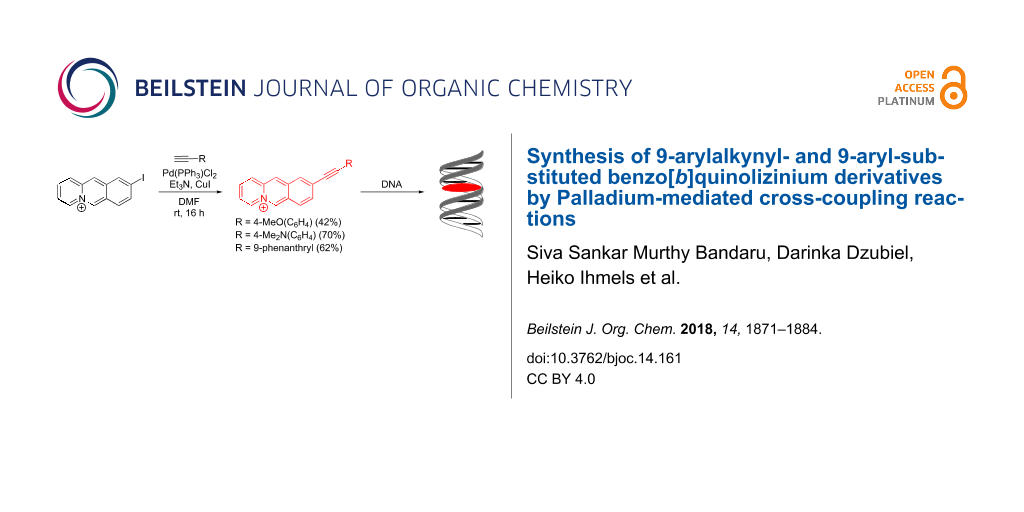
Abstract
9-Arylbenzo[b]quinolizinium derivatives were prepared with base-free Suzuki–Miyaura coupling reactions between benzo[b]quinolizinium-9-trifluoroborate and selected benzenediazonium salts. In addition, the Sonogashira coupling reaction between 9-iodobenzo[b]quinolizinium and the arylalkyne derivatives yielded four novel 9-(arylethynyl)benzo[b]quinolizinium derivatives under relatively mild reaction conditions. The 9-(N,N-dimethylaminophenylethynyl)benzo[b]quinolizinium is only very weakly emitting, but the emission intensity increases by a factor >200 upon protonation, so that this derivative may operate as pH-sensitive light-up probe. Photometric and fluorimetric titrations of duplex and quadruplex DNA to 9-(arylethynyl)benzo[b]quinolizinium derivatives revealed a significant binding affinity of these compounds towards both DNA forms with binding constants of Kb = 0.2–2.2 × 105 M−1.

Graphical Abstract
Introduction
Polycyclic cationic hetarenes are a paradigm of DNA-binding ligands whose association with the nucleic acid may affect the biological activities of the DNA [1-4]. For example, a DNA-bound heterocyclic ligand may interfere with DNA–enzyme recognition events, which are essential for DNA-based cellular processes, e.g., gene replication or transcription [1]. To this end, it was shown that DNA-binding ligands may operate as chemotherapeutic anticancer, antiviral or antibacterial drugs, for example as topoisomerase inhibitors [5]. More recently, much interest in this research area is focused on the non-canonical quadruplex DNA (G4-DNA) [6-8]. Mostly based on the principles and requirements of ligands that bind to duplex DNA, numerous G4-DNA ligands have been developed to study their selectivity and binding properties towards G4-DNA because of the biological importance of G4-DNA [9-13]. Along these lines, we and others have established the class of annelated quinolizinium derivatives as versatile ligands that bind to duplex, triplex and quadruplex DNA depending on their shape and size [14-18] and whose interaction with the nucleic acid may be used for fluorimetric detection of the latter [19,20].
To further exploit the DNA-binding properties of this specific class of cationic hetarenes, synthetic routes to novel derivatives with the desired substitution pattern and functionalization are necessary. In this context, Palladium-mediated cross-coupling reactions provide a powerful tool [21-27]; specifically, as these C–C coupling reactions have been demonstrated to be very useful for the introduction of various substituents to quinolizinium [28-33], benzo[b]quinolizinium [34,35] and naphthoquinolizinium [36] derivatives.
Unfortunately, in the case of benzo[b]quinolizinium substrates, the presence of strong nucleophiles, and for that matter bases in general, often interferes with the Pd-mediated reaction because of the competing addition of the nucleophile at the 6-position of the substrate and subsequent ring-opening reaction [37,38]. Considering this impediment and the additional difficulties that may occur during purification of these cationic hetarenes the reaction and work-up conditions of Pd-mediated coupling reactions of benzo[b]quinolizinium derivatives have to be optimized [34,35]. Accordingly, we extended our studies to improve the conditions of the Suzuki–Miyaura coupling towards biaryl-type benzo[b]quinolizinium derivatives 1a–d (Figure 1), namely to apply the alternative base-free Suzuki–Miyaura coupling reaction [39-42] between the benzo[b]quinolizinium-9-trifluoroborate (3b) and aryldiazonium salts. We focused our attention on derivatives 1a–d because in these cases a direct comparison with the already reported synthesis with a Suzuki–Miyaura reaction is possible. As we are particularly interested in benzo[b]quinolizinium derivatives with a large extension of the π-system, which should provide promising properties as G4-DNA ligands, we also focused our attention on the Sonogashira reaction as synthetic route to arylalkynyl-substituted derivatives. In this case, we aimed at donor-substituted derivatives such as 2b–d since they were proposed to have ideal photophysical and DNA-binding properties. Herein, we present the successful Suzuki–Miyaura and Sonogashira coupling reactions of benzo[b]quinolizinium substrates. In addition, the absorption and emission properties of the novel arylalkynylbenzo[b]quinolizinium derivatives 2a–d are reported (Figure 1), along with preliminary studies of their duplex and quadruplex DNA-binding properties.
![[1860-5397-14-161-1]](/bjoc/content/figures/1860-5397-14-161-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of 9-substituted benzo[b]quinolizinium derivatives 1 and 2.
Figure 1: Structures of 9-substituted benzo[b]quinolizinium derivatives 1 and 2.
Results
Synthesis
Synthesis of 9-aryl-substituted benzo[b]quinolizinium derivatives 1a–d
The 9-aryl-substituted benzo[b]quinolizinium derivatives 1a–d were prepared under base-free conditions by the Pd-catalyzed Suzuki–Miyaura reaction of the aryldiazonium salts 4a–d with benzo[b]quinolizinium-9-trifluoroborate (3b). The latter substrate was obtained as analytically pure product in moderate yield by the reaction of benzo[b]quinolizinium-9-boronic acid (3a) [34] with NaBF4 (Scheme 1).
![[1860-5397-14-161-i1]](/bjoc/content/inline/1860-5397-14-161-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of benzo[b]quinolizinium-9-trifluoroborate (3b) and 9-arylbenzo[b]quinolizinium derivatives 1a–d (see Table 1 for assignment of indices a–d and reaction conditions).
Scheme 1: Synthesis of benzo[b]quinolizinium-9-trifluoroborate (3b) and 9-arylbenzo[b]quinolizinium derivativ...
To identify appropriate reaction conditions for the base-free synthesis of derivatives 1a–d, different catalysts and solvents were tested for the cross-coupling reaction of benzo[b]quinolizinium-9-trifluoroborate (3b) and benzenediazonium salt 4a (Scheme 1, Table 1). With Pd(dppf)2Cl2·CH2Cl2 or Pd(PPh3)4 as catalyst, no conversion was observed, whereas the reaction could be achieved with Pd(OAc)2 as a catalyst and water as a solvent (Table 1, entries 1–5). Thus, the latter reaction conditions were used for the synthesis of 9-arylbenzo[b]quinolizinium derivatives 1b–d (Scheme 1, Table 1). The methoxyphenyl- and dimethylaminophenyl-substituted derivatives 1b and 1c were obtained in a moderate to good yield, but only trace amounts of the pyridyl-substituted derivative 1d were formed as shown by the 1H NMR spectroscopic analysis of the reaction mixture (Table 1, entries 6–8). Nevertheless, the 9-pyridinyl derivative 1d was obtained in low yield by the reaction of the trifluoroborate 3b with the diazonium salt 4d at 80 °C in DMF with Pd(PPh3)4 as a catalyst (Table 1, entry 9). It should be noted that some of these Suzuki–Miyaura coupling reactions require relatively long reaction times (Table 1, entries 6–9), which is a disadvantage considering the competing decomposition of the aryldiazonium ions under the reaction conditions. Thus, the corresponding diazonium salt was added in portions in intervals of 24 h until all of the substrate was consumed.
Table 1: Reaction conditions for the synthesis of 9-arylbenzo[b]quinolizinium derivatives 1a–d according to Scheme 1.
| Entry | Solvent | Catalyst | t (h) | Product | Yield (%) |
|---|---|---|---|---|---|
| 1 | H2O | Pd(OAc)2 | 48 | 1a, R = Ph | 43 |
| 2 | DME/H2O/MeOH | Pd(dppf)2Cl2·CH2Cl2a | 24 | 1a, R = Ph | n.r.b |
| 3 | DMF | Pd(dppf)2Cl2·CH2Cl2a | 24 | 1a, R = Ph | n.r.b |
| 4 | CH3CN | Pd(OAc)2 | 24 | 1a, R = Ph | n.r.b |
| 5 | CH3CN | Pd(PPh3)4 | 24 | 1a, R = Ph | n.r.b |
| 6 | H2O | Pd(OAc)2 | 168 | 1b, R = 4-MeO(C6H5) | 95 |
| 7 | H2O | Pd(OAc)2 | 144 | 1c, R = 4-Me2N(C6H5) | 44 |
| 8 | H2O | Pd(OAc)2 | 168 | 1d, 4-pyridyl | <2 |
| 9 | DMF | Pd(PPh3)4 | 168 | 1d, 4-pyridyl | 16 |
adppf = 1,1’-bis(diphenylphosphino)ferrocene. bNo reaction.
Synthesis of 9-(arylethynyl)benzo[b]quinolizinium derivatives 2a–d
The 9-(arylethynyl)benzo[b]quinolizinium derivatives 2a–d were prepared by Pd-mediated Sonogashira coupling reactions of 9-iodobenzo[b]quinolizinium bromide (5) [43] with arylacetylene derivatives (Scheme 2). To suppress the ring opening of the benzo[b]quinolizinium ring by nucleophilic attack at the 6-position [34,35] two methods were used that avoid the addition or formation of strong nucleophiles during the reaction. In the first approach, (phenylethynyl)copper (6) [44] was prepared separately and subsequently made to react with the substrate 5 to provide derivative 2a as hexafluorophosphate salt in moderate yield (Scheme 2). During the preparation of derivatives 2b–d with this method a crude product was isolated that contains the desired compound along with unidentified impurities, as shown by 1H NMR spectroscopic analysis of the product. Unfortunately, the product could not be further purified.
![[1860-5397-14-161-i2]](/bjoc/content/inline/1860-5397-14-161-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 9-(arylethynyl)benzo[b]quinolizinium derivatives 2a–d.
Scheme 2: Synthesis of 9-(arylethynyl)benzo[b]quinolizinium derivatives 2a–d.
In the second approach, the copper acetylide was formed in situ by the reaction of the acetylene derivative with triethylamine in the presence of Cu+ salts. Hence, the reaction of iodobenzo[b]quinolizinium 5 with arylacetylenes 7b–d in the presence of one equivalent of triethylamine and CuI under anhydrous conditions gave (arylethynyl)benzo[b]quinolizinium derivatives 2b–d in moderate to good yield (Scheme 2).
Several attempts to purify the derivatives 2a–d by column chromatography failed. Apparently, these compounds decompose when in contact with the silica or alumina of the column, so that the pure products were only available by crystallization from appropriate solvents, which resulted in lower yields of these products.
Single crystal X-ray diffraction analysis of 9-(arylethynyl)benzo[b]quinolizinium derivatives 2a and 2b
Single crystals of derivatives 2a and 2b were obtained by crystallization from acetone and CHCl3/MeOH, respectively (Figure 2 and Figure S1, Supporting Information File 1). Derivative 2a crystallizes in the triclinic space group ![[Graphic 1]](/bjoc/content/inline/1860-5397-14-161-i4.svg?max-width=637&scale=1.18182) with two molecules in the unit cell. The crystals were twinned and the compound shows some considerable disorder. The chemical composition, however, was unanimously proven by the data. Derivative 2b crystallizes with one molecule of CHCl3 as lattice solvent in the highly symmetric orthorhombic space group I2/a with 8 molecules in the unit cell. Both cations are essentially planar and π-stacked in an anti-head-to-tail (ht) arrangement in the solid state. A preference of such anti-ht arrangement was observed before in the crystal structures of two series of 9-substituted benzo[b]quinolizinium salts with halides or small alkyl substituents [45,46]. Stratford et al. attributed this observation to repulsion forces between the positively charged nitrogen atoms and π···π donor–acceptor attractions between the phenyl and pyridinium moieties. In our case, the situation is somehow more complex as the novel compounds bear aromatic substituents (via alkyne spacer) in the benzo[b]quinolizinium 9 position. These aromatic substituents now engage in π···π donor–acceptor attractions with the pyridinium moiety (outer most ring of the tricyclic moiety) and the two positively charged nitrogen atoms are per se much further apart due to the larger intramolecular separation between the intermolecularly interacting π-systems. In addition, the aromatic character of the substituent and its engagement in the π···π interaction also brings the two phenyl rings of adjacent benzo[b]quinolizinium moieties in close proximity, which can now also interact in an off-set π···π fashion. The contribution of the charge repulsion has, hence, to be less significant here and the preference for the anti-ht arrangement must be dominated by the π···π attractions. Centroid distances between the aromatic 9-substituent and the pyridinium moiety are 3.619 Å for 2a (C16 → C21; C5 → C9, N1) and 3.676 Å for 2b (C16 → C21; C1 → C5, N1), respectively (Figure S1, Supporting Information File 1). These separations are comparably short as the reported ones range from 3.69 Å to 3.99 Å [46]. The π-system separation between the centroids of the two benzo[b]quinolizinium phenyl rings are 3.803 Å for 2a (C1, C2, C3, C11, C12, C13) and 3.599 Å for 2b (C7 → C12), respectively. Notably, for 2b the phenyl to phenyl π···π interaction of one molecule is not with the same neighbor as the π···π donor–acceptor attraction between the phenyl and pyridinium rings. Therefore, these two distinct π-system-based attractions alternate and form infinite chains of molecules roughly protruding along the a axis. In 2a both interactions are with the same neighbor leading to distinct dimeric associates.
with two molecules in the unit cell. The crystals were twinned and the compound shows some considerable disorder. The chemical composition, however, was unanimously proven by the data. Derivative 2b crystallizes with one molecule of CHCl3 as lattice solvent in the highly symmetric orthorhombic space group I2/a with 8 molecules in the unit cell. Both cations are essentially planar and π-stacked in an anti-head-to-tail (ht) arrangement in the solid state. A preference of such anti-ht arrangement was observed before in the crystal structures of two series of 9-substituted benzo[b]quinolizinium salts with halides or small alkyl substituents [45,46]. Stratford et al. attributed this observation to repulsion forces between the positively charged nitrogen atoms and π···π donor–acceptor attractions between the phenyl and pyridinium moieties. In our case, the situation is somehow more complex as the novel compounds bear aromatic substituents (via alkyne spacer) in the benzo[b]quinolizinium 9 position. These aromatic substituents now engage in π···π donor–acceptor attractions with the pyridinium moiety (outer most ring of the tricyclic moiety) and the two positively charged nitrogen atoms are per se much further apart due to the larger intramolecular separation between the intermolecularly interacting π-systems. In addition, the aromatic character of the substituent and its engagement in the π···π interaction also brings the two phenyl rings of adjacent benzo[b]quinolizinium moieties in close proximity, which can now also interact in an off-set π···π fashion. The contribution of the charge repulsion has, hence, to be less significant here and the preference for the anti-ht arrangement must be dominated by the π···π attractions. Centroid distances between the aromatic 9-substituent and the pyridinium moiety are 3.619 Å for 2a (C16 → C21; C5 → C9, N1) and 3.676 Å for 2b (C16 → C21; C1 → C5, N1), respectively (Figure S1, Supporting Information File 1). These separations are comparably short as the reported ones range from 3.69 Å to 3.99 Å [46]. The π-system separation between the centroids of the two benzo[b]quinolizinium phenyl rings are 3.803 Å for 2a (C1, C2, C3, C11, C12, C13) and 3.599 Å for 2b (C7 → C12), respectively. Notably, for 2b the phenyl to phenyl π···π interaction of one molecule is not with the same neighbor as the π···π donor–acceptor attraction between the phenyl and pyridinium rings. Therefore, these two distinct π-system-based attractions alternate and form infinite chains of molecules roughly protruding along the a axis. In 2a both interactions are with the same neighbor leading to distinct dimeric associates.
![[1860-5397-14-161-2]](/bjoc/content/figures/1860-5397-14-161-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular structures of derivatives 2a (top) and 2b (bottom) in the solid state. Ellipsoids are shown at the 50% probability level. The counter anions and solvent molecule were omitted for clarity.
Figure 2: Molecular structures of derivatives 2a (top) and 2b (bottom) in the solid state. Ellipsoids are sho...
In the individual molecules, the C–C bond lengths of the alkyne unit are 1.24 Å (C14–C15) for the triple bond and 1.41 Å (C13–C14 and C15–C16) for the single bonds in compound 2a, while in derivative 2b they are 1.18 Å (C14–C15) and 1.45 Å (C9–C14 and C15–C16), respectively. Moreover, the π-surface of derivative 2b deviates slightly more from the mean plane as compared with 2a, i.e., as the torsion angle C8–C9–C16–C17 is −12.0° whereas it is 5.6° (C12–C13–C16–C21) in 2a. These data indicate a slightly more pronounced delocalization of π-electrons within the diarylalkyne unit of compound 2a, at least in the solid state.
Absorption and emission properties of 9-(arylethynyl)benzo[b]quinolizinium derivatives 2a–d
In general, compounds 2a–d have a low solubility in water and derivative 2d is moderately soluble in DMSO. The absorption spectra of 9-(arylethynyl)benzo[b]quinolizinium derivatives 2a,b,d show two low-energy maxima between 380 nm and 450 nm which resemble the ones of similar aryl-substituted benzo[b]quinolizinium [34] and naphthoquinolizinium [36] derivatives (Figure 3, Table 2). As a notable exception, the derivative 2c has a broad absorption band with maximum wavelength depending on the solvent, namely at 470 nm in MeOH, 515 nm in CH2Cl2 and 505 nm in CHCl3 (Figure 3C). At low pH, the broad long wavelength absorption band of 2c disappeared and a new absorption band was formed (λmax = 418 nm) that is similar to that of the parent compound 2a (Figure 3C). It should be noted that the benzo[b]quinolizinium derivatives 2a–d have lower absorbance and significantly broadened spectra in less polar solvents, presumably due to their low solubility and the resulting aggregation in these media.
![[1860-5397-14-161-3]](/bjoc/content/figures/1860-5397-14-161-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Absorption spectra of derivatives 2a (A), 2b (B), 2c (C) and, 2d (D); c = 20 μM; solvents: H2O (magenta), MeOH (black), CHCl3 (blue), CH2Cl2 (red), DMSO (green) and 1 N HCl (orange).
Figure 3: Absorption spectra of derivatives 2a (A), 2b (B), 2c (C) and, 2d (D); c = 20 μM; solvents: H2O (mag...
Table 2: Absorption and emission properties of benzo[b]quinolizinium derivatives 2a–d.
| 2a | 2b | 2c | 2d | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Solventa | λabsb / nm | λflc / nm | Φfld / 10−2 | λabsb / nm | λflc / nm | λabsb / nm | λflc / nm | λabsb / nm | λflc / nm | Φfld / 10−2 |
| H2O | 419 | 460 | < 1 | 422 | 562 | 434 | n.d.e | 422 | 572 | <1 |
| MeOH | 419 | 462 | 64 | 426 | 545 | 470 | n.d.e | 428 | 571 | 6 |
| EtOH | 420 | 462 | 39 | 428 | 558 | 481 | n.d.e | 430 | 570 | 6 |
| MeCN | 418 | 460 | 40 | 423 | n.d.e | 472 | n.d.e | 428 | 578 | 5 |
| DMSO | 423 | n.d.e | n.d.e | 428 | n.d.e | 473 | n.d.e | 432 | 570 | <1 |
| aceton | 419 | 460 | 34 | 424 | n.d.e | 472 | n.d.e | 429 | 580 | 2 |
| CH2Cl2 | 428 | 470 | 44 | 435 | 554 | 515 | 497 | 439 | 560 | 4 |
| CHCl3 | 427 | 470 | 43 | 438 | 485 | 505 | 501 | 443 | 460 | 2 |
aSolvents in order of decreasing ET values [47]. bLong-wavelength absorption maximum; c = 20 μM. cFluorescence emission maximum (Abs. = 0.10 at excitation wavelength); λex = 375 nm. dFluorescence quantum yield relative to coumarin 1 [47,48]; estimated error for Φfl: ± 10%. eNot determined.
Except for the derivative 2a the arylethynylbenzoquinolizinium derivatives have low emission quantum yields (Table 2, Figure 4). The derivative 2a has a moderate to high fluorescence intensity with slight deviations of the emission maxima in different solvents (Table 2, Figure 4A). In chloroform, it has two emission maxima at 446 and 470 nm. The derivative 2d has a weak fluorescence intensity in different solvents (Φfl: 0.02–0.06). In chloroform, it shows an emission maximum at 460 nm, while in other solvents it has emission maxima between 560 and 580 nm with a shoulder at 430 nm (Figure 4C). On the other hand, derivatives 2b and 2c exhibit very weak fluorescence intensity in different solvents (Φfl < 0.02). Derivative 2c shows only a weak emission (Φfl = 0.02) in 1 N HCl with significantly blue-shifted emission maxima at 427 and 454 nm (Figure 4B).
![[1860-5397-14-161-4]](/bjoc/content/figures/1860-5397-14-161-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Emission spectra of derivatives 2a (A), 2c (B) and 2d (C); c = 20 μM; λex = 375 nm; solvents: H2O (magenta), MeOH (black), CHCl3 (blue), CH2Cl2 (red), DMSO (green) and 1 N HCl (orange); λex = 375 nm.
Figure 4: Emission spectra of derivatives 2a (A), 2c (B) and 2d (C); c = 20 μM; λex = 375 nm; solvents: H2O (...
To further assess the effect of the pH on the absorption and emission properties of derivative 2c, photometric and fluorimetric acid–base titrations of 2c were performed (Figure 5). With decreasing pH of the solution (pH 7.3–1.1), new absorption bands developed at λmax = 418 nm, 395 nm and 322 nm, along with the disappearance of the initial broad long wavelength absorption (Figure 5A). The emission intensity of derivative 2c increased by a factor of 250 with decreasing pH value (Figure 5B). The pKa value of the protonated amine 2c in water was determined from the titration curve to be 3.1 which is in the same range as the ones of 9-(p-amino)phenylacridinium ions (pKa = 2.5–3.5) [49,50] and the dimethylaminophenyl-substituted benzo[b]quinolizinium ion [34] (pKa = 3.8).
![[1860-5397-14-161-5]](/bjoc/content/figures/1860-5397-14-161-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Photometric (A) and fluorimetric (B) acid-base titration of 2c; c = 20 μM in Britton–Robinson buffer; λex = 375 nm. Arrows indicate the development of absorption or emission bands with decreasing pH value. Inset: Plot of the absorption (A) at 360 nm (black rectangle) and 322 nm (white rectangle) and emission (B) at 427 nm (black triangle) versus pH. Lines denote the best fit of experimental data to the theoretical model.
Figure 5: Photometric (A) and fluorimetric (B) acid-base titration of 2c; c = 20 μM in Britton–Robinson buffe...
Photometric and fluorimetric DNA titrations of 9-(arylethynyl)benzo[b]quinolizinium derivatives 2a–d
The interactions of the arylethynylbenzoquinolizinium derivatives 2a–d with ct DNA and G4-DNA 22AG [d(AG3T2AG3T2AG3T2AG3)] were investigated with photometric and fluorimetric titrations (Figures 6–9, Table 3). In general, a hypochromic effect and a bathochromic shift were observed by the addition of DNA. For example, the addition of ct DNA and G4-DNA 22AG to derivative 2a led to the evolution of a new maximum at 437 nm and 423 nm, respectively, with an isosbestic point at 325 nm. However, during the titration of DNA to the derivatives 2b–d isosbestic points were not formed. In the case of 2d, only a hypochromic effect was observed upon the addition of ct DNA (Figure 6D). In contrast, the addition of 22AG to 2d resulted in a red shift with Δλabs = 16 nm. Notably, the addition of DNA to derivative 2c led to the largest bathochromic shifts and hypochromic effect (ct DNA: Δλabs = 42 nm; 22AG: Δλabs = 58 nm). Only the data extracted from the photometric titration of 22AG to derivatives 2b and 2c could be used to deduce the binding constant Kb (Figure S2, Table 3).
![[1860-5397-14-161-6]](/bjoc/content/figures/1860-5397-14-161-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Photometric titration of 2a (A), 2b (B), 2c (C), and 2d (D) with ct DNA in BPE buffer (16 mM Na+; 5% DMSO; pH 7.0); cL = 20.0 μM. Arrows indicate the development of bands with increasing DNA concentration. Inset: Plot of the absorption at long wavelength versus DNA concentration.
Figure 6: Photometric titration of 2a (A), 2b (B), 2c (C), and 2d (D) with ct DNA in BPE buffer (16 mM Na+; 5...
![[1860-5397-14-161-7]](/bjoc/content/figures/1860-5397-14-161-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Photometric titration of 2a (A), 2b (B), 2c (C) and 2d (D) with 22AG in potassium phosphate buffer (95 mM K+; 5% DMSO; pH 7.0); cL = 20.0 μM. Arrows indicate the development of bands with increasing DNA concentration. Inset: Plot of the absorption at long wavelength versus DNA concentration.
Figure 7: Photometric titration of 2a (A), 2b (B), 2c (C) and 2d (D) with 22AG in potassium phosphate buffer ...
Table 3: Absorption and emission properties of ligands 2a–d upon the addition of DNA, and binding constants Kb.
| Ligand | ct DNA | 22AG | ||||||
|---|---|---|---|---|---|---|---|---|
| λabsa / nm | Δλabsb / nm | I/I0c | Kbd / 104 M−1 | λabsa / nm | Δλabsb / nm | I/I0c | Kbd / 104 M−1 | |
| 2a | 437 | 18 | 0.14 | 14 | 432 | 19 | 0.05 | 22 |
| 2b | 443 | 21 | 3 | 1.5 | 440 | 18 | n.d.e | 2.6f |
| 2c | 476 | 42 | n.d.e | n.d.e | 492 | 58 | n.d.e | 1.6f |
| 2d | 422 | 0 | 0.38 | n.d.e | 438 | 16 | 0.19 | 3.0 |
aLong-wavelength absorption maximum of the DNA-bound ligand. bShift of the long-wavelength absorption maximum between free and bound ligand. cRelative emission intensity, I/I0 (I = emission intensity of DNA-bound ligand at saturation, I0 = emission of unbound ligand). dBinding constant of ligand–DNA complex, Kb, determined from fluorimetric titrations. eNot determined. fKb determined from photometric titrations; DNA concentration in base pairs for ct DNA and in oligonucleotide for 22AG.
The addition of ct DNA to the derivative 2a led to quenching of the emission intensity (Figure 8A). In contrast, a light-up effect with a factor of 3 was observed upon the addition of ct DNA to derivative 2b (Figure 8B, Table 2). Notably, the emission intensity of derivative 2d at λfl = 572 nm decreased at the beginning of the titration with ct DNA at a ligand–DNA ratio (LDR) > 8. With further addition of ct DNA, however, the emission intensity increased slightly at the same emission wavelength (Figure 8C). The binding constants, Kb, between ct DNA and derivatives 2a (1.4 × 105 M−1) and 2b (1.5 × 104 M−1) were determined from the fluorimetric titrations by fitting the resulting binding isotherms to the theoretical model (insets in Figure 8, Table 3) [51]. Unfortunately, the data obtained from the fluorimetric titration of 2d with ct DNA could not be fitted to the theoretical model. The low emission intensity of the derivative 2c was not affected by the addition of ct DNA or 22AG.
![[1860-5397-14-161-8]](/bjoc/content/figures/1860-5397-14-161-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Fluorimetric titration of 2a (A), 2b (B) and 2d (C) with ct DNA in potassium phosphate buffer (95 mM K+; 5% DMSO; pH 7.0); cLigand = 20.0 μM. Arrows indicate the development of the bands with increasing DNA concentration. Inset: Plot of the relative emission intensity, I/I0 versus cDNA/cL. Lines denote the best fit of experimental data to the theoretical model; λex = 335 nm (A), 420 nm (B) and 380 nm (C).
Figure 8: Fluorimetric titration of 2a (A), 2b (B) and 2d (C) with ct DNA in potassium phosphate buffer (95 m...
The emission intensity of 2a was quenched upon addition of G4-DNA 22AG (Figure 9A). Remarkably, the addition of 22AG to derivative 2d resulted in a decrease of the emission intensity at λfl = 572 nm and a new weak emission band evolved at λfl = 425 nm. The emission intensity of 2b was not influenced significantly by the addition of 22AG. The binding constants Kb between 22AG and derivatives 2a (2.2 × 105 M−1) and 2d (3.0 × 104 M−1) were determined from the fluorimetric data by fitting the binding isotherms to the theoretical model (insets in Figure 9, Table 2) [51].
![[1860-5397-14-161-9]](/bjoc/content/figures/1860-5397-14-161-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Fluorimetric titration of 2a (A) and 2d (B) with 22AG in potassium phosphate buffer (95 mM K+; 5% DMSO; pH 7.0); cLigand = 20.0 μM. Arrows indicate the development of the bands with increasing DNA concentration. Inset: Plot of the relative emission intensity, I/I0 versus cDNA/cL. Lines denote the best fit of experimental data to the theoretical model; λex = 335 nm (A) and 380 nm (B).
Figure 9: Fluorimetric titration of 2a (A) and 2d (B) with 22AG in potassium phosphate buffer (95 mM K+; 5% D...
Discussion
Pd-mediated coupling reactions of halogenobenzo[b]quinolizinium derivatives
Although it was shown in this work that in particular cases appropriately substituted benzo[b]quinolizinium substrates can be functionalized as aryl- or alkynyl-substituted derivatives by Sonogashira and base-free Suzuki–Miyaura coupling reactions, it is obvious that this synthetic approach has its limitations. As compared with the corresponding quinolizinium substrates, that can be used for a variety of metal-mediated coupling reactions [28-32], the benzo[b]quinolizinium core appears to be very sensitive towards the reaction conditions, leading to serious side or secondary reactions. All experimental results indicate that the "usual" experimental protocols cannot be applied due to the high susceptibility of the benzo[b]quinolizinium ring towards nucleophilic attack at 6-position that leads to ring opening [37,38]. Thus, the Sonogashira reaction of 5 requires either the separate generation of copper acetylide or strict water-free conditions to avoid the formation of hydroxide ions. To avoid the potential interference of bases, we attempted to improve of the conditions for the Suzuki–Miyaura coupling in the base-free variant using aryldiazonium reagents [39]. Although the coupling reactions between aryldiazonium salts and arylboronic acids or esters with base-free conditions are known [39,42], in our hands the reaction of benzo[b]quinolizinium-9-boronic acid (3a) with benzenediazonium tetrafluoroborate 4a only resulted in the formation of the benzo[b]quinolizinium-9-trifluoroborate (3b). Consequently, we used the latter substrate for subsequent synthesis, as it has been reported that organotrifluoroborates may also be employed as starting materials in Suzuki–Miyaura coupling reactions of aryl halides [52,53]. Indeed, starting from benzo[b]quinolizinium-9-trifluoroborate (3b) and the corresponding aryldiazonium ions the 9-arylbenzo[b]quinolizinium derivatives 1a–d were available in yields that are comparable, or even slightly higher, than the ones obtained with the Suzuki–Miyaura reaction of benzo[b]quinolizinium-9-boronic acid (3a) with bromoarenes [34].
In our previous attempts to synthesize the corresponding benzo[b]quinolizinium-9-trifluoroborate (3b), the reaction of benzo[b]quinolizinium-9-boronic acid (3a) with KHF2 only resulted in a partly contaminated product [34]. In this work, we used NaBF4 as reagent, as we have rather accidentally observed that it can be used for the synthesis of the trifluoroborate 3b (see above); however, with lower yield (Scheme 3). Interestingly, to the best of our knowledge, there is only one report in the literature about the explicit use of NaBF4 as fluorinating reagent for boronic acids [54], and we have not investigated the general applicability of this reaction so far. Nevertheless, this approach appears to be a useful, complementary method to the usual fluorination with KHF2. And it may be suggested that this simple procedure might be used as a general straightforward method for the generation of the synthetically highly useful aryltrifluoroborates.
![[1860-5397-14-161-i3]](/bjoc/content/inline/1860-5397-14-161-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Photoinduced charge transfer upon the excitation of derivative 2d.
Scheme 3: Photoinduced charge transfer upon the excitation of derivative 2d.
Absorption and emission properties of 9-(arylethynyl)benzo[b]quinolizinium derivatives
The absorption maxima of the 9-(arylethynyl)benzo[b]quinolizinium derivatives 2a–c are slightly red-shifted as compared to the corresponding 9-arylbenzo[b]quinolizinium compounds 1a–c [34]. And in similar analogy, the extent of the red shift (H < OMe < NMe2) corresponds well with the strength of the donor–acceptor interplay between the electron-donating aryl substituent and the benzo[b]quinolizinium chromophore. The absorption properties depend only slightly on the solvent properties indicating that the corresponding ground state and vertical excited states are stabilized by the solvents to the same degree. As the only exception, larger red shifts of the absorption maxima of derivatives 2a–d were observed in CHCl3 and CH2Cl2, which is presumably caused by the high polarizability of these solvents, as frequently observed with cationic dyes [34,46,55,56]. In the case of alkaline compound 2c, the protonation of the amino group changes the ammonium-substituted aryl substituent to an electron acceptor which leads to a blue shift of the absorption maxima (Figure 3C).
The emission properties of the phenyl-substituted derivative 2a do not depend significantly on the solvent properties, which indicate the absence of specific stabilization or destabilization of the excited molecule, even after solvent relaxation. In contrast, the phenanthryl-substituted derivative 2d shows fluorosolvatochromism, specifically indicated by the strong blue shift in CHCl3. This effect is presumably caused by a charge shift (CS) or, more likely, by a charge transfer (CT) in the excited state from the electron-donating aryl unit to the excited quinolizinium (Scheme 3) [57], which has been proposed also to take place in structurally resembling excited biaryl-type acridinium, benzo[b]quinolizinium and naphtho[b]quinolizinium derivatives [36,58-64]. The CS/CT leads to an intermediate excited molecule with a charge neutral quinolizinyl radical and the radical cation of the phenanthryl unit (Scheme 3) that is well stabilized in polar solvents after solvent relaxation. At the same time, less polar solvents such as CHCl3 cannot stabilize this intermediate so that emission occurs from the energetically higher first local excited (LE) state which results in the blue-shifted emission. It should be noted that this blue-shifted emission band was also observed in polar solvents though with less intensity (Figure 4), which indicates that the emission from the LC state can compete with the charge shift and solvent relaxation, leading to dual emission.
Remarkably, the emission quantum yields of the methoxy- and amino-substituted derivatives 2b and 2c are very low (Φfl < 0.02). Such low emission intensities have been observed also for donor-substituted 9-arylbenzo[b]quinolizinium derivatives and explained either with a radiationless deactivation of the excited state by torsional relaxation or by a photoinduced electron transfer [33,49,65,66]. The effect of the donor substituent on the emission quenching was supported by the strong increase of the emission quantum yield of 2c upon protonation of the amino group, that is, by the transformation of the donor to an acceptor substituent (Figure 5) [34]. Considering the water solubility of compound 2c, though just moderate, the emission light-up effect may be used for fluorimetric detection of slightly acidic aqueous media.
Interactions with DNA
The spectrometric titrations of DNA to compounds 2a–d revealed the characteristic spectroscopic features of ligand–DNA interactions, namely a hypochromic effect and red shift of the absorption bands as well as emission quenching or enhancement upon addition of the nucleic acid. Moreover, the binding constants Kb, as determined from the resulting binding isotherms, are in the same range (Kb = 2.0–22 × 104 M−1, Table 3) of known DNA-intercalating benzo[b]quinolizinium derivatives [52,67,68], so that it may be concluded that the derivatives 2a–d bind to DNA in a similar binding mode. Notably, a pronounced decrease of the long-wavelength absorption followed by the development of new band at longer wavelength was observed during the photometric titrations (Figure 6 and Figure 7), and only in the titration of the phenylethynyl-substituted derivative 2a an isosbestic point was formed. These observations clearly show that the ligands bind in at least two different binding modes to DNA. Considering the low solubility of these compounds in water it is assumed that at the beginning of the titration, i.e., with large ligand–DNA ratio and a paucity of DNA binding sites, the ligand forms aggregates along the DNA backbone. With increasing DNA concentration more binding sites are available such that the ligands can intercalate. In the case of quadruplex DNA, the derivatives 2a–d show a typical titration signature for ligands that bind to the quadruplex by terminal π-stacking [14]; however, in analogy to the binding to duplex DNA the derivatives 2b–d form aggregates along the DNA backbone at large ligand–DNA ratio, i.e., at the beginning of the titration.
The fluorescence intensity of the derivatives 2a and 2d is significantly quenched by the addition of DNA, respectively (Figure 8 and Figure 9). This observation usually indicates a photoinduced electron transfer between the excited molecules and the DNA bases [69]. By contrast, the association of ct DNA with the methoxy-substituted derivative 2b led to an increase of the low emission intensity by a factor of 3 (Figure 8B). Although this effect is rather small, it indicates the suppression of a deactivation pathway in the excited state upon the accommodation of 2b in a constrained binding site of ct DNA, presumably due to the restriction of the conformational flexibility inside the binding site [65].
Conclusion
In summary, different synthetic approaches toward the Pd-mediated coupling reactions of benzo[b]quinolizinium derivatives were assessed that enable the functionalization and further development of this useful class of compounds. In particular, we demonstrated that optimized base-free Suzuki–Miyaura and Sonogashira coupling reactions can be used for the synthesis of aryl- and arylalkynyl-substituted benzo[b]quinolizinium derivatives in moderate to good yields. Therefore, the optimized protocol for Pd-mediated reactions may be employed for other base-sensitive substrates as well.
The photophysical properties as well as the DNA-binding properties of the (arylethynyl)benzo[b]quinolizinium derivatives were studied. It was demonstrated that derivatives 2a–d bind to duplex and quadruplex DNA with binding constants Kb of 0.2–2.2 × 105 M−1. Unfortunately, a differentiation between duplex and quadruplex DNA by derivatives 2a–d was not observed. Therefore, future work has to focus on further functionalizations that lead to selective binding of the ligands to particular DNA forms, e.g., by fine tuning of the stereoelectronic or steric properties of substituents.
Supporting Information
| Supporting Information File 1: Additional spectral data, detailed description of the experiments performed, 1H NMR of the derivatives 2a–d and crystallographic data. | ||
| Format: PDF | Size: 898.0 KB | Download |
References
-
Pett, L.; Hartley, J.; Kiakos, K. Curr. Top. Med. Chem. 2015, 15, 1293–1322. doi:10.2174/1568026615666150413155431
Return to citation in text: [1] [2] -
Rescifina, A.; Zagni, C.; Varrica, M. G.; Pistarà, V.; Corsaro, A. Eur. J. Med. Chem. 2014, 74, 95–115. doi:10.1016/j.ejmech.2013.11.029
Return to citation in text: [1] -
Banerjee, S.; Veale, E. B.; Phelan, C. M.; Murphy, S. A.; Tocci, G. M.; Gillespie, L. J.; Frimannsson, D. O.; Kelly, J. M.; Gunnlaugsson, T. Chem. Soc. Rev. 2013, 42, 1601–1618. doi:10.1039/c2cs35467e
Return to citation in text: [1] -
Pazos, E.; Mosquera, J.; Vázquez, M. E.; Mascareñas, J. L. ChemBioChem 2011, 12, 1958–1973. doi:10.1002/cbic.201100247
Return to citation in text: [1] -
Pommier, Y. ACS Chem. Biol. 2013, 8, 82–95. doi:10.1021/cb300648v
Return to citation in text: [1] -
Neidle, S. Nat. Rev. Chem. 2017, 1, 41. doi:10.1038/s41570-017-0041
Return to citation in text: [1] -
Neidle, S. J. Med. Chem. 2016, 59, 5987–6011. doi:10.1021/acs.jmedchem.5b01835
Return to citation in text: [1] -
Balasubramanian, S.; Hurley, L. H.; Neidle, S. Nat. Rev. Drug Discovery 2011, 10, 261–275. doi:10.1038/nrd3428
Return to citation in text: [1] -
Xu, Y. Chem. Soc. Rev. 2011, 40. doi:10.1039/c0cs00134a
Return to citation in text: [1] -
Murat, P.; Balasubramanian, S. Curr. Opin. Genet. Dev. 2014, 25, 22–29. doi:10.1016/j.gde.2013.10.012
Return to citation in text: [1] -
Xiong, Y.-X.; Huang, Z.-S.; Tan, J.-H. Eur. J. Med. Chem. 2015, 97, 538–551. doi:10.1016/j.ejmech.2014.11.021
Return to citation in text: [1] -
Maji, B.; Bhattacharya, S. Chem. Commun. 2014, 50, 6422–6438. doi:10.1039/C4CC00611A
Return to citation in text: [1] -
Ohnmacht, S. A.; Neidle, S. Bioorg. Med. Chem. Lett. 2014, 24, 2602–2612. doi:10.1016/j.bmcl.2014.04.029
Return to citation in text: [1] -
Granzhan, A.; Ihmels, H. Synlett 2016, 27, 1775–1793. doi:10.1055/s-0035-1561445
Return to citation in text: [1] [2] -
Manna, S. K.; Mandal, A.; Mondal, S. K.; Adak, A. K.; Jana, A.; Das, S.; Chattopadhyay, S.; Roy, S.; Ghorai, S. K.; Samanta, S.; Hossain, M.; Baidya, M. Org. Biomol. Chem. 2015, 13, 8037–8047. doi:10.1039/C5OB01082A
Return to citation in text: [1] -
Abarca, B.; Custodio, R.; Cuadro, A. M.; Sucunza, D.; Domingo, A.; Mendicuti, F.; Alvarez-Builla, J.; Vaquero, J. J. Org. Lett. 2014, 16, 3464–3467. doi:10.1021/ol5013668
Return to citation in text: [1] -
Suárez, R. M.; Bosch, P.; Sucunza, D.; Cuadro, A. M.; Domingo, A.; Mendicuti, F.; Vaquero, J. J. Org. Biomol. Chem. 2015, 13, 527–538. doi:10.1039/C4OB01465K
Return to citation in text: [1] -
Miskolczy, Z.; Megyesi, M.; Biczók, L.; Görner, H. Photochem. Photobiol. Sci. 2011, 10, 592–600. doi:10.1039/c0pp00367k
Return to citation in text: [1] -
Ma, D.-L.; He, H.-Z.; Leung, K.-H.; Zhong, H.-J.; Chan, D. S.-H.; Leung, C.-H. Chem. Soc. Rev. 2013, 42, 3427–3440. doi:10.1039/c2cs35472a
Return to citation in text: [1] -
Granzhan, A.; Ihmels, H.; Tian, M. ARKIVOC 2015, No. vi, 494–523.
Return to citation in text: [1] -
Gildner, P. G.; Colacot, T. J. Organometallics 2015, 34, 5497–5508. doi:10.1021/acs.organomet.5b00567
Return to citation in text: [1] -
Cano, R.; Schmidt, A. F.; McGlacken, G. P. Chem. Sci. 2015, 6, 5338–5346. doi:10.1039/C5SC01534K
Return to citation in text: [1] -
Cho, S. H.; Kim, J. Y.; Kwak, J.; Chang, S. Chem. Soc. Rev. 2011, 40, 5068–5083. doi:10.1039/c1cs15082k
Return to citation in text: [1] -
Beletskaya, I. P.; Cheprakov, A. V. Organometallics 2012, 31, 7753–7808. doi:10.1021/om300683c
Return to citation in text: [1] -
Johansson Seechurn, C. C. C.; Kitching, M. O.; Colacot, T. J.; Snieckus, V. Angew. Chem., Int. Ed. 2012, 51, 5062–5085. doi:10.1002/anie.201107017
Return to citation in text: [1] -
Fortman, G. C.; Nolan, S. P. Chem. Soc. Rev. 2011, 40, 5151–5169. doi:10.1039/c1cs15088j
Return to citation in text: [1] -
de Meijere, A.; Diederich, F. Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/9783527619535
Return to citation in text: [1] -
Cañeque, T.; Cuadro, A. M.; Alvarez-Builla, J.; Vaquero, J. J. Tetrahedron Lett. 2009, 50, 1419–1422. doi:10.1016/j.tetlet.2009.01.040
Return to citation in text: [1] [2] -
García-Cuadrado, D.; Cuadro, A. M.; Alvarez-Builla, J.; Vaquero, J. J. Synlett 2002, 1904–1906. doi:10.1055/s-2002-34903
Return to citation in text: [1] [2] -
Barchín, B. M.; Valenciano, J.; Cuadro, A. M.; Vaquero, J. J. Org. Lett. 1999, 1, 545–548. doi:10.1021/ol990626y
Return to citation in text: [1] [2] -
García-Cuadrado, D.; Cuadro, A. M.; Barchín, B. M.; Nuñez, A.; Cañeque, T.; Alvarez-Builla, J.; Vaquero, J. J. J. Org. Chem. 2006, 71, 7989–7995. doi:10.1021/jo060634+
Return to citation in text: [1] [2] -
García, D.; Cuadro, A. M.; Alvarez-Builla, J.; Vaquero, J. J. Org. Lett. 2004, 6, 4175–4178. doi:10.1021/ol048368e
Return to citation in text: [1] [2] -
Sucunza, D.; Cuadro, A. M.; Alvarez-Builla, J.; Vaquero, J. J. J. Org. Chem. 2016, 81, 10126–10135. doi:10.1021/acs.joc.6b01092
Return to citation in text: [1] [2] -
Tian, M.; Ihmels, H. Synthesis 2009, 4226–4234. doi:10.1055/s-0029-1217060
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] -
Tian, M.; Ihmels, H.; Ye, S. Org. Biomol. Chem. 2012, 10, 3010–3018. doi:10.1039/c2ob06948b
Return to citation in text: [1] [2] [3] -
Pithan, P. M.; Decker, D.; Sardo, M. S.; Viola, G.; Ihmels, H. Beilstein J. Org. Chem. 2016, 12, 854–862. doi:10.3762/bjoc.12.84
Return to citation in text: [1] [2] [3] -
Deiseroth, H.-J.; Granzhan, A.; Ihmels, H.; Schlosser, M.; Tian, M. Org. Lett. 2008, 10, 757–760. doi:10.1021/ol702792r
Return to citation in text: [1] [2] -
Krapcho, A. P.; Cadamuro, S. A.; Macnee, L. ARKIVOC 2007, No. ix, 28–44.
Return to citation in text: [1] [2] -
Colleville, A. P.; Horan, R. A. J.; Tomkinson, N. C. O. Org. Process Res. Dev. 2014, 18, 1128–1136. doi:10.1021/op5002353
Return to citation in text: [1] [2] [3] -
Oger, N.; Felpin, F.-X. ChemCatChem 2016, 8, 1998–2009. doi:10.1002/cctc.201600134
Return to citation in text: [1] -
Mahanta, A.; Raul, P. K.; Saikia, S.; Bora, U.; Thakur, A. J. Appl. Organomet. Chem. 2018, 32, e4192. doi:10.1002/aoc.4192
Return to citation in text: [1] -
Bonin, H.; Delbrayelle, D.; Demonchaux, P.; Gras, E. Chem. Commun. 2010, 46, 2677–2679. doi:10.1039/b926547n
Return to citation in text: [1] [2] -
Bradsher, C. K.; Sherer, J. P.; Parham, J. H. J. Chem. Eng. Data 1965, 10, 180–183. doi:10.1021/je60025a036
Return to citation in text: [1] -
Gallego, D.; Brück, A.; Irran, E.; Meier, F.; Kaupp, M.; Driess, M.; Hartwig, J. F. J. Am. Chem. Soc. 2013, 135, 15617–15626. doi:10.1021/ja408137t
Return to citation in text: [1] -
Ihmels, H.; Leusser, D.; Pfeiffer, M.; Stalke, D. J. Org. Chem. 1999, 64, 5715–5718. doi:10.1021/jo990465b
Return to citation in text: [1] -
Stratford, S. A.; Arhangelskis, M.; Bučar, D.-K.; Jones, W. CrystEngComm 2014, 16, 10830–10836. doi:10.1039/c4ce01622j
Return to citation in text: [1] [2] [3] -
Jones, G.; Jackson, W. R.; Choi, C. Y.; Bergmark, W. R. J. Phys. Chem. 1985, 89, 294–300. doi:10.1021/j100248a024
Return to citation in text: [1] [2] -
Crosby, G. A.; Demas, J. N. J. Phys. Chem. 1971, 75, 991–1024. doi:10.1021/j100678a001
Return to citation in text: [1] -
Jonker, S. A.; Ariese, F.; Verhoeven, J. W. Recl. Trav. Chim. Pays-Bas 1989, 108, 109–115. doi:10.1002/recl.19891080307
Return to citation in text: [1] [2] -
Jonker, S. A.; van Dijk, S. I.; Goubitz, K.; Reiss, C. A.; Schuddeboom, W.; Verhoeven, J. W. Mol. Cryst. Liq. Cryst. 1990, 183, 273–282. doi:10.1080/15421409008047464
Return to citation in text: [1] -
Stootman, F. H.; Fisher, D. M.; Rodger, A.; Aldrich-Wright, J. R. Analyst 2006, 131, 1145–1151. doi:10.1039/b604686j
Return to citation in text: [1] [2] -
Molander, G. A.; Canturk, B.; Kennedy, L. E. J. Org. Chem. 2009, 74, 973–980. doi:10.1021/jo802590b
Return to citation in text: [1] [2] -
Molander, G. A.; Petrillo, D. E. Org. Lett. 2008, 10, 1795–1798. doi:10.1021/ol800357c
Return to citation in text: [1] -
Gott, A. L.; Piers, W. E.; McDonald, R.; Parvez, M. Inorg. Chim. Acta 2011, 369, 180–189. doi:10.1016/j.ica.2010.12.030
Return to citation in text: [1] -
Granzhan, A.; Ihmels, H.; Viola, G. J. Am. Chem. Soc. 2007, 129, 1254–1267. doi:10.1021/ja0668872
Return to citation in text: [1] -
van den Berg, O.; Jager, W. F.; Picken, S. J. J. Org. Chem. 2006, 71, 2666–2676. doi:10.1021/jo052441c
Return to citation in text: [1] -
Bendig, J.; Geppert, B.; Helm, S.; Kreysig, D. Theor. Exp. Chem. 1978, 14, 488–495. doi:10.1007/BF01004352
Return to citation in text: [1] -
Kotani, H.; Ohkubo, K.; Fukuzumi, S. Faraday Discuss. 2012, 155, 89–102. doi:10.1039/C1FD00084E
Return to citation in text: [1] -
Verhoeven, J. W.; van Ramesdonk, H. J.; Groeneveld, M. M.; Benniston, A. C.; Harriman, A. ChemPhysChem 2005, 6, 2251–2260. doi:10.1002/cphc.200500029
Return to citation in text: [1] -
Horng, M.; Dahl, K.; Jones, G., II; Maroncelli, M. Chem. Phys. Lett. 1999, 315, 363–370. doi:10.1016/s0009-2614(99)01258-0
Return to citation in text: [1] -
Jones, G., II; Farahat, M. S.; Greenfield, S. R.; Gosztola, D. J.; Wasielewski, M. R. Chem. Phys. Lett. 1994, 229, 40–46. doi:10.1016/0009-2614(94)00996-1
Return to citation in text: [1] -
Li, X.; Liang, M.; Chakraborty, A.; Kondo, M.; Maroncelli, M. J. Phys. Chem. B 2011, 115, 6592–6607. doi:10.1021/jp200339e
Return to citation in text: [1] -
Hu, J.; Xia, B.; Bao, D.; Ferreira, A.; Wan, J.; Jones, G.; Vullev, V. I. J. Phys. Chem. A 2009, 113, 3096–3107. doi:10.1021/jp810909v
Return to citation in text: [1] -
Jones, G.; Yan, D.-X.; Gosztola, D. J.; Greenfield, S. R.; Wasielewski, M. R. J. Am. Chem. Soc. 1999, 121, 11016–11017. doi:10.1021/ja9927319
Return to citation in text: [1] -
Bortolozzi, R.; Ihmels, H.; Thomas, L.; Tian, M.; Viola, G. Chem. – Eur. J. 2013, 19, 8736–8741. doi:10.1002/chem.201301164
Return to citation in text: [1] [2] -
de Silva, A. P.; Gunaratne, H. Q. N.; Gunnlaugsson, T.; Huxley, A. J. M.; McCoy, C. P.; Rademacher, J. T.; Rice, T. E. Chem. Rev. 1997, 97, 1515–1566. doi:10.1021/cr960386p
Return to citation in text: [1] -
Faulhaber, K.; Granzhan, A.; Ihmels, H.; Otto, D.; Thomas, L.; Wells, S. Photochem. Photobiol. Sci. 2011, 10, 1535–1545. doi:10.1039/c1pp05106g
Return to citation in text: [1] -
Ihmels, H.; Faulhaber, K.; Vedaldi, D.; Dall’Acqua, F.; Viola, G. Photochem. Photobiol. 2005, 81, 1107–1115. doi:10.1562/2005-01-25-ir-427
Return to citation in text: [1] -
Juskowiak, B. Anal. Bioanal. Chem. 2011, 399, 3157–3176. doi:10.1007/s00216-010-4304-5
Return to citation in text: [1]
| 51. | Stootman, F. H.; Fisher, D. M.; Rodger, A.; Aldrich-Wright, J. R. Analyst 2006, 131, 1145–1151. doi:10.1039/b604686j |
| 28. | Cañeque, T.; Cuadro, A. M.; Alvarez-Builla, J.; Vaquero, J. J. Tetrahedron Lett. 2009, 50, 1419–1422. doi:10.1016/j.tetlet.2009.01.040 |
| 29. | García-Cuadrado, D.; Cuadro, A. M.; Alvarez-Builla, J.; Vaquero, J. J. Synlett 2002, 1904–1906. doi:10.1055/s-2002-34903 |
| 30. | Barchín, B. M.; Valenciano, J.; Cuadro, A. M.; Vaquero, J. J. Org. Lett. 1999, 1, 545–548. doi:10.1021/ol990626y |
| 31. | García-Cuadrado, D.; Cuadro, A. M.; Barchín, B. M.; Nuñez, A.; Cañeque, T.; Alvarez-Builla, J.; Vaquero, J. J. J. Org. Chem. 2006, 71, 7989–7995. doi:10.1021/jo060634+ |
| 32. | García, D.; Cuadro, A. M.; Alvarez-Builla, J.; Vaquero, J. J. Org. Lett. 2004, 6, 4175–4178. doi:10.1021/ol048368e |
| 37. | Deiseroth, H.-J.; Granzhan, A.; Ihmels, H.; Schlosser, M.; Tian, M. Org. Lett. 2008, 10, 757–760. doi:10.1021/ol702792r |
| 38. | Krapcho, A. P.; Cadamuro, S. A.; Macnee, L. ARKIVOC 2007, No. ix, 28–44. |
| 1. | Pett, L.; Hartley, J.; Kiakos, K. Curr. Top. Med. Chem. 2015, 15, 1293–1322. doi:10.2174/1568026615666150413155431 |
| 2. | Rescifina, A.; Zagni, C.; Varrica, M. G.; Pistarà, V.; Corsaro, A. Eur. J. Med. Chem. 2014, 74, 95–115. doi:10.1016/j.ejmech.2013.11.029 |
| 3. | Banerjee, S.; Veale, E. B.; Phelan, C. M.; Murphy, S. A.; Tocci, G. M.; Gillespie, L. J.; Frimannsson, D. O.; Kelly, J. M.; Gunnlaugsson, T. Chem. Soc. Rev. 2013, 42, 1601–1618. doi:10.1039/c2cs35467e |
| 4. | Pazos, E.; Mosquera, J.; Vázquez, M. E.; Mascareñas, J. L. ChemBioChem 2011, 12, 1958–1973. doi:10.1002/cbic.201100247 |
| 9. | Xu, Y. Chem. Soc. Rev. 2011, 40. doi:10.1039/c0cs00134a |
| 10. | Murat, P.; Balasubramanian, S. Curr. Opin. Genet. Dev. 2014, 25, 22–29. doi:10.1016/j.gde.2013.10.012 |
| 11. | Xiong, Y.-X.; Huang, Z.-S.; Tan, J.-H. Eur. J. Med. Chem. 2015, 97, 538–551. doi:10.1016/j.ejmech.2014.11.021 |
| 12. | Maji, B.; Bhattacharya, S. Chem. Commun. 2014, 50, 6422–6438. doi:10.1039/C4CC00611A |
| 13. | Ohnmacht, S. A.; Neidle, S. Bioorg. Med. Chem. Lett. 2014, 24, 2602–2612. doi:10.1016/j.bmcl.2014.04.029 |
| 6. | Neidle, S. Nat. Rev. Chem. 2017, 1, 41. doi:10.1038/s41570-017-0041 |
| 7. | Neidle, S. J. Med. Chem. 2016, 59, 5987–6011. doi:10.1021/acs.jmedchem.5b01835 |
| 8. | Balasubramanian, S.; Hurley, L. H.; Neidle, S. Nat. Rev. Drug Discovery 2011, 10, 261–275. doi:10.1038/nrd3428 |
| 43. | Bradsher, C. K.; Sherer, J. P.; Parham, J. H. J. Chem. Eng. Data 1965, 10, 180–183. doi:10.1021/je60025a036 |
| 34. | Tian, M.; Ihmels, H. Synthesis 2009, 4226–4234. doi:10.1055/s-0029-1217060 |
| 46. | Stratford, S. A.; Arhangelskis, M.; Bučar, D.-K.; Jones, W. CrystEngComm 2014, 16, 10830–10836. doi:10.1039/c4ce01622j |
| 55. | Granzhan, A.; Ihmels, H.; Viola, G. J. Am. Chem. Soc. 2007, 129, 1254–1267. doi:10.1021/ja0668872 |
| 56. | van den Berg, O.; Jager, W. F.; Picken, S. J. J. Org. Chem. 2006, 71, 2666–2676. doi:10.1021/jo052441c |
| 34. | Tian, M.; Ihmels, H. Synthesis 2009, 4226–4234. doi:10.1055/s-0029-1217060 |
| 35. | Tian, M.; Ihmels, H.; Ye, S. Org. Biomol. Chem. 2012, 10, 3010–3018. doi:10.1039/c2ob06948b |
| 1. | Pett, L.; Hartley, J.; Kiakos, K. Curr. Top. Med. Chem. 2015, 15, 1293–1322. doi:10.2174/1568026615666150413155431 |
| 39. | Colleville, A. P.; Horan, R. A. J.; Tomkinson, N. C. O. Org. Process Res. Dev. 2014, 18, 1128–1136. doi:10.1021/op5002353 |
| 40. | Oger, N.; Felpin, F.-X. ChemCatChem 2016, 8, 1998–2009. doi:10.1002/cctc.201600134 |
| 41. | Mahanta, A.; Raul, P. K.; Saikia, S.; Bora, U.; Thakur, A. J. Appl. Organomet. Chem. 2018, 32, e4192. doi:10.1002/aoc.4192 |
| 42. | Bonin, H.; Delbrayelle, D.; Demonchaux, P.; Gras, E. Chem. Commun. 2010, 46, 2677–2679. doi:10.1039/b926547n |
| 54. | Gott, A. L.; Piers, W. E.; McDonald, R.; Parvez, M. Inorg. Chim. Acta 2011, 369, 180–189. doi:10.1016/j.ica.2010.12.030 |
| 28. | Cañeque, T.; Cuadro, A. M.; Alvarez-Builla, J.; Vaquero, J. J. Tetrahedron Lett. 2009, 50, 1419–1422. doi:10.1016/j.tetlet.2009.01.040 |
| 29. | García-Cuadrado, D.; Cuadro, A. M.; Alvarez-Builla, J.; Vaquero, J. J. Synlett 2002, 1904–1906. doi:10.1055/s-2002-34903 |
| 30. | Barchín, B. M.; Valenciano, J.; Cuadro, A. M.; Vaquero, J. J. Org. Lett. 1999, 1, 545–548. doi:10.1021/ol990626y |
| 31. | García-Cuadrado, D.; Cuadro, A. M.; Barchín, B. M.; Nuñez, A.; Cañeque, T.; Alvarez-Builla, J.; Vaquero, J. J. J. Org. Chem. 2006, 71, 7989–7995. doi:10.1021/jo060634+ |
| 32. | García, D.; Cuadro, A. M.; Alvarez-Builla, J.; Vaquero, J. J. Org. Lett. 2004, 6, 4175–4178. doi:10.1021/ol048368e |
| 33. | Sucunza, D.; Cuadro, A. M.; Alvarez-Builla, J.; Vaquero, J. J. J. Org. Chem. 2016, 81, 10126–10135. doi:10.1021/acs.joc.6b01092 |
| 36. | Pithan, P. M.; Decker, D.; Sardo, M. S.; Viola, G.; Ihmels, H. Beilstein J. Org. Chem. 2016, 12, 854–862. doi:10.3762/bjoc.12.84 |
| 52. | Molander, G. A.; Canturk, B.; Kennedy, L. E. J. Org. Chem. 2009, 74, 973–980. doi:10.1021/jo802590b |
| 53. | Molander, G. A.; Petrillo, D. E. Org. Lett. 2008, 10, 1795–1798. doi:10.1021/ol800357c |
| 21. | Gildner, P. G.; Colacot, T. J. Organometallics 2015, 34, 5497–5508. doi:10.1021/acs.organomet.5b00567 |
| 22. | Cano, R.; Schmidt, A. F.; McGlacken, G. P. Chem. Sci. 2015, 6, 5338–5346. doi:10.1039/C5SC01534K |
| 23. | Cho, S. H.; Kim, J. Y.; Kwak, J.; Chang, S. Chem. Soc. Rev. 2011, 40, 5068–5083. doi:10.1039/c1cs15082k |
| 24. | Beletskaya, I. P.; Cheprakov, A. V. Organometallics 2012, 31, 7753–7808. doi:10.1021/om300683c |
| 25. | Johansson Seechurn, C. C. C.; Kitching, M. O.; Colacot, T. J.; Snieckus, V. Angew. Chem., Int. Ed. 2012, 51, 5062–5085. doi:10.1002/anie.201107017 |
| 26. | Fortman, G. C.; Nolan, S. P. Chem. Soc. Rev. 2011, 40, 5151–5169. doi:10.1039/c1cs15088j |
| 27. | de Meijere, A.; Diederich, F. Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/9783527619535 |
| 37. | Deiseroth, H.-J.; Granzhan, A.; Ihmels, H.; Schlosser, M.; Tian, M. Org. Lett. 2008, 10, 757–760. doi:10.1021/ol702792r |
| 38. | Krapcho, A. P.; Cadamuro, S. A.; Macnee, L. ARKIVOC 2007, No. ix, 28–44. |
| 19. | Ma, D.-L.; He, H.-Z.; Leung, K.-H.; Zhong, H.-J.; Chan, D. S.-H.; Leung, C.-H. Chem. Soc. Rev. 2013, 42, 3427–3440. doi:10.1039/c2cs35472a |
| 20. | Granzhan, A.; Ihmels, H.; Tian, M. ARKIVOC 2015, No. vi, 494–523. |
| 39. | Colleville, A. P.; Horan, R. A. J.; Tomkinson, N. C. O. Org. Process Res. Dev. 2014, 18, 1128–1136. doi:10.1021/op5002353 |
| 14. | Granzhan, A.; Ihmels, H. Synlett 2016, 27, 1775–1793. doi:10.1055/s-0035-1561445 |
| 15. | Manna, S. K.; Mandal, A.; Mondal, S. K.; Adak, A. K.; Jana, A.; Das, S.; Chattopadhyay, S.; Roy, S.; Ghorai, S. K.; Samanta, S.; Hossain, M.; Baidya, M. Org. Biomol. Chem. 2015, 13, 8037–8047. doi:10.1039/C5OB01082A |
| 16. | Abarca, B.; Custodio, R.; Cuadro, A. M.; Sucunza, D.; Domingo, A.; Mendicuti, F.; Alvarez-Builla, J.; Vaquero, J. J. Org. Lett. 2014, 16, 3464–3467. doi:10.1021/ol5013668 |
| 17. | Suárez, R. M.; Bosch, P.; Sucunza, D.; Cuadro, A. M.; Domingo, A.; Mendicuti, F.; Vaquero, J. J. Org. Biomol. Chem. 2015, 13, 527–538. doi:10.1039/C4OB01465K |
| 18. | Miskolczy, Z.; Megyesi, M.; Biczók, L.; Görner, H. Photochem. Photobiol. Sci. 2011, 10, 592–600. doi:10.1039/c0pp00367k |
| 34. | Tian, M.; Ihmels, H. Synthesis 2009, 4226–4234. doi:10.1055/s-0029-1217060 |
| 35. | Tian, M.; Ihmels, H.; Ye, S. Org. Biomol. Chem. 2012, 10, 3010–3018. doi:10.1039/c2ob06948b |
| 39. | Colleville, A. P.; Horan, R. A. J.; Tomkinson, N. C. O. Org. Process Res. Dev. 2014, 18, 1128–1136. doi:10.1021/op5002353 |
| 42. | Bonin, H.; Delbrayelle, D.; Demonchaux, P.; Gras, E. Chem. Commun. 2010, 46, 2677–2679. doi:10.1039/b926547n |
| 45. | Ihmels, H.; Leusser, D.; Pfeiffer, M.; Stalke, D. J. Org. Chem. 1999, 64, 5715–5718. doi:10.1021/jo990465b |
| 46. | Stratford, S. A.; Arhangelskis, M.; Bučar, D.-K.; Jones, W. CrystEngComm 2014, 16, 10830–10836. doi:10.1039/c4ce01622j |
| 34. | Tian, M.; Ihmels, H. Synthesis 2009, 4226–4234. doi:10.1055/s-0029-1217060 |
| 35. | Tian, M.; Ihmels, H.; Ye, S. Org. Biomol. Chem. 2012, 10, 3010–3018. doi:10.1039/c2ob06948b |
| 57. | Bendig, J.; Geppert, B.; Helm, S.; Kreysig, D. Theor. Exp. Chem. 1978, 14, 488–495. doi:10.1007/BF01004352 |
| 44. | Gallego, D.; Brück, A.; Irran, E.; Meier, F.; Kaupp, M.; Driess, M.; Hartwig, J. F. J. Am. Chem. Soc. 2013, 135, 15617–15626. doi:10.1021/ja408137t |
| 36. | Pithan, P. M.; Decker, D.; Sardo, M. S.; Viola, G.; Ihmels, H. Beilstein J. Org. Chem. 2016, 12, 854–862. doi:10.3762/bjoc.12.84 |
| 58. | Kotani, H.; Ohkubo, K.; Fukuzumi, S. Faraday Discuss. 2012, 155, 89–102. doi:10.1039/C1FD00084E |
| 59. | Verhoeven, J. W.; van Ramesdonk, H. J.; Groeneveld, M. M.; Benniston, A. C.; Harriman, A. ChemPhysChem 2005, 6, 2251–2260. doi:10.1002/cphc.200500029 |
| 60. | Horng, M.; Dahl, K.; Jones, G., II; Maroncelli, M. Chem. Phys. Lett. 1999, 315, 363–370. doi:10.1016/s0009-2614(99)01258-0 |
| 61. | Jones, G., II; Farahat, M. S.; Greenfield, S. R.; Gosztola, D. J.; Wasielewski, M. R. Chem. Phys. Lett. 1994, 229, 40–46. doi:10.1016/0009-2614(94)00996-1 |
| 62. | Li, X.; Liang, M.; Chakraborty, A.; Kondo, M.; Maroncelli, M. J. Phys. Chem. B 2011, 115, 6592–6607. doi:10.1021/jp200339e |
| 63. | Hu, J.; Xia, B.; Bao, D.; Ferreira, A.; Wan, J.; Jones, G.; Vullev, V. I. J. Phys. Chem. A 2009, 113, 3096–3107. doi:10.1021/jp810909v |
| 64. | Jones, G.; Yan, D.-X.; Gosztola, D. J.; Greenfield, S. R.; Wasielewski, M. R. J. Am. Chem. Soc. 1999, 121, 11016–11017. doi:10.1021/ja9927319 |
| 33. | Sucunza, D.; Cuadro, A. M.; Alvarez-Builla, J.; Vaquero, J. J. J. Org. Chem. 2016, 81, 10126–10135. doi:10.1021/acs.joc.6b01092 |
| 49. | Jonker, S. A.; Ariese, F.; Verhoeven, J. W. Recl. Trav. Chim. Pays-Bas 1989, 108, 109–115. doi:10.1002/recl.19891080307 |
| 65. | Bortolozzi, R.; Ihmels, H.; Thomas, L.; Tian, M.; Viola, G. Chem. – Eur. J. 2013, 19, 8736–8741. doi:10.1002/chem.201301164 |
| 66. | de Silva, A. P.; Gunaratne, H. Q. N.; Gunnlaugsson, T.; Huxley, A. J. M.; McCoy, C. P.; Rademacher, J. T.; Rice, T. E. Chem. Rev. 1997, 97, 1515–1566. doi:10.1021/cr960386p |
| 51. | Stootman, F. H.; Fisher, D. M.; Rodger, A.; Aldrich-Wright, J. R. Analyst 2006, 131, 1145–1151. doi:10.1039/b604686j |
| 47. | Jones, G.; Jackson, W. R.; Choi, C. Y.; Bergmark, W. R. J. Phys. Chem. 1985, 89, 294–300. doi:10.1021/j100248a024 |
| 48. | Crosby, G. A.; Demas, J. N. J. Phys. Chem. 1971, 75, 991–1024. doi:10.1021/j100678a001 |
| 65. | Bortolozzi, R.; Ihmels, H.; Thomas, L.; Tian, M.; Viola, G. Chem. – Eur. J. 2013, 19, 8736–8741. doi:10.1002/chem.201301164 |
| 49. | Jonker, S. A.; Ariese, F.; Verhoeven, J. W. Recl. Trav. Chim. Pays-Bas 1989, 108, 109–115. doi:10.1002/recl.19891080307 |
| 50. | Jonker, S. A.; van Dijk, S. I.; Goubitz, K.; Reiss, C. A.; Schuddeboom, W.; Verhoeven, J. W. Mol. Cryst. Liq. Cryst. 1990, 183, 273–282. doi:10.1080/15421409008047464 |
| 36. | Pithan, P. M.; Decker, D.; Sardo, M. S.; Viola, G.; Ihmels, H. Beilstein J. Org. Chem. 2016, 12, 854–862. doi:10.3762/bjoc.12.84 |
| 14. | Granzhan, A.; Ihmels, H. Synlett 2016, 27, 1775–1793. doi:10.1055/s-0035-1561445 |
| 47. | Jones, G.; Jackson, W. R.; Choi, C. Y.; Bergmark, W. R. J. Phys. Chem. 1985, 89, 294–300. doi:10.1021/j100248a024 |
| 69. | Juskowiak, B. Anal. Bioanal. Chem. 2011, 399, 3157–3176. doi:10.1007/s00216-010-4304-5 |
| 46. | Stratford, S. A.; Arhangelskis, M.; Bučar, D.-K.; Jones, W. CrystEngComm 2014, 16, 10830–10836. doi:10.1039/c4ce01622j |
| 52. | Molander, G. A.; Canturk, B.; Kennedy, L. E. J. Org. Chem. 2009, 74, 973–980. doi:10.1021/jo802590b |
| 67. | Faulhaber, K.; Granzhan, A.; Ihmels, H.; Otto, D.; Thomas, L.; Wells, S. Photochem. Photobiol. Sci. 2011, 10, 1535–1545. doi:10.1039/c1pp05106g |
| 68. | Ihmels, H.; Faulhaber, K.; Vedaldi, D.; Dall’Acqua, F.; Viola, G. Photochem. Photobiol. 2005, 81, 1107–1115. doi:10.1562/2005-01-25-ir-427 |
© 2018 Bandaru et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)