Abstract
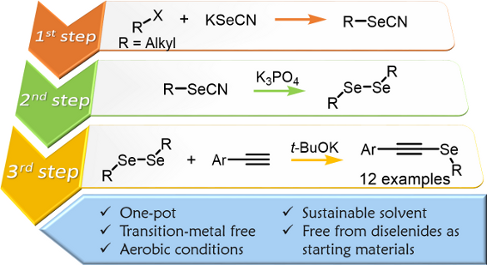
Alkynyl selenides were synthesized by a straightforward one-pot and three-step methodology, without the need of diselenides as starting reagents, under an oxygen atmosphere and using PEG 200 as the solvent. This procedure involves the in situ generation of dialkyl diselenides through a K3PO4-assisted reaction of an alkyl selenocyanate obtained by a nucleophilic substitution reaction between KSeCN and alkyl halides. Successive reaction with terminal alkynes in the presence of t-BuOK affords the corresponding alkyl alkynyl selenide in moderate to good yields. Finally, this methodology allowed the synthesis of 2-alkylselanyl-substituted benzofuran and indole derivatives starting from convenient 2-substituted acetylenes.
Graphical Abstract
Introduction
Alkynyl selenides, as many other selenium compounds, have potential anti-oxidant activities, and may play a role in certain diseases such as cancer, heart diseases, inflammatory processes, arthritis, and skin damage caused by exposure to UV radiation [1-3]. However, all these properties belong to organoselenides due to their ability to engage in enzymatic processes [4,5] and alkynyl selenides have demonstrated only limited biological properties [6,7]. On the other hand, these compounds are widely applied in organic chemistry due to the presence of an alkynyl group [8,9] and an organoselenium moiety [10] in one molecule both comprising versatile building blocks or intermediates in organic synthesis. Some of the most outstanding synthetic applications of alkynyl selenides are electrophilic addition reactions, many of them modulated by transition-metal catalysts [11]. Some examples are: hydroboration which results in a vinylborane selenide used in Pd-catalyzed Suzuki cross-coupling reactions [12], the addition to tributyltin hydride in the presence of Pd and Cu catalysts to afford (E)-α-selenylstannanes for the synthesis of trisubstituted alkenes [13] and hydrozirconation with further replacement of Zr atom by hydrogen or halogen [14] or their use in the preparation of α-seleno-α,β-unsaturated ketones [15]. Besides, hydrogen halide-addition reactions to alkynyl selenides in the absence of transition metals [16] and the addition of p-toluenesulfonic acid [17] are applied to obtain vinyl organometallic compounds and key selenoester intermediates. Finally, electrophilic cyclizations [18,19] of systems bearing a selenide alkynyl group allow the synthesis of seleno-functionalized heterocycles [20,21]. Recently, the use of alkynyl selenides as substrates for Pd-catalyzed Suzuki, Negishi, Kumada and Sonogashira cross-coupling reactions has been reported with good yields [22].
Due to the synthetic relevance of alkynyl selenides several methodologies for their synthesis have been developed. Among them are reactions between lithium or sodium acetylides and electrophilic selenium reactants [23]. The use of hypervalent iodine(III) species [24] or alkynyl bromides with RSeLi [25] as nucleophilic selenium species or the reaction of alkynyl bromides under Cu catalysis [26-29], from terminal alkynes in the presence of bases and Cu [30-33], Fe [34,35] or In [36] catalysis, or with t-BuOK without of transition metal [37] have been reported. In general, these methodologies require selenyl halides or diselenides as starting materials, which limit their application. Furthermore, in most cases these procedures involve the use of reducing agents, such as elemental Mg or Zn, transition-metal catalysis, inert atmosphere, controlled temperatures, and long reaction times.
Recently, we have reported the synthesis of vinyl selenides 1 by a three-step one-pot procedure using KSeCN (2), alkyl (3) and styryl halides (4) with good yields (Scheme 1, A) [38]. This reaction proceeds through an initial nucleophilic substitution reaction between the alkyl halide and KSeCN to afford the corresponding alkyl selenocyanate (RSeCN). The treatment of the latter with K3PO4 generates the alkyl selenolate anion (RSe−) which reacts with another RSeCN molecule to give the corresponding diselenide R2Se2. Reduction of the latter diselenide by treatment with NaBH4 affords an alkyl selenolate anion, which reacts with styryl halide to give the desired styryl selenide 1. During optimization of the reaction conditions to 1 and in the presence of only t-BuOK, the corresponding alkynyl selenide 5 was obtained together with dialkyl selenides R2Se and R2Se2. In view of the important synthetic application of alkynyl selenides, we envisioned the possibility of obtaining alkynyl analogues based on our methodology developed for the synthesis of vinyl selenides.
![[1860-5397-13-92-i1]](/bjoc/content/inline/1860-5397-13-92-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: One-pot synthesis of vinyl and alkynyl selenides.
Scheme 1: One-pot synthesis of vinyl and alkynyl selenides.
We herein report the optimized reaction conditions for the one-pot and three-step synthesis of alkynyl selenides 5 using KSeCN as selenium source in PEG 200 as the solvent under an oxygen atmosphere. The reaction does not require diselenides or selenyl halides as starting electrophilic reagents, thus offering important advantages over the methods currently reported in the literature (Scheme 1, B).
Results and Discussion
Optimization of conditions
During a base screening for the synthesis of alkyl styryl selenides (1) [38], we observed that the addition of K3PO4 to RSeCN, obtained in situ by reaction of RX with KSeCN, afforded exclusively the corresponding R2Se2. Based on this result we started seeking the best conditions to afford alkynyl selenide 5a using styryl bromide (4a), 2 and n-octyl bromide (3a). Thus, we applied the optimized conditions for the synthesis of 1 (Scheme 1, A), replacing the reducing agent by t-BuOK in the third step. When 1.5 equiv of t-BuOK with regard to 4a were employed in the reaction, alkyne 5a was obtained in 24%, while the use of 2.0 and 3.0 equiv increased the yield to 61% and 53%, respectively (Scheme 2). Using more than two equiv of base resulted in a lower yield of alkyne 5a and the formation of styryl selenide 1a could be detected.
![[1860-5397-13-92-i2]](/bjoc/content/inline/1860-5397-13-92-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Effect of t-BuOK on the formation of n-octyl alkynyl selenide 5a.
Scheme 2: Effect of t-BuOK on the formation of n-octyl alkynyl selenide 5a.
Afterward we performed some control experiments. Thus, the reaction of styryl bromide (4a) with 1 equiv of t-BuOK afforded 64% yield of phenylacetylene (6a), while the elimination reaction was quantitative with 2 equiv of the base. These results suggested that 6a is a key intermediate in the reaction. Furthermore, the fact that in the presence of 1 equiv of base the elimination product was obtained in approximately 50% yield implies that the deprotonation of 6a is faster than the elimination from 4a.
Since 6a is an intermediate compound of the reaction, we optimized the reaction conditions using terminal alkyne 6a as the substrate instead of styryl bromide (4a), following a three-step one-pot procedure (see Scheme 2). First, we explored the reactivity of KSeCN (2), n-octyl bromide (3a) and acetylene 6a in DMF as solvent under different atmospheric conditions (Table 1, entries 1–3). Conducting the reaction under an inert atmosphere, compound 5a was obtained with a yield of 38%. A slightly higher performance was observed when the reaction was carried out under an air atmosphere (41%) and a remarkably improved yield of 71% was obtained under an oxygen atmosphere with continuous bubbling. These results show that the presence of oxygen in the reaction medium contributes to the formation of the expected product.
Table 1: Solvent screening for the one-pot synthesis of selenide 5a.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-92-i8.svg?max-width=637&scale=1.0)
|
||
| entry | solvent | yield % 5ab |
|---|---|---|
| 1 | DMF | 71 |
| 2c | DMF | 38 |
| 3d | DMF | 41 |
| 4 | MeCN | 47 |
| 5 | NMP | 9 |
| 6 | DMSO | 10 |
| 7 | dioxane | <5 |
| 8 | toluene | 18 |
| 9 | ethanol | 43 |
| 10 | isopropanol | 65 |
| 11 | water | nd |
| 12 | PEG 300 | 63 |
| 13 | PEG 200 | 71 |
aThe reaction was performed by using: 0.25 mmol of 6a, 1.35 equiv of 2, 3a and K3PO4 and 1.1 equiv of t-BuOK in 2 mL of solvent under O2 atmosphere, unless otherwise indicated. bQuantified by GC with internal standard. nd: not detected. cUnder N2. dOpen to air.
Next, we performed a screening of solvents under oxygen atmosphere (Table 1). Best yields were obtained with DMF, isopropanol or polyethylene glycol (PEG) as the solvents (Table 1, entries 1, 10, 12 and 13), whereas only moderate yields below 50% were obtained when the reaction was conducted in acetonitrile or ethanol (Table 1, entries 4 and 9). Poor yields below 20% were observed by using N-methylpyrrolidone (NMP), dimethyl sulfoxide (DMSO), dioxane or toluene (Table 1, entries 5–8). Finally, product 5a was not detectable at all when performing the reaction in water. As similar yields were found with DMF and PEG 200, we chose PEG 200 as the solvent for subsequent reactions, which is a non-toxic and inexpensive biodegradable material also used as a sustainable and alternative solvent due to its reusability [39].
Table 2 summarizes the results for the screening and concentration of the base used. Formation of 5a increases as the volume of solvent increases from 1 mL to 3 mL, with yields of 61%, 71% and 84%, respectively (Table 2, entries 1–3); no considerable change (80%) was observed in 4 mL of PEG 200 (Table 2, entry 4). To understand this trend, the reaction between 6a and Ph2Se2 was performed, in which the concentration of reactants was twelve times higher than that under standard conditions, Scheme 3, observing the formation of phenyl (2-phenylethynyl)selane [24] and the addition product 1-phenyl-2-(phenylselanyl)vinylselanylbenzene [40] (27% and 73% yields of relative area by GC, respectively). This result shows that under the conditions used, the Ph2Se2 addition reaction to the triple bond competes with Se-alkynylation. This side reaction is less efficient when the concentration decreases, favoring the reaction of interest when the amount of solvent increases.
Table 2: Concentration effects and base screening for the one-pot synthesis of selenide 5a.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-92-i9.svg?max-width=637&scale=1.0)
|
|||
| entry | base | PEG 200 (mL) | yield % 5ab |
|---|---|---|---|
| 1 | t-BuOK | 1 | 61 |
| 2 | t-BuOK | 2 | 71 |
| 3 | t-BuOK | 3 | 84 |
| 4 | t-BuOK | 4 | 80 |
| 5 | KOH | 3 | 37 |
| 6 | NaOMe | 3 | 55 |
| 7c | CuI/Et3N | 3 | 31 |
| 8d | t-BuOK | 3 | 93 (78)e |
| 9d,f | t-BuOK | 3 | (56)e,g |
| 10f,h | t-BuOK | 3 | 26i |
aThe reaction was performed by using: 0.25 mmol of 6a, 1.35 equiv of 2, 3a and K3PO4 and 1.1 equiv of t-BuOK in PEG 200 as solvent under O2 atmosphere, unless otherwise indicated. bQuantified by GC with internal standard. cCuI (10 mol %), 1.1 equiv of Et3N at 100 °C for 24 h under N2 atmosphere. d0.25 mmol of 6a, 1.0 equiv of 2, 3a and K3PO4, and 2.0 equiv of t-BuOK; 6a and t-BuOK were added in the last step. eIsolated yield. fWithout addition of K3PO4. gTogether with R2Se and R2Se2 (35% with a ratio of 1:6). hn-Octyl selenocyanate, prepared by reaction of 3a (0.25 mmol) and 2 (0.25 mmol) in 1 mL of PEG 200, was dropped to a solution of alkynyl anion, obtained by reaction of 6a (0.25 mmol) and t-BuOK (0.25 mmol) in 2 mL of PEG 200 under N2 atmosphere, and heated at 100 °C for 2 h. iTogether with a mixture of R2Se and R2Se2 (74% with a ratio of 1:3).
![[1860-5397-13-92-i3]](/bjoc/content/inline/1860-5397-13-92-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Effect of reactants concentration on alkynyl selenide formation.
Scheme 3: Effect of reactants concentration on alkynyl selenide formation.
Subsequently, a set of bases able to deprotonate the alkyne in the last step was tested. By using KOH or NaOMe, 5a was generated in 37% and 55% yield, respectively (Table 2 entries 5 and 6). The CuI/Et3N system, a commonly used combination to activate alkynes in Sonogashira reactions, was also tested, giving 31% yield of 5a (Table 2, entry 7). However, t-BuOK afforded the best results and therefore selected for the subsequent reactions. Next, the ratio of reactants was further investigated in order to obtain the best possible product yield. Thus, by using 6a/2/3a/K3PO4/t-BuOK at a 1:1:1:1:2 ratio, respectively, and by adding 6a with t-BuOK in the last step, the yield of selenide 5a was 93% quantified by GC and 78% isolated yield (Table 2, entry 8). As a control experiment, the reaction was repeated under optimized conditions but without the addition of K3PO4 (Table 2, entry 9). As previously observed [38], the reaction occurred with the formation of di-n-octyl selenide and di-n-octyl diselenide as byproducts, decreasing the yield of 5a to 56% and hampering its isolation. Similar results were obtained when adding n-octyl selenocyanate to a solution of alkynyl anion under N2 atmosphere followed by heating at 100 °C for 2 h (Table 2, entry 10).
With the optimized reaction conditions at hand, we explored the scope and limitation of this methodology (Scheme 1B) for the synthesis of alkyl alkynyl selenides 5 from various substituted terminal arylacetylenes 6, synthesized according to known procedures [41,42], and different alkyl halides 3.
Table 3 comprises the results obtained from the reaction of phenylacetylene (6a) with different alkyl halides 3. The reaction of primary alkyl bromides afforded the corresponding alkynyl selenide 5 in moderate to good yields. It is worth mentioning, that replacing n-octyl bromide by the corresponding tosylated compound, the yield of 5a decreased from 78% to 67% (Table 3, entries 1 and 2). A comparable reactivity was found for the reactions with methyl iodide and n-butyl bromide, affording 5b and 5c in 77% and 79% yields, respectively (Table 3, entries 3 and 4). However, with 6-bromohex-1-ene the yield of compound 5d dropped to 41% (Table 3, entry 5) and 2-methyltetrahydro-2H-selenopyran was detected by GC–MS. This indicates a possible competitive intramolecular addition reaction between the selenium atom-centered radical and the alkenyl moiety.
Table 3: One-pot synthesis of alkynyl selenides 5.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-92-i10.svg?max-width=637&scale=1.0)
|
|||
| entry | R–X | yield % 5b | |
|---|---|---|---|
| 1 | n-OctBr |
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-92-i11.svg?max-width=637&scale=1.0)
5a |
78 |
| 2 | n-OctOTs |
![[Graphic 5]](/bjoc/content/inline/1860-5397-13-92-i12.svg?max-width=637&scale=1.0)
5a |
67 |
| 3 | MeI |
![[Graphic 6]](/bjoc/content/inline/1860-5397-13-92-i13.svg?max-width=637&scale=1.0)
5b |
77 |
| 4 | n-BuBr |
![[Graphic 7]](/bjoc/content/inline/1860-5397-13-92-i14.svg?max-width=637&scale=1.0)
5c |
79 |
| 5 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-13-92-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 9]](/bjoc/content/inline/1860-5397-13-92-i16.svg?max-width=637&scale=1.0)
5d |
41 |
| 6 | c-HexCH2Br |
![[Graphic 10]](/bjoc/content/inline/1860-5397-13-92-i17.svg?max-width=637&scale=1.0)
5e |
56 |
| 7 | c-HexBr |
![[Graphic 11]](/bjoc/content/inline/1860-5397-13-92-i18.svg?max-width=637&scale=1.0)
5f |
nd |
| 8 | t-BuBr |
![[Graphic 12]](/bjoc/content/inline/1860-5397-13-92-i19.svg?max-width=637&scale=1.0)
5g |
nd |
| 9 | PhCH2Br |
![[Graphic 13]](/bjoc/content/inline/1860-5397-13-92-i20.svg?max-width=637&scale=1.0)
5h |
nd |
aThe reaction was performed by using: 0.25 mmol of 6a, 1.0 equiv of 2, 3 and K3PO4 and 2.0 equiv of t-BuOK, in 3 mL PEG 200 as solvent under an O2 atmosphere; 6a and t-BuOK were added in the last step. bIsolated yield, nd: not detected.
The reaction with (bromomethyl)cyclohexane afforded 5e in 56% yield (Table 3, entry 6). The difference in reactivity relative to 5a is probably due to steric hindrance caused by the cyclohexyl ring to the alkynyl anion attack. The basic reaction conditions were responsible for promoting elimination reactions (E2) when secondary and tertiary alkyl halides were used, preventing formation of 5f and 5g products, and this is a limitation of the procedure reported herein (Table 3, entries 7 and 8). By employing a benzylic halide such as benzyl bromide, the expected product 5h (Table 3, entry 9) was not detected. Again, the basic environment and moderate acidity of the methylene protons in benzyl selenocyanate (7h) or dibenzyl diselenide (8h) may afford intermediates prone to decomposition, directly affecting formation of 5h.
Furthermore, the electronic and steric effects offered by different substituents on the aryl moieties attached to the acetylenic group were also studied (Table 4). Thus, the p-methyl-substituted derivative 6b afforded the selenide 5i in comparable yields as with the unsubstituted alkyne 6a (Table 4, entries 1 and 2). The presence of a strong electron donor such as a methoxy group (compound 6c), decreased the yield of product 5j to 52%. This is ascribed to a lower acidity of the acetylenic proton in 6c in relation to the unsubstituted alkyne 6a (Table 4, entry 3). For phenylethynyl derivatives substituted in para or ortho position by halides, the yields of the corresponding products 5k and 5l were 64% and 61%, respectively (Table 4, entries 4 and 5). The ortho-substituent showed no remarkable steric hindrance since the reactive center was located relatively far from this group.
Table 4: One-pot synthesis of alkynyl selenide 5.a
![[Graphic 14]](/bjoc/content/inline/1860-5397-13-92-i21.svg?max-width=637&scale=1.0)
|
|||
| entry | R, 6 | product 5 | yield %b |
|---|---|---|---|
| 1 |
Ph
6a |
![[Graphic 15]](/bjoc/content/inline/1860-5397-13-92-i22.svg?max-width=637&scale=1.0)
5a |
78 |
| 2 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-13-92-i23.svg?max-width=637&scale=1.0)
6b |
![[Graphic 17]](/bjoc/content/inline/1860-5397-13-92-i24.svg?max-width=637&scale=1.0)
5i |
81 |
| 3 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-13-92-i25.svg?max-width=637&scale=1.0)
6c |
![[Graphic 19]](/bjoc/content/inline/1860-5397-13-92-i26.svg?max-width=637&scale=1.0)
5j |
52 |
| 4 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-13-92-i27.svg?max-width=637&scale=1.0)
6d |
![[Graphic 21]](/bjoc/content/inline/1860-5397-13-92-i28.svg?max-width=637&scale=1.0)
5k |
64 |
| 5 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-13-92-i29.svg?max-width=637&scale=1.0)
6e |
![[Graphic 23]](/bjoc/content/inline/1860-5397-13-92-i30.svg?max-width=637&scale=1.0)
5l |
61 |
| 6 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-13-92-i31.svg?max-width=637&scale=1.0)
6f |
![[Graphic 25]](/bjoc/content/inline/1860-5397-13-92-i32.svg?max-width=637&scale=1.0)
5m |
nd |
| 7 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-13-92-i33.svg?max-width=637&scale=1.0)
6g |
![[Graphic 27]](/bjoc/content/inline/1860-5397-13-92-i34.svg?max-width=637&scale=1.0)
5n |
53 |
| 8 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-13-92-i35.svg?max-width=637&scale=1.0)
6h |
![[Graphic 29]](/bjoc/content/inline/1860-5397-13-92-i36.svg?max-width=637&scale=1.0)
5o |
nd |
| 9c |
![[Graphic 30]](/bjoc/content/inline/1860-5397-13-92-i37.svg?max-width=637&scale=1.0)
6i |
![[Graphic 31]](/bjoc/content/inline/1860-5397-13-92-i38.svg?max-width=637&scale=1.0)
5p |
<5d |
| 10c |
![[Graphic 32]](/bjoc/content/inline/1860-5397-13-92-i39.svg?max-width=637&scale=1.0)
6j |
![[Graphic 33]](/bjoc/content/inline/1860-5397-13-92-i40.svg?max-width=637&scale=1.0)
5q |
51 |
aThe reaction was performed by using: 0.25 mmol of 6, 1.0 equiv of 2, 3a and K3PO4 and 2.0 equiv of t-BuOK, in 3 mL of PEG 200 as solvent under an O2 atmosphere; 6 and t-BuOK were added in the last step. bIsolated yield, nd: not detected. c3.0 equiv of t-BuOK and stirring at 100 °C for 6 h were used in the last step. dThe main product was 1-ethyl-2-(octylselanyl)-1H-indole (9) in 62% isolated yield.
On the other hand, in the presence of a strong electron-withdrawing substituent such as a nitro group, the corresponding product 5m was not detected (Table 4, entry 6). This result indicates that although the acidity of acetylenic proton in 6f is higher than that for the unsubstituted one 6a, the resulting anion is highly stabilized by this group, thus reducing its nucleophilic character and being unreactive against di(n-octyl) diselenide (8a). Alkyne 6g, having a styryl group afforded compound 5n in 53% yield (as a mixture of E and Z isomers in a 5:1 ratio, respectively; Table 4, entry 7) and the aliphatic terminal alkyne 6h failed to react under the selected conditions (Table 4, entry 8).
In order to apply the present methodology to the one-pot synthesis of heterocycles derived from indole and benzofuran, convenient ortho-substituted phenylacetylenes were also tested. To our delight, the o-diethylamino-substituted derivative 6i afforded N-ethyl-2-(n-octylselanyl)-1H-indole (9) in 62% isolated yield (Scheme 4). The primary selenyl-substituted product 5p was only detected in traces by GC–MS suggesting that this product cyclized in the reaction medium to afford indole 9 (Table 4, entry 9). On the other hand, when o-MeO-substituted phenylacetylene 6j was employed the corresponding selenyl-substituted product 5q was isolated in 51% yield (Table 4, entry 10). The subsequent electrophilic addition of I2 to selenide 5q afforded 3-iodo-2-(n-octylselanyl)benzofuran (10) in excellent yield (93% isolated yield, Scheme 4).
![[1860-5397-13-92-i4]](/bjoc/content/inline/1860-5397-13-92-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of N-ethyl-2-(n-octylselanyl)-1H-indole (9) and 3-iodo-2-(n-octylselanyl)benzofuran (10).
Scheme 4: Synthesis of N-ethyl-2-(n-octylselanyl)-1H-indole (9) and 3-iodo-2-(n-octylselanyl)benzofuran (10).
Finally, we compared our method with syntheses reported in the literature that mainly rely on commercially available selenium compounds such as n-Bu2Se2, Me2Se2 or Ph2Se2. Our procedure does not require transition-metal catalysts, it uses PEG 200 as solvent within shorter or comparable reaction times and it has the advantage of in situ generating R2Se2 from commercially available and cheaper salts, that are easier to handle and have a much less unpleasant odor than the aliphatic diselenides (R2Se2). Thus the global 79% yield of 5c obtained by our method, involves the generation of R2Se2 over two steps followed by reaction with phenylacetylene. The other methods reported comparable or smaller isolated yields of 5c (Table 5).
Table 5: Comparison of the present method with other reported procedures for the synthesis of 5c from 6a and n-Bu2Se2.
| entry | conditions | yield % 5ca | reference |
|---|---|---|---|
| 1 | NpsFe3O4 (10 mol %), K2CO3, DMF, 80 °C, 14 h | 51 | [34] |
| 2 | NpsCuO (10 mol %), K2CO3, DMSO, 80 °C, 14 h | 79 | [32] |
| 3 | InCl3 (10 mol %), Cs2CO3, DMSO, 80 °C, 12 h | 64 | [36] |
| 4b | CuI (1 equiv), HMPA, N2, rt, 2 h | 70 | [26] |
| 5b | BuSeLi, THF, N2, 0 °C, 1 h | 60 | [25] |
| 6c | K3PO4, t-BuOK, PEG 200, O2, 100 °C, 3 h 10 min | 79 | this work |
aIsolated yield. bThe reaction was performed by using 1-bromo-2-phenylacetylene as starting material. cThe reaction was performed by using KSeCN (1.0 equiv) and n-BuBr (1.0 equiv) for the in situ generation of n-Bu2Se2.
Proposed mechanism
In order to assess the mechanism of the three-step one-pot synthesis of 5, the reaction between the commercial reagents 6a and 0.5 equiv of diphenyl diselenide (as analogue to the dialkyl diselenides 8 proposed as intermediates) was carried out in the presence of 1 equiv t-BuOK at 100 °C for 1 h under air atmosphere. As a result, phenyl(2-phenylethynyl)selane [34] was isolated in 64% yield. Repeating the reaction under oxygen the conversion to the alkynyl selenide increased to 90% (Scheme 5A). After work-up a positive test with I2/starch indicator of the aqueous layer confirmed the presence of H2O2 [43] resulting from the superoxide radical anion (O2·−) generated during reaction under oxygen atmosphere. It is also possible that the superoxide radical anion oxidizes the alkyl selenide anion (11) to the corresponding alkyl selenide radical and the formed O22− affords H2O2 after work-up. On the other hand, when the same reaction was performed under a nitrogen atmosphere and after one hour quenched with MeI, alkynyl selenide was formed in 47% yield together with methyl(phenyl)selane in 50% yield, determined as relative areas by GC. The latter result confirms that under inert gas atmosphere the phenyl selenide anion remains unoxidized and can be trapped by methylation. Furthermore, under the current reaction conditions no reduction of Se–Se bonds by the used base t-BuOK as proposed in the literature takes place (Scheme 5B) [44].
![[1860-5397-13-92-i5]](/bjoc/content/inline/1860-5397-13-92-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Control reactions and mechanistic study.
Scheme 5: Control reactions and mechanistic study.
To account for the results obtained we propose the following mechanism for the formation of alkynyl selenides (5) (Scheme 6). In the first step a nucleophilic substitution reaction between the alkyl halide 3 with KSeCN (2) yields alkyl selenocyanate 7, which, after addition of K3PO4, quantitatively affords dialkyl diselenide 8 [38]. Subsequently, the arylacetylene 6 and t-BuOK are added with concomitant generation of the corresponding arylacetylene anion. The anion of 6, once formed, attacks the diselenide intermediate 8, leading to the expected product 5 and an alkyl selenolate anion (11). Given the fact, that the reaction occurs under air atmosphere, the presence of oxygen promotes the oxidation of anion 11 to 8 [45], continuing the cycle until complete consumption of 11.
![[1860-5397-13-92-i6]](/bjoc/content/inline/1860-5397-13-92-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Proposed mechanism for the formation of selenides 5.
Scheme 6: Proposed mechanism for the formation of selenides 5.
Due to the aerobic conditions and the presence of oxidative species such as O2·−, O22− or H2O2, the oxidative retro-alkylation is favored [46-49]. Therefore, we can explain the formation of indole 9 by oxidation of 5p, generated in situ as described above, to afford the iminium intermediate 12. Hydrolysis of the latter liberates the secondary amine 13 (Scheme 7). This intermediate was detected by GC–MS in traces. Finally, the presence of base in excess and the correct arrangement and electronic properties of both the amine and alkynyl groups promote a spontaneous cyclization reaction to form indole 9 in good yields [50].
![[1860-5397-13-92-i7]](/bjoc/content/inline/1860-5397-13-92-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Proposed mechanism for the formation of indole 9.
Scheme 7: Proposed mechanism for the formation of indole 9.
Conclusion
We have developed a novel one-pot procedure for the synthesis of alkyl alkynyl selenides in moderate to good yields. The reaction does not require selenolate anions as starting materials and proceeds through the in situ formation of diselenides from commercially available potassium selenocyanate. Furthermore, the reaction readily takes place in PEG 200, a sustainable solvent, in short reaction times and under an oxygen atmosphere. The method is also applicable to the synthesis of functionalized heterocycles in good to excellent yields.
Supporting Information
| Supporting Information File 1: Experimental details, characterization data and copies of 1H, 13C and 77Se NMR spectra for products 5a–e, 5i–l, 5n, 5q, 9 and 10. | ||
| Format: PDF | Size: 2.7 MB | Download |
References
-
Mugesh, G.; du Mont, W.-W.; Sies, H. Chem. Rev. 2001, 101, 2125–2179. doi:10.1021/cr000426w
Return to citation in text: [1] -
Nogueira, C. W.; Zeni, G.; Rocha, J. B. T. Chem. Rev. 2004, 104, 6255–6285. doi:10.1021/cr0406559
Return to citation in text: [1] -
Chasteen, T. G.; Bentley, R. Chem. Rev. 2003, 103, 1–26. doi:10.1021/cr010210+
Return to citation in text: [1] -
Morgenstern, R.; Cotgreave, I. A.; Engman, L. Chem.-Biol. Interact. 1992, 84, 77–84. doi:10.1016/0009-2797(92)90122-2
Return to citation in text: [1] -
May, S. W.; Wang, L.; Gill-Woznichak, M. M.; Browner, R. F.; Ogonowski, A. A.; Smith, J. B.; Pollock, S. H. J. Pharmacol. Exp. Ther. 1997, 283, 470–477.
Return to citation in text: [1] -
Diaz, P.; Gendre, F.; Bernardon, J.-M. Tetrahedron Lett. 1998, 39, 9003–9006. doi:10.1016/S0040-4039(98)02038-3
Return to citation in text: [1] -
Bolzan, R. C.; Folmer, V.; Farina, M.; Zeni, G.; Nogueira, C. W.; Rocha, J. B. T.; Emanuelli, T. Basic Clin. Pharmacol. Toxicol. 2002, 90, 214–219. doi:10.1034/j.1600-0773.2002.900408.x
Return to citation in text: [1] -
Trost, B. M.; Li, C. J. Modern Alkyne Chemistry. Catalytic and Atom-Economic Transformations; Wiley-VCH Verlag GmbH & Co: Weinheim, Germany, 2014.
Return to citation in text: [1] -
Diederich, F.; Stang, P. J.; Tykwinski, R. R. Acetylene Chemistry: Chemistry, Biology and Material Science; Wiley-VCH: Weinheim, Germany, 2005.
Return to citation in text: [1] -
Rappoport, Z., Ed. The Chemistry of Organic Selenium and Tellurium Compounds; Wiley and Sons: Chichester, U.K., 2014; Vol. 4.
Return to citation in text: [1] -
Alonso, F.; Beletskaya, I. P.; Yus, M. Chem. Rev. 2004, 104, 3079–3159. doi:10.1021/cr0201068
Return to citation in text: [1] -
Yang, D. Y.; Huang, X. J. Organomet. Chem. 1997, 543, 165–169. doi:10.1016/S0022-328X(97)00147-2
Return to citation in text: [1] -
Huang, X.; Ma, Y. Synthesis 1997, 417–419. doi:10.1055/s-1997-1203
Return to citation in text: [1] -
Dabdoub, M. J.; Begnini, M. L.; Guerrero, P. G., Jr. Tetrahedron 1998, 54, 2371–2400. doi:10.1016/S0040-4020(98)00014-3
Return to citation in text: [1] -
Sun, A.; Huang, X. Synthesis 2000, 775–777. doi:10.1055/s-2000-6272
Return to citation in text: [1] -
Comasseto, J. V.; Menezes, P. H.; Stefani, H. A.; Zeni, G.; Braga, A. L. Tetrahedron 1996, 52, 9687–9702. doi:10.1016/0040-4020(96)00505-4
Return to citation in text: [1] -
Tiecco, M.; Testaferri, L.; Temperini, A.; Bagnoli, L.; Marini, F.; Santi, C.; Terlizzi, R. Eur. J. Org. Chem. 2004, 3447–3458. doi:10.1002/ejoc.200400177
Return to citation in text: [1] -
Godoi, B.; Schumacher, R. F.; Zeni, G. Chem. Rev. 2011, 111, 2937–2980. doi:10.1021/cr100214d
Return to citation in text: [1] -
Gabriele, B.; Mancuso, R.; Larock, R. C. Curr. Org. Chem. 2014, 18, 341–358. doi:10.2174/13852728113179990034
Return to citation in text: [1] -
Manarin, F.; Roehrs, J. A.; Gay, R. M.; Brandão, R.; Menezes, P. H.; Nogueira, C. W.; Zeni, G. J. Org. Chem. 2009, 74, 2153–2162. doi:10.1021/jo802736e
Return to citation in text: [1] -
Godoi, B.; Speranca, A.; Back, D. F.; Brandao, R.; Nogueira, C. W.; Zeni, G. J. Org. Chem. 2009, 74, 3469–3477. doi:10.1021/jo900307k
Return to citation in text: [1] -
Stein, A. L.; Bilheri, F. N.; Zeni, G. Chem. Commun. 2015, 51, 15522–15525. doi:10.1039/C5CC06347G
Return to citation in text: [1] -
Cook, D. J.; Hill, A. F.; Wilson, D. J. J. Chem. Soc., Dalton Trans. 1998, 1171–1173. doi:10.1039/a708647d
Return to citation in text: [1] -
Das, J. P.; Roy, U. K.; Roy, S. Organometallics 2005, 24, 6136–6140. doi:10.1021/om050504b
Return to citation in text: [1] [2] -
Dahdoub, M. J.; Comasseto, J. V.; Braga, A. L. Synth. Commun. 1988, 18, 1979–1983. doi:10.1080/00397918808068265
Return to citation in text: [1] [2] -
Braga, A. L.; Reckziegel, A.; Menezes, P. H.; Stefani, H. A. Tetrahedron Lett. 1993, 34, 393–394. doi:10.1016/0040-4039(93)85084-A
Return to citation in text: [1] [2] -
Kundu, A.; Roy, S. Organometallics 2000, 19, 105–107. doi:10.1021/om9907977
Return to citation in text: [1] -
Sharma, A.; Schwab, R. S.; Braga, A. L.; Barcellos, T.; Paixão, M. W. Tetrahedron Lett. 2008, 49, 5172–5174. doi:10.1016/j.tetlet.2008.06.071
Return to citation in text: [1] -
Ahammed, S.; Bhadra, S.; Kundu, D.; Sreedhar, B.; Ranu, B. C. Tetrahedron 2012, 68, 10542–10549. doi:10.1016/j.tet.2012.08.046
Return to citation in text: [1] -
Bieber, L. W.; da Silva, M. F.; Menezes, P. H. Tetrahedron Lett. 2004, 45, 2735–2737. doi:10.1016/j.tetlet.2004.02.042
Return to citation in text: [1] -
Mohammadi, E.; Movassagh, B. Tetrahedron Lett. 2014, 55, 1613–1615. doi:10.1016/j.tetlet.2014.01.088
Return to citation in text: [1] -
Godoi, M.; Ricardo, E. W.; Frizon, T. E.; Rocha, M. S. T.; Singh, D.; Paixão, M. W.; Braga, A. L. Tetrahedron 2012, 68, 10426–10430. doi:10.1016/j.tet.2012.08.086
Return to citation in text: [1] [2] -
Mohan, B.; Hwang, S.; Woo, H.; Park, K. H. Synthesis 2015, 47, 3741–3745. doi:10.1055/s-0035-1560458
Return to citation in text: [1] -
Godoi, M.; Liz, D. G.; Ricardo, E. W.; Rocha, M. S. T.; Azeredo, J. B.; Braga, A. L. Tetrahedron 2014, 70, 3349–3354. doi:10.1016/j.tet.2013.09.095
Return to citation in text: [1] [2] [3] -
Mohan, B.; Park, J. C.; Park, K. H. ChemCatChem 2016, 8, 2345–2350. doi:10.1002/cctc.201600280
Return to citation in text: [1] -
Rampon, D. S.; Giovenardi, R.; Silva, T. L.; Rambo, R. S.; Merlo, A. A.; Schneider, P. H. Eur. J. Org. Chem. 2011, 7066–7070. doi:10.1002/ejoc.201101245
Return to citation in text: [1] [2] -
Movassagh, B.; Navidi, M. Chin. Chem. Lett. 2012, 23, 1035–1038. doi:10.1016/j.cclet.2012.06.036
Return to citation in text: [1] -
Heredia, A. A.; Peñéñory, A. B. RSC Adv. 2015, 5, 105699–105706. doi:10.1039/C5RA20883A
Return to citation in text: [1] [2] [3] [4] -
Kerton, F.; Marriott, R. Alternative Solvents for Green Chemistry. Liquid Polymers; Royal Society of Chemistry: Cambridge, U.K., 2009; pp 170–187.
Return to citation in text: [1] -
Perin, G.; Borges, E. L.; Alves, D. Tetrahedron Lett. 2012, 53, 2066–2069. doi:10.1016/j.tetlet.2012.02.028
Return to citation in text: [1] -
Michel, P.; Gennet, D.; Rassat, A. Tetrahedron Lett. 1999, 40, 8575–8578. doi:10.1016/S0040-4039(99)01830-4
Return to citation in text: [1] -
Hijfte, L. V.; Kolb, M.; Witz, P. Tetrahedron Lett. 1989, 30, 3655–3656. doi:10.1016/S0040-4039(01)80473-1
Return to citation in text: [1] -
Turrens, J. F. J. Physiol. 2003, 552, 335–344. doi:10.1113/jphysiol.2003.049478
Return to citation in text: [1] -
Kumar, A.; Bhakuni, B. S.; Prasad, C. D.; Kumar, S.; Kumar, S. Tetrahedron 2013, 69, 5383–5392. doi:10.1016/j.tet.2013.04.113
Return to citation in text: [1] -
Xu, X.-H.; Lu, R. L.; Zhang, Q.-L.; Chen, X.; Li, Y.-J.; Zhang, Q.-L. Gaodeng Xuexiao Huaxue Xuebao 2005, 26, 267–269.
Return to citation in text: [1] -
Feula, A.; Fossey, J. S. RSC Adv. 2013, 3, 5370–5373. doi:10.1039/c3ra40228b
Return to citation in text: [1] -
Nouri-Nigjeh, E.; Permentier, H. P.; Bischoff, R.; Bruins, A. P. Anal. Chem. 2010, 82, 7625–7633. doi:10.1021/ac101364s
Return to citation in text: [1] -
Shaw, S.; Jayatilleke, E.; Herbert, V.; Colman, N. Biochem. J. 1989, 257, 277–280. doi:10.1042/bj2570277
Return to citation in text: [1] -
Vázquez-Vivar, J.; Whitsett, J.; Martásek, P.; Hogg, N.; Kalyanaraman, B. Free Radical Biol. Med. 2001, 31, 975–985. doi:10.1016/S0891-5849(01)00680-3
Return to citation in text: [1] -
Koradin, C.; Dohle, W.; Rodriguez, A. L.; Schmid, B.; Knochel, P. Tetrahedron 2003, 59, 1571–1587. doi:10.1016/S0040-4020(03)00073-5
Return to citation in text: [1]
| 40. | Perin, G.; Borges, E. L.; Alves, D. Tetrahedron Lett. 2012, 53, 2066–2069. doi:10.1016/j.tetlet.2012.02.028 |
| 38. | Heredia, A. A.; Peñéñory, A. B. RSC Adv. 2015, 5, 105699–105706. doi:10.1039/C5RA20883A |
| 41. | Michel, P.; Gennet, D.; Rassat, A. Tetrahedron Lett. 1999, 40, 8575–8578. doi:10.1016/S0040-4039(99)01830-4 |
| 42. | Hijfte, L. V.; Kolb, M.; Witz, P. Tetrahedron Lett. 1989, 30, 3655–3656. doi:10.1016/S0040-4039(01)80473-1 |
| 1. | Mugesh, G.; du Mont, W.-W.; Sies, H. Chem. Rev. 2001, 101, 2125–2179. doi:10.1021/cr000426w |
| 2. | Nogueira, C. W.; Zeni, G.; Rocha, J. B. T. Chem. Rev. 2004, 104, 6255–6285. doi:10.1021/cr0406559 |
| 3. | Chasteen, T. G.; Bentley, R. Chem. Rev. 2003, 103, 1–26. doi:10.1021/cr010210+ |
| 10. | Rappoport, Z., Ed. The Chemistry of Organic Selenium and Tellurium Compounds; Wiley and Sons: Chichester, U.K., 2014; Vol. 4. |
| 22. | Stein, A. L.; Bilheri, F. N.; Zeni, G. Chem. Commun. 2015, 51, 15522–15525. doi:10.1039/C5CC06347G |
| 43. | Turrens, J. F. J. Physiol. 2003, 552, 335–344. doi:10.1113/jphysiol.2003.049478 |
| 8. | Trost, B. M.; Li, C. J. Modern Alkyne Chemistry. Catalytic and Atom-Economic Transformations; Wiley-VCH Verlag GmbH & Co: Weinheim, Germany, 2014. |
| 9. | Diederich, F.; Stang, P. J.; Tykwinski, R. R. Acetylene Chemistry: Chemistry, Biology and Material Science; Wiley-VCH: Weinheim, Germany, 2005. |
| 23. | Cook, D. J.; Hill, A. F.; Wilson, D. J. J. Chem. Soc., Dalton Trans. 1998, 1171–1173. doi:10.1039/a708647d |
| 44. | Kumar, A.; Bhakuni, B. S.; Prasad, C. D.; Kumar, S.; Kumar, S. Tetrahedron 2013, 69, 5383–5392. doi:10.1016/j.tet.2013.04.113 |
| 6. | Diaz, P.; Gendre, F.; Bernardon, J.-M. Tetrahedron Lett. 1998, 39, 9003–9006. doi:10.1016/S0040-4039(98)02038-3 |
| 7. | Bolzan, R. C.; Folmer, V.; Farina, M.; Zeni, G.; Nogueira, C. W.; Rocha, J. B. T.; Emanuelli, T. Basic Clin. Pharmacol. Toxicol. 2002, 90, 214–219. doi:10.1034/j.1600-0773.2002.900408.x |
| 18. | Godoi, B.; Schumacher, R. F.; Zeni, G. Chem. Rev. 2011, 111, 2937–2980. doi:10.1021/cr100214d |
| 19. | Gabriele, B.; Mancuso, R.; Larock, R. C. Curr. Org. Chem. 2014, 18, 341–358. doi:10.2174/13852728113179990034 |
| 25. | Dahdoub, M. J.; Comasseto, J. V.; Braga, A. L. Synth. Commun. 1988, 18, 1979–1983. doi:10.1080/00397918808068265 |
| 4. | Morgenstern, R.; Cotgreave, I. A.; Engman, L. Chem.-Biol. Interact. 1992, 84, 77–84. doi:10.1016/0009-2797(92)90122-2 |
| 5. | May, S. W.; Wang, L.; Gill-Woznichak, M. M.; Browner, R. F.; Ogonowski, A. A.; Smith, J. B.; Pollock, S. H. J. Pharmacol. Exp. Ther. 1997, 283, 470–477. |
| 20. | Manarin, F.; Roehrs, J. A.; Gay, R. M.; Brandão, R.; Menezes, P. H.; Nogueira, C. W.; Zeni, G. J. Org. Chem. 2009, 74, 2153–2162. doi:10.1021/jo802736e |
| 21. | Godoi, B.; Speranca, A.; Back, D. F.; Brandao, R.; Nogueira, C. W.; Zeni, G. J. Org. Chem. 2009, 74, 3469–3477. doi:10.1021/jo900307k |
| 34. | Godoi, M.; Liz, D. G.; Ricardo, E. W.; Rocha, M. S. T.; Azeredo, J. B.; Braga, A. L. Tetrahedron 2014, 70, 3349–3354. doi:10.1016/j.tet.2013.09.095 |
| 14. | Dabdoub, M. J.; Begnini, M. L.; Guerrero, P. G., Jr. Tetrahedron 1998, 54, 2371–2400. doi:10.1016/S0040-4020(98)00014-3 |
| 16. | Comasseto, J. V.; Menezes, P. H.; Stefani, H. A.; Zeni, G.; Braga, A. L. Tetrahedron 1996, 52, 9687–9702. doi:10.1016/0040-4020(96)00505-4 |
| 36. | Rampon, D. S.; Giovenardi, R.; Silva, T. L.; Rambo, R. S.; Merlo, A. A.; Schneider, P. H. Eur. J. Org. Chem. 2011, 7066–7070. doi:10.1002/ejoc.201101245 |
| 17. | Tiecco, M.; Testaferri, L.; Temperini, A.; Bagnoli, L.; Marini, F.; Santi, C.; Terlizzi, R. Eur. J. Org. Chem. 2004, 3447–3458. doi:10.1002/ejoc.200400177 |
| 26. | Braga, A. L.; Reckziegel, A.; Menezes, P. H.; Stefani, H. A. Tetrahedron Lett. 1993, 34, 393–394. doi:10.1016/0040-4039(93)85084-A |
| 12. | Yang, D. Y.; Huang, X. J. Organomet. Chem. 1997, 543, 165–169. doi:10.1016/S0022-328X(97)00147-2 |
| 34. | Godoi, M.; Liz, D. G.; Ricardo, E. W.; Rocha, M. S. T.; Azeredo, J. B.; Braga, A. L. Tetrahedron 2014, 70, 3349–3354. doi:10.1016/j.tet.2013.09.095 |
| 11. | Alonso, F.; Beletskaya, I. P.; Yus, M. Chem. Rev. 2004, 104, 3079–3159. doi:10.1021/cr0201068 |
| 32. | Godoi, M.; Ricardo, E. W.; Frizon, T. E.; Rocha, M. S. T.; Singh, D.; Paixão, M. W.; Braga, A. L. Tetrahedron 2012, 68, 10426–10430. doi:10.1016/j.tet.2012.08.086 |
| 26. | Braga, A. L.; Reckziegel, A.; Menezes, P. H.; Stefani, H. A. Tetrahedron Lett. 1993, 34, 393–394. doi:10.1016/0040-4039(93)85084-A |
| 27. | Kundu, A.; Roy, S. Organometallics 2000, 19, 105–107. doi:10.1021/om9907977 |
| 28. | Sharma, A.; Schwab, R. S.; Braga, A. L.; Barcellos, T.; Paixão, M. W. Tetrahedron Lett. 2008, 49, 5172–5174. doi:10.1016/j.tetlet.2008.06.071 |
| 29. | Ahammed, S.; Bhadra, S.; Kundu, D.; Sreedhar, B.; Ranu, B. C. Tetrahedron 2012, 68, 10542–10549. doi:10.1016/j.tet.2012.08.046 |
| 24. | Das, J. P.; Roy, U. K.; Roy, S. Organometallics 2005, 24, 6136–6140. doi:10.1021/om050504b |
| 38. | Heredia, A. A.; Peñéñory, A. B. RSC Adv. 2015, 5, 105699–105706. doi:10.1039/C5RA20883A |
| 25. | Dahdoub, M. J.; Comasseto, J. V.; Braga, A. L. Synth. Commun. 1988, 18, 1979–1983. doi:10.1080/00397918808068265 |
| 45. | Xu, X.-H.; Lu, R. L.; Zhang, Q.-L.; Chen, X.; Li, Y.-J.; Zhang, Q.-L. Gaodeng Xuexiao Huaxue Xuebao 2005, 26, 267–269. |
| 46. | Feula, A.; Fossey, J. S. RSC Adv. 2013, 3, 5370–5373. doi:10.1039/c3ra40228b |
| 47. | Nouri-Nigjeh, E.; Permentier, H. P.; Bischoff, R.; Bruins, A. P. Anal. Chem. 2010, 82, 7625–7633. doi:10.1021/ac101364s |
| 48. | Shaw, S.; Jayatilleke, E.; Herbert, V.; Colman, N. Biochem. J. 1989, 257, 277–280. doi:10.1042/bj2570277 |
| 49. | Vázquez-Vivar, J.; Whitsett, J.; Martásek, P.; Hogg, N.; Kalyanaraman, B. Free Radical Biol. Med. 2001, 31, 975–985. doi:10.1016/S0891-5849(01)00680-3 |
| 39. | Kerton, F.; Marriott, R. Alternative Solvents for Green Chemistry. Liquid Polymers; Royal Society of Chemistry: Cambridge, U.K., 2009; pp 170–187. |
| 24. | Das, J. P.; Roy, U. K.; Roy, S. Organometallics 2005, 24, 6136–6140. doi:10.1021/om050504b |
| 38. | Heredia, A. A.; Peñéñory, A. B. RSC Adv. 2015, 5, 105699–105706. doi:10.1039/C5RA20883A |
| 38. | Heredia, A. A.; Peñéñory, A. B. RSC Adv. 2015, 5, 105699–105706. doi:10.1039/C5RA20883A |
| 36. | Rampon, D. S.; Giovenardi, R.; Silva, T. L.; Rambo, R. S.; Merlo, A. A.; Schneider, P. H. Eur. J. Org. Chem. 2011, 7066–7070. doi:10.1002/ejoc.201101245 |
| 37. | Movassagh, B.; Navidi, M. Chin. Chem. Lett. 2012, 23, 1035–1038. doi:10.1016/j.cclet.2012.06.036 |
| 30. | Bieber, L. W.; da Silva, M. F.; Menezes, P. H. Tetrahedron Lett. 2004, 45, 2735–2737. doi:10.1016/j.tetlet.2004.02.042 |
| 31. | Mohammadi, E.; Movassagh, B. Tetrahedron Lett. 2014, 55, 1613–1615. doi:10.1016/j.tetlet.2014.01.088 |
| 32. | Godoi, M.; Ricardo, E. W.; Frizon, T. E.; Rocha, M. S. T.; Singh, D.; Paixão, M. W.; Braga, A. L. Tetrahedron 2012, 68, 10426–10430. doi:10.1016/j.tet.2012.08.086 |
| 33. | Mohan, B.; Hwang, S.; Woo, H.; Park, K. H. Synthesis 2015, 47, 3741–3745. doi:10.1055/s-0035-1560458 |
| 50. | Koradin, C.; Dohle, W.; Rodriguez, A. L.; Schmid, B.; Knochel, P. Tetrahedron 2003, 59, 1571–1587. doi:10.1016/S0040-4020(03)00073-5 |
| 34. | Godoi, M.; Liz, D. G.; Ricardo, E. W.; Rocha, M. S. T.; Azeredo, J. B.; Braga, A. L. Tetrahedron 2014, 70, 3349–3354. doi:10.1016/j.tet.2013.09.095 |
| 35. | Mohan, B.; Park, J. C.; Park, K. H. ChemCatChem 2016, 8, 2345–2350. doi:10.1002/cctc.201600280 |
© 2017 Heredia and Peñéñory; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)