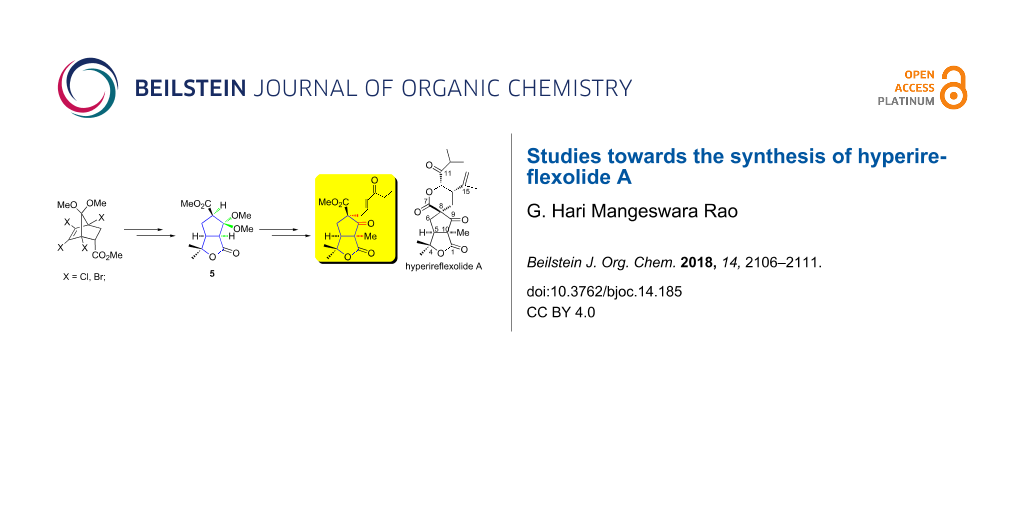
Abstract
The first approach to hyperireflexolide A, based on the synthesis of γ-lactone-fused cyclopentane 5, a functionalized key intermediate, is presented. Compound 5 is involved in hydrolysis, α-allylation at C-8 and α-methylation at C-10 stereoselectively from the convex face. Then it is subjected to cross metathesis to give α,β-unsaturated ketone 11 as precursor in the total synthesis of hyperireflexolide A.

Graphical Abstract
Introduction
Hyperireflexolide A (1) [1] is a spiroterpenoid, isolated from hypericum reflexum, plants of the genus hypericum (Figure 1). Hyperireflexolide A is widely used in folk medicine, displays antifungal [2] and cytotoxic activities [3].
γ-Lactone-fused cyclopentanes are of vital importance in organic synthesis and are the most abundant substructures found in various naturally occurring molecules [4,5]. A cis-cyclopentane ring-fused γ-lactone is the key structural unit of many complex and challenging biologically active natural products [6-19]. The γ-lactone-fused cyclopentane ring system is also an important component for the synthesis of a variety of cyclopentanoid natural and unnatural products [20-24].
In the literature, numerous synthetic methods are reported to attain γ-lactone-fused cyclopentanes [25-31]. Earlier from our lab, we reported a short and efficient methodology for the synthesis of γ-lactone-fused cyclopentane 5 [32]. The cis-ring junction of this carbocylic ring system offers a high degree of selectivity for the assemblage of various substituents on the convex surface. The lactone part can serve as a useful tool to append various side chains.
![[1860-5397-14-185-1]](/bjoc/content/figures/1860-5397-14-185-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
The presence of a γ-lactone-fused cyclopentane moiety in hyperireflexolide A (1) attracted our attention. In fact, the ketal moiety in 5 could not only act as a surrogate for C-9 carbonyl but also facilitate installation of an angular methyl group.
The retrosynthetic analysis for hyperireflexolide A is depicted in Scheme 1. We envisioned that hyperireflexolide A (1) could be synthesized by metal-catalyzed opening of the epoxide 2 with 2-bromopropene followed by lactonization. Enone 3 could be synthesized from 4 by installation of the methyl group at C-10 followed by cross metathesis reaction. Compound 4 could be obtained from the γ-lactone-fused cyclopentane 5 by deprotection of C-9 followed by allylation at C-8.
![[1860-5397-14-185-i1]](/bjoc/content/inline/1860-5397-14-185-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Previously, we reported from our laboratory the synthesis of γ-lactone-fused cyclopentane derivative 5 from the respective Diels–Alder adduct in 5 steps with an overall yield of 29% [32]. Hydrolysis of dimethyl ketal 5 with MeSO3H in 1,2-DCE furnished γ-lactone-fused cyclopentanone 6 in 97% yield. Cyclopentanone 6 exists in its tautomeric enol form 7, observed in the 1H NMR spectrum after column chromatographic purification (6 and 7 were not separated) as represented in Scheme 2 [33].
![[1860-5397-14-185-i2]](/bjoc/content/inline/1860-5397-14-185-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Hydrolysis of dimethyl ketal 5.
Scheme 2: Hydrolysis of dimethyl ketal 5.
In order to check the feasibility of the alkylation reaction of γ-lactone-fused β-ketoester 6, initially a mixture of 6 and 7 was subjected to methylation using 1.1 equiv of K2CO3 in the presence of methyl iodide (MeI). The α-methylated β-ketoester 8 was obtained in good yield. In the 1H NMR, 8 showed a signal at 3.70 ppm as doublet for C-10 (ring junction) proton confirming the selective methylation at C-8. Further, installation of the methyl group at C-10 was achieved by treatment of 8 with 1.1 equiv of K2CO3 in the presence of MeI to give bis-methylated γ-lactone-fused β-ketoester 9 in 72% yield (Scheme 3). These results demonstrated that regioselective alkylation at the two sites were possible. Notably, one diastereomeric product was isolated from these bis-alkylation reactions due to favorable attack from the less hindered convex face of 6 [10] to give α,α'-cis stereochemistry.
![[1860-5397-14-185-i3]](/bjoc/content/inline/1860-5397-14-185-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Alkylation of γ-lactone-fused β-ketoester 6.
Scheme 3: Alkylation of γ-lactone-fused β-ketoester 6.
Now the stage has been set for allylation of γ-lactone fused cyclopentanone 6. Treatment of 6 with K2CO3 in the presence of allyl bromide at 0 °C afforded α-allylated γ-lactone-fused β-ketoester 4 in 88% yield. In the 1H NMR a signal appeared at 3.64 ppm as doublet for ring junction (C-10) proton confirmed that selective allylation occurred at C-8. Compound 4 was then subjected to methylation at C-10 using K2CO3 and MeI to obtain the requisite γ-lactone fused cyclopentanone 10 in excellent yield (Scheme 4). Allylation and methylation both were occurred stereoselectively from the convex face to give α,α'-cis stereochemistry. The allyl derivative 10 was then subjected to cross metathesis reaction with ethyl vinyl ketone. Initially, the reaction performed using Grubbs’ 1st generation catalyst (3–20 mol %) was unsuccessful. Treatment of 10 with ethyl vinyl ketone using Grubbs’ 2nd generation catalyst (3 mol %) in the presence of CH2Cl2 furnished (E)-enone 11 in 77% yield as shown in Scheme 4. In the 1H NMR spectrum, 11 showed a signal at 6.13 ppm as doublet with coupling constant 15.8 Hz (trans-configuration) for the α-proton of enone [34-37].
![[1860-5397-14-185-i4]](/bjoc/content/inline/1860-5397-14-185-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of α,β-unsaturated ketone 11.
Scheme 4: Synthesis of α,β-unsaturated ketone 11.
After successfully synthesizing the side chain via cross-metathesis, our next task was the steresoselective epoxidation of (E)-enone 11. Unfortunately, the stereoselective epoxidation of 11 under basic conditions were unsuccessful [38], which prevented completion of the proposed synthetic sequence.
Conclusion
In conclusion, synthetic studies towards hyperireflexolide A, the synthetic precursor α,β-unsaturated ketone 11 was synthesized. Failure of the stereoselective epoxidation of 11 prevented completion of the proposed synthetic sequence. Future studies will include the stereoselective epoxidation of 11 followed by opening of the epoxide and lactonization or 1,4-nucleophilic addition to the α,β-unsaturated ketone 11 followed by epoxidation of the resulted enolate with subsequent lactonization to achieve hyperireflexolide A (1).
Experimental
General methods
All the reactions were performed in oven dried apparatus and the reaction mixtures were magnetically stirred. Thin-layer chromatography was performed on Acme and Spectrochem Silica gel (Mumbai, India) coated on microscopic slides. Visualization of spots was effected by exposure to iodine or spraying with 4% ethanolic H2SO4 and charring. Column chromatography was performed using Acme's silica gel (100–200 mesh), and ethyl acetate/hexane was used as eluent. Evaporation of solvents was performed at reduced pressure using a Büchi rotary evaporator.
Melting points were recorded on JSGW melting point apparatus and are uncorrected. Infrared spectra were recorded on Perkin-Elmer 1320 and Shimadzu 420 spectrophotometers as KBr pellets (solids), or as thin films on NaCl flats (liquids). 1H NMR spectra were recorded at 400 MHz on a JEOL spectrometer unless otherwise mentioned (500 MHz). Data are reported as follows: (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet; integration; coupling constant(s) in Hz; assignment). Chemical shifts are reported in ppm, and coupling constants are reported in Hz. Proton decoupled 13C NMR spectra were recorded at 100 MHz (125 MHz) on a JEOL spectrometer. Samples for NMR were made in CDCl3. Tetramethylsilane was used as the internal standard.
Commercial grade solvents were distilled before use. Ethyl acetate was distilled over anhydrous sodium carbonate. Dichloromethane and dichloroethane (1,2-DCE) were distilled over phosphorous pentoxide and stored over 4 Å molecular sieves. Acetone was distilled over anhydrous K2CO3. Methanol was refluxed and distilled over magnesium turnings and stored over 4 Å molecular sieves. Distilled water was used for aqueous reactions and aqueous work-up.
Experimental procedures and analytical data
Methyl 1,1,5-trimethyl-3,4-dioxohexahydro-1H-cyclopenta[c]furan-5-carboxylate (8): To a solution of the γ-lactone-fused cyclopentanone 6 (16 mg, 0.07 mmol) in dry acetone (0.4 mL) under argon was added anhydrous K2CO3 (9.7 mg, 0.07 mmol) and methyl iodide (12 mg, 0.08 mmol) at 0 °C. The reaction was stirred at room temperature for 21 h. Completion of the reaction was monitored by tlc. The reaction was diluted with water (3 mL) and the organic layer was extracted with EtOAc (3 × 3 mL). The combined organic layers were washed with brine solution (2 mL) and dried over anhydrous Na2SO4. The solvent was concentrated in vacuo to furnish a residue which was purified by silica gel column chromatography (25% EtOAc/hexane) to afford 8 in 71% yield. Viscous liquid; 1H NMR (400 MHz, CDCl3) δ 3.72 (s, 3H, CO2Me), 3.70 (d, J = 1.9 Hz, 1H), 2.98–2.91 (m, 1H), 2.52 (dd, J = 13.2, 11.2 Hz, 1H), 2.11–2.04 (m, 1H), 1.50 (s, 3H, Me), 1.47 (s, 3H, Me), 1.37 (s, 3H, Me); 13C NMR (100 MHz, CDCl3) δ 203.4 (C=O), 171.0, 167.7, 84.5, 58.0, 55.6, 52.8, 44.9, 35.1, 29.3, 22.3, 18.7; IR (neat): 2900, 1760, 1740, 1440 cm−1; HRMS (m/z): [M]+ calcd for C12H16O5, 240.0998; found, 240.1001.
Methyl 1,1,3a,5-tetramethyl-3,4-dioxohexahydro-1H-cyclopenta[c]furan-5-carboxylate (9): To a solution of the γ-lactone-fused cyclopentanone 8 (15 mg, 0.07 mmol) in dry acetone (0.4 mL) under argon was added anhydrous K2CO3 (9.7 mg, 0.07 mmol) and methyl iodide (12 mg, 0.08 mmol) at 0 °C. The reaction was stirred at room temperature for 22 h. Completion of the reaction was monitored by tlc. The reaction was diluted with water (3 mL) and the organic layer was extracted with EtOAc (3 × 3 mL). The combined organic layers were washed with brine solution (2 mL) and dried over anhydrous Na2SO4. The solvent was concentrated in vacuo to furnish a residue which was purified by silica gel column chromatography (20% EtOAc/hexane) to afford 9 in 72% yield. 1H NMR (400 MHz, CDCl3) δ 3.70 (s, 3H, CO2Me), 2.66 (t, J = 8.6 Hz, 1H), 2.55 (dd, J = 13.9, 8.8 Hz, 1H), 2.05 (dd, J = 13.9, 8.4 Hz, 1H), 1.64 (s, 3H, Me), 1.60 (s, 3H, Me), 1.49 (s, 3H, Me), 1.36 (s, 3H, Me); 13C NMR (100 MHz, CDCl3) δ 201.4 (C=O), 169.3, 168.4, 80.2, 59.2, 56.4, 53.5, 44.4, 36.5, 28.7, 22.6, 19.8, 18.7; IR (neat): 2900, 1760, 1740, 1440 cm−1; HRMS (m/z): [M]+ calcd for C13H18O5, 254.1154; found, 254.1155.
Methyl 5-allyl-1,1-dimethyl-3,4-dioxohexahydro-1H-cyclopenta[c]furan-5-carboxylate (4): To a solution of the γ-lactone-fused cyclopentanone 6 (75 mg, 0.331 mmol) in dry acetone (1 mL) under argon was added anhydrous K2CO3 (55 mg, 0.3972 mmol) and allyl bromide (48 mg, 0.3972 mmol) at 0 °C. The reaction was stirred at 0 °C for 21 h. Completion of the reaction was monitored by tlc. The reaction was diluted with water (4 mL) and the organic layer was extracted with EtOAc (3 × 5 mL). The combined organic layers were washed with brine solution (3 mL) and dried over anhydrous Na2SO4. The solvent was concentrated in vacuo to furnish a residue which was purified by silica gel column chromatography (15% EtOAc/hexane) to afford 4 in 88% yield. Viscous liquid; 1H NMR (500 MHz, CDCl3) δ 5.72–5.64 (m, 1H), 5.18–5.12 (m, 2H), 3.71 (s, 3H, CO2Me), 3.64 (d, J = 7.2 Hz, 1H), 2.86–2.80 (m, 1H), 2.70 (dd, J = 13.8, 6.5 Hz, 1H), 2.50–2.45 (m, 1H), 2.35–2.30 (m, 1H), 2.25–2.20 (m, 1H), 1.50 (s, 3H, Me), 1.44 (s, 3H, Me); 13C NMR (125 MHz, CDCl3) δ 202.6, 169.9, 167.8, 131.8, 120.2, 84.8, 61.8, 56.2, 53.0, 44.5, 37.3, 31.2, 29.4, 22.3; IR (neat): 2900, 1760, 1720, 1420 cm−1; HRMS (m/z): [M]+ calcd for C14H18O5, 266.1154; found, 266.1150.
Methyl 5-allyl-1,1,3a-trimethyl-3,4-dioxohexahydro-1H-cyclopenta[c]furan-5-carboxylate (10): To a solution of the γ-lactone-fused cyclopentanone 4 (29 mg, 0.109 mmol) in dry acetone (0.6 mL) under argon was added anhydrous K2CO3 (16.5 mg, 0.1199 mmol) and methyl iodide (18.5 mg, 0.1308 mmol) at 0 °C. The reaction was stirred at room temperature for 14 h. Completion of the starting material was monitored by tlc. The reaction was diluted with water (3 mL) and the organic layer was extracted with EtOAc (3 × 4 mL). The combined organic layers were washed with brine solution (3 mL) and dried over anhydrous Na2SO4. The solvent was concentrated in vacuo to furnish a residue which was purified by silica gel column chromatography (10% EtOAc/hexane) to afford 10 in 94% yield. Viscous liquid; 1H NMR (500 MHz, CDCl3) δ 5.69–5.61 (m, 1H), 5.17–5.11 (m, 2H), 3.69 (s, 3H, OMe), 2.66 (dd, J = 13.9, 6.5 Hz, 1H), 2.57 (t, J = 8.5 Hz, 1H), 2.49 (dd, J = 14.0, 8.5 Hz, 1H), 2.35 (dd, J = 13.9, 7.5 Hz, 1H), 2.19 (dd, J = 14.0, 8.5 Hz, 1H), 1.59 (s, 3H, Me), 1.53 (s, 3H, Me), 1.50 (s, 3H, Me); 13C NMR (100 MHz, CDCl3) δ 206.0, 171.8, 170.3, 131.9, 120.1, 84.0, 60.7, 60.1, 52.8, 50.4, 37.8, 30.5, 30.2, 23.7, 22.5; IR (neat): 2900, 1760, 1740, 1460 cm−1; HRMS (m/z): [M]+ calcd for C15H20O5, 280.1311; found, 280.1310.
Methyl 1,1,3a-trimethyl-3,4-dioxo-5-((E)-4-oxohex-2-enyl)hexahydro-1H-cyclopenta[c]furan-5-carboxylate (11): To a solution of the compound 10 (110 mg, 0.327 mmol) in CH2Cl2 (0.8 mL) was added ethyl vinyl ketone (31 mg, 0.3597 mmol) and 3 mol % Grubbs’ 2nd generation catalyst (8.5 mg, 0.00981 mmol), then heated the reaction mixture at 40 °C for 8 h (monitored by tlc). The solvent was concentrated in vacuo to furnish a residue which was purified by silica gel column chromatography (15% EtOAc/hexane) to afford 11 in 77% yield. Viscous liquid; 1H NMR (500 MHz, CDCl3) δ 6.67–6.61 (m, 1H), 6.13 (d, J = 15.8 Hz, 1H, -C(O)-CH=C-), 3.70 (s, 3H, CO2Me), 2.80–2.75 (m, 1H), 2.59–2.51 (m, 4H), 2.48–2.43 (m, 1H), 2.12 (dd, J = 13.1, 7.4 Hz, 1H), 1.58 (s, 3H, Me), 1.53 (s, 3H, Me), 1.49 (s, 3H, Me), 1.07 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 205.6, 200.3, 171.5, 169.9, 139.1, 134.0, 84.1, 60.3, 60.1, 53.1, 50.5, 36.1, 33.5, 31.0, 30.3, 23.7, 22.5, 7.8; IR (neat): 2900, 1760, 1720, 1670, 1620, 1420 cm−1; HRMS (m/z): [M]+ calcd for C18H24O6, 336.1573; found, 336.1576.
Supporting Information
| Supporting Information File 1: Copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 419.3 KB | Download |
References
-
Cardona, L.; Pedro, J. R.; Serrano, A.; Muñoz, M. C.; Solans, X. Phytochemistry 1993, 33, 1185–1187. doi:10.1016/0031-9422(93)85046-T
Return to citation in text: [1] -
Décostered, L. A.; Hostettmann, K.; Stoeckli-Evans, H.; Msonthi, J. D. Helv. Chim. Acta 1987, 70, 1694–1702. doi:10.1002/hlca.19870700705
Return to citation in text: [1] -
Jayasuriya, H.; McChesney, J. D.; Swanson, S. M.; Pezzuto, J. M. J. Nat. Prod. 1989, 52, 325–331. doi:10.1021/np50062a018
Return to citation in text: [1] -
Morita, H.; Fujiwara, M.; Naotoshi, Y.; Kobayashi, J. Tetrahedron 2000, 56, 5801–5805. doi:10.1016/S0040-4020(00)00530-5
Return to citation in text: [1] -
Miyaoka, H.; Tamura, M.; Yamada, Y. Tetrahedron 2000, 56, 8083–8094. doi:10.1016/S0040-4020(00)00730-4
Return to citation in text: [1] -
Huang, J.-m.; Yokoyama, R.; Yang, C.-s.; Fukuyama, Y. Tetrahedron Lett. 2000, 41, 6111–6114. doi:10.1016/S0040-4039(00)01023-6
Return to citation in text: [1] -
Mehta, G.; Singh, S. R. Angew. Chem., Int. Ed. 2006, 45, 953–955. doi:10.1002/anie.200503618
Return to citation in text: [1] -
Inoue, M.; Sato, T.; Hirama, M. J. Am. Chem. Soc. 2003, 125, 10772–10773. doi:10.1021/ja036587+
Return to citation in text: [1] -
Shi, L.; Meyer, K.; Greaney, M. F. Angew. Chem. 2010, 122, 9436–9439. doi:10.1002/ange.201005156
Return to citation in text: [1] -
He, W.; Huang, J.; Sun, X.; Frontier, A. J. J. Am. Chem. Soc. 2008, 130, 300–308. doi:10.1021/ja0761986
Return to citation in text: [1] [2] -
Birman, V. B.; Danishefsky, S. J. J. Am. Chem. Soc. 2002, 124, 2080–2081. doi:10.1021/ja012495d
Return to citation in text: [1] -
Meng, Z.; Danishefsky, S. J. Angew. Chem. 2005, 117, 1535–1537. doi:10.1002/ange.200462509
Return to citation in text: [1] -
Kouno, I.; Mori, K.; Kawano, N.; Sato, S. Tetrahedron Lett. 1989, 30, 7451–7454. doi:10.1016/S0040-4039(00)70722-2
Return to citation in text: [1] -
Kouno, I.; Mori, K.; Okamoto, S.; Sato, S. Chem. Pharm. Bull. 1990, 38, 3060–3063. doi:10.1248/cpb.38.3060
Return to citation in text: [1] -
Wang, R.; Shimizu, Y. J. Chem. Soc., Chem. Commun. 1990, 413–414. doi:10.1039/c39900000413
Return to citation in text: [1] -
Ghosh, S.; Sinha, S.; Drew, M. G. B. Org. Lett. 2006, 8, 3781–3784. doi:10.1021/ol061377y
Return to citation in text: [1] -
Crimmins, M. T.; Jung, D. K.; Gray, J. L. J. Am. Chem. Soc. 1992, 114, 5445–5447. doi:10.1021/ja00039a077
Return to citation in text: [1] -
Corey, E. J.; Su, W. G. J. Am. Chem. Soc. 1987, 109, 7534–7536. doi:10.1021/ja00258a050
Return to citation in text: [1] -
Corey, E. J.; Su, W.-g. Tetrahedron Lett. 1988, 29, 3423–3426. doi:10.1016/0040-4039(88)85179-7
Return to citation in text: [1] -
Scarborough, R. M., Jr.; Toder, B. H.; Smith, A. B., III. J. Am. Chem. Soc. 1980, 102, 3904–3913. doi:10.1021/ja00531a037
Return to citation in text: [1] -
Smith, A. B., III; Boschelli, D. J. Org. Chem. 1983, 48, 1217–1226. doi:10.1021/jo00156a015
Return to citation in text: [1] -
Hudlicky, T.; Govindan, S. V.; Frazier, J. O. J. Org. Chem. 1985, 50, 4166–4171. doi:10.1021/jo00221a043
Return to citation in text: [1] -
Wolnisky, J.; Wolf, H.; Gibson, T. J. Org. Chem. 1963, 28, 274–275. doi:10.1021/jo01036a541
Return to citation in text: [1] -
Niwa, H.; Wakamastu, K.; Hida, T.; Niiyama, K.; Kigoshi, H.; Yamada, M.; Nagase, H.; Suzuki, M.; Yamada, K. J. Am. Chem. Soc. 1984, 106, 4547–4552. doi:10.1021/ja00328a041
Return to citation in text: [1] -
Ernst, A. B.; Fristad, W. E. Tetrahedron Lett. 1985, 26, 3761–3764. doi:10.1016/S0040-4039(00)89244-8
Return to citation in text: [1] -
Curran, D. P.; Chang, C. T. J. Org. Chem. 1989, 54, 3140–3157. doi:10.1021/jo00274a034
Return to citation in text: [1] -
Kraus, G. A.; Landgrebe, K. Tetrahedron 1985, 41, 4039–4046. doi:10.1016/S0040-4020(01)97182-0
Return to citation in text: [1] -
Lange, J. H. M.; Klunder, A. J. H.; Zwanenburg, B. Tetrahedron Lett. 1989, 30, 127–130. doi:10.1016/S0040-4039(01)80342-7
Return to citation in text: [1] -
Isawa, S.; Yamamoto, M.; Kohmoto, S.; Yamada, K. J. Org. Chem. 1991, 56, 2849–2853. doi:10.1021/jo00008a048
Return to citation in text: [1] -
Hibbs, D. E.; Hursthouse, M. B.; Jones, I. G.; Jones, W.; Malik, K. M. A.; North, M. J. Org. Chem. 1999, 64, 5413–5421. doi:10.1021/jo990140v
Return to citation in text: [1] -
Mandal, S. K.; Amin, S. R.; Crowe, W. E. J. Am. Chem. Soc. 2001, 123, 6457–6458. doi:10.1021/ja005568m
Return to citation in text: [1] -
Khan, F. A.; Dash, J.; Sahu, N.; Sudheer, C. J. Org. Chem. 2002, 67, 3783–3787. doi:10.1021/jo025521e
Return to citation in text: [1] [2] -
Rao, G. H. M.; Khan, F. A. Synth. Commun. 2018, 48, 318–322. doi:10.1080/00397911.2017.1401638
Return to citation in text: [1] -
Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. doi:10.1021/ja0214882
Return to citation in text: [1] -
Chatterjee, A. K.; Morgan, J. P.; Scholl, M.; Grubbs, R. H. J. Am. Chem. Soc. 2000, 122, 3783–3784. doi:10.1021/ja9939744
Return to citation in text: [1] -
Galan, B. R.; Kalbarczyk, K. P.; Szczepankiewicz, S.; Keister, J. B.; Diver, S. T. Org. Lett. 2007, 9, 1203–1206. doi:10.1021/ol0631399
Return to citation in text: [1] -
Abbas, M.; Leitgeb, A.; Slugovc, C. Synlett 2013, 24, 1193–1196. doi:10.1055/s-0033-1338425
Return to citation in text: [1] -
Epoxidation of enone 11 with 30% aq H2O2 solution using NaHCO3 afforded a 6:1 of two diastereomers (determined by 1H NMR analysis of the crude reaction mixture) of α,β-epoxy ketone in very low yield (10%). Changing the base from NaHCO3 to aq NaOH solution (1 N) did not improve the yield. Then the diastereomeric mixture of α,β-epoxy ketone was subjected to nucleophilic addition of Grignard reagent derived from 2-bromopropene in the presence of CuI led to the formation of an intractable mixture.
Return to citation in text: [1]
| 1. | Cardona, L.; Pedro, J. R.; Serrano, A.; Muñoz, M. C.; Solans, X. Phytochemistry 1993, 33, 1185–1187. doi:10.1016/0031-9422(93)85046-T |
| 6. | Huang, J.-m.; Yokoyama, R.; Yang, C.-s.; Fukuyama, Y. Tetrahedron Lett. 2000, 41, 6111–6114. doi:10.1016/S0040-4039(00)01023-6 |
| 7. | Mehta, G.; Singh, S. R. Angew. Chem., Int. Ed. 2006, 45, 953–955. doi:10.1002/anie.200503618 |
| 8. | Inoue, M.; Sato, T.; Hirama, M. J. Am. Chem. Soc. 2003, 125, 10772–10773. doi:10.1021/ja036587+ |
| 9. | Shi, L.; Meyer, K.; Greaney, M. F. Angew. Chem. 2010, 122, 9436–9439. doi:10.1002/ange.201005156 |
| 10. | He, W.; Huang, J.; Sun, X.; Frontier, A. J. J. Am. Chem. Soc. 2008, 130, 300–308. doi:10.1021/ja0761986 |
| 11. | Birman, V. B.; Danishefsky, S. J. J. Am. Chem. Soc. 2002, 124, 2080–2081. doi:10.1021/ja012495d |
| 12. | Meng, Z.; Danishefsky, S. J. Angew. Chem. 2005, 117, 1535–1537. doi:10.1002/ange.200462509 |
| 13. | Kouno, I.; Mori, K.; Kawano, N.; Sato, S. Tetrahedron Lett. 1989, 30, 7451–7454. doi:10.1016/S0040-4039(00)70722-2 |
| 14. | Kouno, I.; Mori, K.; Okamoto, S.; Sato, S. Chem. Pharm. Bull. 1990, 38, 3060–3063. doi:10.1248/cpb.38.3060 |
| 15. | Wang, R.; Shimizu, Y. J. Chem. Soc., Chem. Commun. 1990, 413–414. doi:10.1039/c39900000413 |
| 16. | Ghosh, S.; Sinha, S.; Drew, M. G. B. Org. Lett. 2006, 8, 3781–3784. doi:10.1021/ol061377y |
| 17. | Crimmins, M. T.; Jung, D. K.; Gray, J. L. J. Am. Chem. Soc. 1992, 114, 5445–5447. doi:10.1021/ja00039a077 |
| 18. | Corey, E. J.; Su, W. G. J. Am. Chem. Soc. 1987, 109, 7534–7536. doi:10.1021/ja00258a050 |
| 19. | Corey, E. J.; Su, W.-g. Tetrahedron Lett. 1988, 29, 3423–3426. doi:10.1016/0040-4039(88)85179-7 |
| 4. | Morita, H.; Fujiwara, M.; Naotoshi, Y.; Kobayashi, J. Tetrahedron 2000, 56, 5801–5805. doi:10.1016/S0040-4020(00)00530-5 |
| 5. | Miyaoka, H.; Tamura, M.; Yamada, Y. Tetrahedron 2000, 56, 8083–8094. doi:10.1016/S0040-4020(00)00730-4 |
| 3. | Jayasuriya, H.; McChesney, J. D.; Swanson, S. M.; Pezzuto, J. M. J. Nat. Prod. 1989, 52, 325–331. doi:10.1021/np50062a018 |
| 38. | Epoxidation of enone 11 with 30% aq H2O2 solution using NaHCO3 afforded a 6:1 of two diastereomers (determined by 1H NMR analysis of the crude reaction mixture) of α,β-epoxy ketone in very low yield (10%). Changing the base from NaHCO3 to aq NaOH solution (1 N) did not improve the yield. Then the diastereomeric mixture of α,β-epoxy ketone was subjected to nucleophilic addition of Grignard reagent derived from 2-bromopropene in the presence of CuI led to the formation of an intractable mixture. |
| 2. | Décostered, L. A.; Hostettmann, K.; Stoeckli-Evans, H.; Msonthi, J. D. Helv. Chim. Acta 1987, 70, 1694–1702. doi:10.1002/hlca.19870700705 |
| 32. | Khan, F. A.; Dash, J.; Sahu, N.; Sudheer, C. J. Org. Chem. 2002, 67, 3783–3787. doi:10.1021/jo025521e |
| 10. | He, W.; Huang, J.; Sun, X.; Frontier, A. J. J. Am. Chem. Soc. 2008, 130, 300–308. doi:10.1021/ja0761986 |
| 32. | Khan, F. A.; Dash, J.; Sahu, N.; Sudheer, C. J. Org. Chem. 2002, 67, 3783–3787. doi:10.1021/jo025521e |
| 34. | Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. doi:10.1021/ja0214882 |
| 35. | Chatterjee, A. K.; Morgan, J. P.; Scholl, M.; Grubbs, R. H. J. Am. Chem. Soc. 2000, 122, 3783–3784. doi:10.1021/ja9939744 |
| 36. | Galan, B. R.; Kalbarczyk, K. P.; Szczepankiewicz, S.; Keister, J. B.; Diver, S. T. Org. Lett. 2007, 9, 1203–1206. doi:10.1021/ol0631399 |
| 37. | Abbas, M.; Leitgeb, A.; Slugovc, C. Synlett 2013, 24, 1193–1196. doi:10.1055/s-0033-1338425 |
| 25. | Ernst, A. B.; Fristad, W. E. Tetrahedron Lett. 1985, 26, 3761–3764. doi:10.1016/S0040-4039(00)89244-8 |
| 26. | Curran, D. P.; Chang, C. T. J. Org. Chem. 1989, 54, 3140–3157. doi:10.1021/jo00274a034 |
| 27. | Kraus, G. A.; Landgrebe, K. Tetrahedron 1985, 41, 4039–4046. doi:10.1016/S0040-4020(01)97182-0 |
| 28. | Lange, J. H. M.; Klunder, A. J. H.; Zwanenburg, B. Tetrahedron Lett. 1989, 30, 127–130. doi:10.1016/S0040-4039(01)80342-7 |
| 29. | Isawa, S.; Yamamoto, M.; Kohmoto, S.; Yamada, K. J. Org. Chem. 1991, 56, 2849–2853. doi:10.1021/jo00008a048 |
| 30. | Hibbs, D. E.; Hursthouse, M. B.; Jones, I. G.; Jones, W.; Malik, K. M. A.; North, M. J. Org. Chem. 1999, 64, 5413–5421. doi:10.1021/jo990140v |
| 31. | Mandal, S. K.; Amin, S. R.; Crowe, W. E. J. Am. Chem. Soc. 2001, 123, 6457–6458. doi:10.1021/ja005568m |
| 20. | Scarborough, R. M., Jr.; Toder, B. H.; Smith, A. B., III. J. Am. Chem. Soc. 1980, 102, 3904–3913. doi:10.1021/ja00531a037 |
| 21. | Smith, A. B., III; Boschelli, D. J. Org. Chem. 1983, 48, 1217–1226. doi:10.1021/jo00156a015 |
| 22. | Hudlicky, T.; Govindan, S. V.; Frazier, J. O. J. Org. Chem. 1985, 50, 4166–4171. doi:10.1021/jo00221a043 |
| 23. | Wolnisky, J.; Wolf, H.; Gibson, T. J. Org. Chem. 1963, 28, 274–275. doi:10.1021/jo01036a541 |
| 24. | Niwa, H.; Wakamastu, K.; Hida, T.; Niiyama, K.; Kigoshi, H.; Yamada, M.; Nagase, H.; Suzuki, M.; Yamada, K. J. Am. Chem. Soc. 1984, 106, 4547–4552. doi:10.1021/ja00328a041 |
| 33. | Rao, G. H. M.; Khan, F. A. Synth. Commun. 2018, 48, 318–322. doi:10.1080/00397911.2017.1401638 |
© 2018 Rao; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)