D. A.; Ennis, M. D.; Mathre, D. J. J. Am. Chem. Soc. 1982, 104, 1737–1739. doi:10.1021/ja00370a050 |
| 1. | Porter, N. A.; Giese, B.; Curran, D. P. Acc. Chem. Res. 1991, 24, 296–304. doi:10.1021/ar00010a003 |
| 2. | Smadja, W. Synlett 1994, 1–26. doi:10.1055/s-1994-22728 |
| 3. | Curran, D. P.; Porter, N. A.; Giese, B. Stereochemistry of Radical Reactions; VCH: Weinheim, 1996. |
| 4. | Sibi, M. P.; Porter, N. A. Acc. Chem. Res. 1999, 32, 163–171. doi:10.1021/ar9600547 |
| 5. | Sibi, M. P.; Manyem, S.; Zimmerman, J. Chem. Rev. 2003, 103, 3263–3296. doi:10.1021/cr020044l |
| 6. | Srikanth, G. S. C.; Castle, S. L. Tetrahedron 2005, 61, 10377–10441. doi:10.1016/j.tet.2005.07.077 |
| 7. | Subramanian, H.; Landais, Y.; Sibi, M. P. Radical Addition Reactions. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P.; Molander, G. A., Eds.; Elsevier, 2014; Vol. 4, pp 699–741. |
| 8. | Yang, Y.-H.; Sibi, M. P. Stereoselective Radical Reactions. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu, C.; Studer, A., Eds.; John Wiley and Sons, 2012; Vol. 2, pp 655–691. |
| 9. | Guindon, Y.; Godin, F.; Mochirian, P. A.; Prevost, M. Selected diastereoselective reactions: free radical additions and cyclizations. In Comprehensive Chirality; Carreira, E. M.; Yamamoto, H., Eds.; Elsevier, 2012; Vol. 2, pp 472–503. doi:10.1016/B978-0-08-095167-6.00215-9 |
| 15. |
Klotz-Berendes, B.; Schäfer, H. J.; Grehl, M.; Fröhlich, R. Angew. Chem. 1995, 107, 218–220. doi:10.1002/ange.19951070217
Angew. Chem., Int. Ed. Engl. 1995, 34, 189–191. doi:10.1002/anie.199501891 |
| 16. | Schäfer, H. J. In Electroorganic Chemistry, 5th ed.; Hammerich, O.; Speiser, B., Eds.; CRC Press, 2016; pp 705–773. |
| 26. | Dickinson, T.; Wynne-Jones, W. F. K. Trans. Faraday Soc. 1962, 58, 382–387. doi:10.1039/TF9625800382 |
| 20. | Buschmann, H.; Scharf, H. D. Synthesis 1988, 827–830. doi:10.1055/s-1988-27724 |
| 21. | Červinka, O.; Svatoš, A.; Masojídková, M. Collect. Czech. Chem. Commun. 1990, 55, 491–498. doi:10.1135/cccc19900491 |
| 12. | Wessig, P.; Wettstein, P.; Giese, B.; Neuburger, M.; Zehnder, M. Helv. Chim. Acta 1994, 77, 829–837. doi:10.1002/hlca.19940770322 |
| 13. | Wessig, P.; Lindemann, U.; Neuburger, M.; Zehnder, M. J. Inf. Rec. 1996, 23, 19–21. |
| 14. |
Lomölder, R.; Schäfer, H. J. Angew. Chem. 1987, 99, 1282–1283. doi:10.1002/ange.19870991211
Angew. Chem., Int. Ed. Engl. 1987, 26, 1253–1254. doi:10.1002/anie.198712531 |
| 23. | Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC-101327. Copies of the data can be obtained free of charge on application to The Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: int. code +44(1223)336-033, e-mail: deposit@ccdc.cam.ac.uk]. |
| 10. | Eichin, K.-H.; Beckhaus, H.-D.; Hellmann, S.; Fritz, H.; Peters, E.-M.; Peters, K.; von Schnering, H.-G.; Rüchardt, C. Chem. Ber. 1983, 116, 1787–1821. doi:10.1002/cber.19831160509 |
| 11. | Peyman, A.; Beckhaus, H.-D.; Kramer, D.; Peters, K.; von Schnering, H.-G.; Rüchardt, C. Chem. Ber. 1991, 124, 1989–1996. doi:10.1002/cber.19911240918 |
| 24. | Johnes, G. B.; Chapman, B. J. Synthesis 1995, 475–497. doi:10.1055/s-1995-3957 |
| 51. | Nguyen, P. Q., Master Thesis, University of Münster, 1998, p. 39. The experimental procedure for the conversion of 30c into 31c and 3 is given in Supporting Information File 1. |
| 10. | Eichin, K.-H.; Beckhaus, H.-D.; Hellmann, S.; Fritz, H.; Peters, E.-M.; Peters, K.; von Schnering, H.-G.; Rüchardt, C. Chem. Ber. 1983, 116, 1787–1821. doi:10.1002/cber.19831160509 |
| 11. | Peyman, A.; Beckhaus, H.-D.; Kramer, D.; Peters, K.; von Schnering, H.-G.; Rüchardt, C. Chem. Ber. 1991, 124, 1989–1996. doi:10.1002/cber.19911240918 |
| 12. | Wessig, P.; Wettstein, P.; Giese, B.; Neuburger, M.; Zehnder, M. Helv. Chim. Acta 1994, 77, 829–837. doi:10.1002/hlca.19940770322 |
| 13. | Wessig, P.; Lindemann, U.; Neuburger, M.; Zehnder, M. J. Inf. Rec. 1996, 23, 19–21. |
| 14. |
Lomölder, R.; Schäfer, H. J. Angew. Chem. 1987, 99, 1282–1283. doi:10.1002/ange.19870991211
Angew. Chem., Int. Ed. Engl. 1987, 26, 1253–1254. doi:10.1002/anie.198712531 |
| 15. |
Klotz-Berendes, B.; Schäfer, H. J.; Grehl, M.; Fröhlich, R. Angew. Chem. 1995, 107, 218–220. doi:10.1002/ange.19951070217
Angew. Chem., Int. Ed. Engl. 1995, 34, 189–191. doi:10.1002/anie.199501891 |
| 16. | Schäfer, H. J. In Electroorganic Chemistry, 5th ed.; Hammerich, O.; Speiser, B., Eds.; CRC Press, 2016; pp 705–773. |
| 25. | Effenberger, F.; Kottmann, K. Tetrahedron 1985, 41, 4171–4182. doi:10.1016/S0040-4020(01)97192-3 |
| 51. | Nguyen, P. Q., Master Thesis, University of Münster, 1998, p. 39. The experimental procedure for the conversion of 30c into 31c and 3 is given in Supporting Information File 1. |
| 20. | Buschmann, H.; Scharf, H. D. Synthesis 1988, 827–830. doi:10.1055/s-1988-27724 |
| 48. | Hawkes, G. E.; Utley, J. H. P.; Yates, G. B. J. Chem. Soc., Perkin Trans. 2 1976, 1709–1716. doi:10.1039/p29760001709 |
| 49. | Utley, J. H. P.; Yates, G. B. J. Chem. Soc., Perkin Trans. 2 1978, 395–400. doi:10.1039/p29780000395 |
| 19. | 8-p-Anisylmenthol (4) was synthesized following parts of the procedures given in [20,21]. The diastereomeric menthols were separated by HPLC. |
| 23. | Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC-101327. Copies of the data can be obtained free of charge on application to The Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: int. code +44(1223)336-033, e-mail: deposit@ccdc.cam.ac.uk]. |
| 50. | Solladié-Cavallo, A.; Csaky, A. G.; Gantz, I.; Suffert, J. J. Org. Chem. 1994, 59, 5343–5346. doi:10.1021/jo00097a041 |
| 18. | Krapcho, A. P.; Jahngen, E. G. E., Jr.; Kashdan, D. S. Tetrahedron Lett. 1974, 15, 2721–2723. doi:10.1016/S0040-4039(01)91723-X |
| 43. | Eberson, L. Electrochim. Acta 1967, 12, 1473–1478. doi:10.1016/0013-4686(67)80061-6 |
| 44. | Eberson, L.; Gränse, S.; Olofsson, B. Acta Chem. Scand. 1968, 22, 2462–2470. doi:10.3891/acta.chem.scand.22-2462 |
| 45. | Eberson, L. Acta Chem. Scand. 1963, 17, 2004–2018. doi:10.3891/acta.chem.scand.17-2004 |
| 17. | Reiffers, S.; Wynberg, H.; Strating, J. Tetrahedron Lett. 1971, 32, 3001–3004. doi:10.1016/S0040-4039(01)97075-3 |
| 21. | Červinka, O.; Svatoš, A.; Masojídková, M. Collect. Czech. Chem. Commun. 1990, 55, 491–498. doi:10.1135/cccc19900491 |
| 46. | Eberson, L.; Ryde-Petterson, G. Acta Chem. Scand. 1973, 27, 1159–1161. doi:10.3891/acta.chem.scand.27-1159 |
| 47. | Eberson, L.; Nyberg, K.; Servin, R. Acta Chem. Scand., Ser. B 1976, 30, 906–907. doi:10.3891/acta.chem.scand.30b-0906 |
| 28. | Bednarski, M.; Danishefsky, S. J. Am. Chem. Soc. 1986, 108, 7060–7067. doi:10.1021/ja00282a035 |
| 23. | Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC-101327. Copies of the data can be obtained free of charge on application to The Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: int. code +44(1223)336-033, e-mail: deposit@ccdc.cam.ac.uk]. |
| 15. |
Klotz-Berendes, B.; Schäfer, H. J.; Grehl, M.; Fröhlich, R. Angew. Chem. 1995, 107, 218–220. doi:10.1002/ange.19951070217
Angew. Chem., Int. Ed. Engl. 1995, 34, 189–191. doi:10.1002/anie.199501891 |
| 35. | Klotz-Berendes, B. Studien zur diastereoselektiven Kupplung anodisch erzeugter Radikale. Ph.D. Thesis, Universität Münster, 1994. |
| 33. | Ihara, M.; Takahashi, M.; Taniguchi, N.; Yagui, K.; Niitsuma, H.; Fukumoto, K. J. Chem. Soc., Perkin Trans. 1 1991, 525–535. doi:10.1039/P19910000525 |
| 34. | Zhang, A.; Mohan, R. M.; Cook, L.; Kazanis, S.; Peisach, D.; Foxman, B. M.; Snider, B. B. J. Org. Chem. 1993, 58, 7640–7651. doi:10.1021/jo00079a006 |
| 31. | Comins, D. L.; Guerra-Weltzien, L. Tetrahedron Lett. 1996, 37, 3807–3810. doi:10.1016/0040-4039(96)00709-5 |
| 32. | Siering, C.; Grimme, S.; Waldvogel, S. R. Chem. – Eur. J. 2005, 11, 1877–1888. doi:10.1002/chem.200401002 |
| 29. | Whitesell, J. K.; Bhattacharya, A.; Buchanan, C. M.; Chen, H. H.; Deyo, D.; James, D.; Liu, C.-L.; Minton, M. A. Tetrahedron 1986, 42, 2993–3001. doi:10.1016/S0040-4020(01)90590-3 |
| 30. | Oppolzer, W.; Löhner, H. J. Helv. Chim. Acta 1981, 64, 2808–2811. doi:10.1002/hlca.19810640842 |
© 2017 Letzel et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)