Abstract
The syntheses of various pyrimidinones as potentially bioactive products by means of the highly controlled continuous-flow retro-Diels–Alder reaction of condensed pyrimidinone derivatives are presented. Noteworthy, the use of this approach allowed us to rapidly screen a selection of conditions and quickly confirm the viability of preparing the desired pyrimidinones in short reaction times. Yields typically higher than those published earlier using conventional batch or microwave processes were achieved.

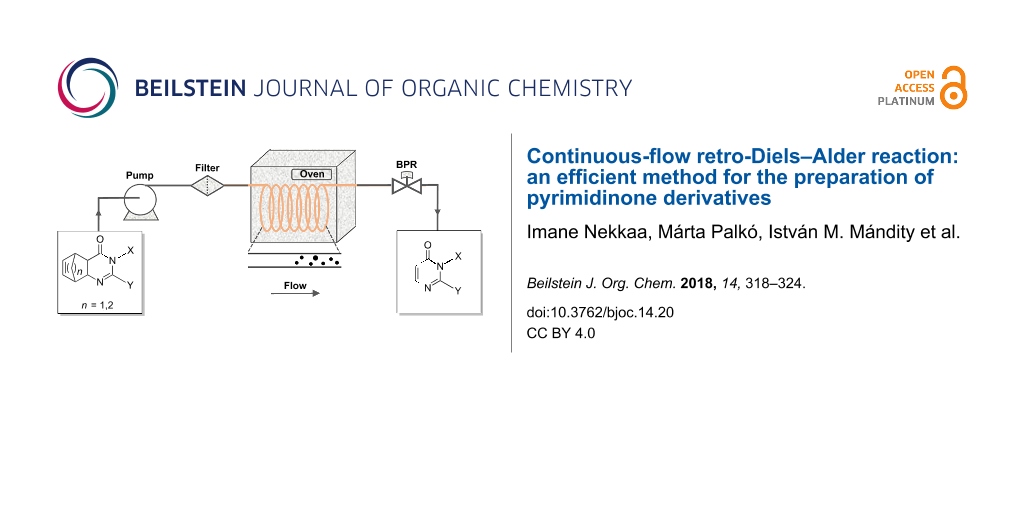
Graphical Abstract
Introduction
The continuous-flow (CF) technology has gained significant importance in modern synthetic chemistry [1-3] and becomes a core technology in the pharmaceutical, agrochemical, and fine chemical industries [4,5]. The use of this technology opens a new door to a quick optimization, acceleration [6], and easy scale-up with a wide and growing range of chemical transformations in combination with an inherently safe and green nature [7-12]. Advantageously, safety issues are complied with excellent mixing and heat transfer [7-14]. These allow the access to elevated temperatures and pressures accredited to superheating of organic solvents in a controlled and safe fashion [6,14-17]. The accurate tuning of residence time can further broaden the versatility of CF processes by governing the outcome of chemical reactions, determining the reaction rate and the conversion and by influencing product selectivities [17-19]. Thus, flow chemistry has long been selected to provide a simple means to use more rigorous reaction conditions and revisit difficult reactions that have been neglected in the past [21].
The retro-Diels–Alder (rDA) reaction has become an important tool for synthetic chemists in their search towards the synthesis and design of novel heterocyclic scaffolds. This pyrolytic dissociation arises when one or both fragments are notably stable [22]. The unsaturation present in the original starting material is produced in the DA addition, and the same atoms are involved in both the bond formation and cleavage steps [23-25]. The rDA process is an efficient technique for the introduction of a double bond into a heterocyclic skeleton [26] as well as for the enantiodivergent [27] and the enantiocontrolled [28] syntheses of heterocyclic compounds. The rDA products can be gained, due to a thermal [4 + 2]-cycloreversion, by distillation under reduced pressure [29], boiling in solvent [30,31], and applying microwave irradiation [32-35] or flash vacuum pyrolysis [35,36]. rDA reactions under mild conditions have been widely examined and discussed for the laboratory preparation of heteromonocycles or condensed-ring heterocycles [37-40]. However, the CF rDA method was introduced when Meyers’ group performed the preparation of a precursor intermediate for the construction of diverse tetracycline antibiotics [41]. Our aim in the present study was to synthesize functionalized pyrimidinone systems through rDA reactions. Many of these products are of high importance in drug design due to their diverse biological properties including antimicrobial, antiviral, antioxidant and antitumor activities. In addition, they are present in several natural frameworks [42-44].
We wanted to exploit the benefits of flow processing for reaction optimization and synthesis and develop novel sustainable synthetic methodologies with possible useful applications for the pharmaceutical industry. Our results show that the developed CF technology is superior to existing conventional batch technologies.
Results and Discussion
The starting materials, i.e., fused tricyclic or tetracyclic pyrimidinones 1–8 have been previously prepared by literature methods [26,45-50]. Cyclization of the corresponding di-exo- or di-endo-amino acids or esters with ethyl p-chlorobenzimidate resulted in tricyclic pyrimidinones 1, 2a and 2b [26,45-49]. Methanopyrrolo-, methanopyrido- and methanoazepino[2,1-b]quinazolinones 3–6 were prepared by ring enlargement of di-exo-norbornene-fused azetidinones with lactim ethers [50]. For the preparation of 2-thioxopyrimidinones 7, 8a and 8b, the most common method is the reaction of the appropriate amino esters with phenyl isothiocyanate, followed by cyclization of the resulting thiourea with hydrogen chloride under reflux [45,49]. The starting materials were selected to comprise molecules where good (>80%), medium (70–80%) and no conversion was observed under batch rDA conditions. Batch reactions were carried out by the following ways: heating under neat conditions, refluxing in solvents having a high boiling point [chlorobenzene (CB) or 1,2-dichlorobenzene (DCB)], and under microwave (MW) conditions in DCB.
In order to provide a rapid and efficient access to the desired pyrimidinones 9–14 (Scheme 1), we reinvestigated these rDA reactions by using another method involving flow chemistry. Therefore, a modular flow system was designed, equipped with a heated 304 stainless steel coil and an adjustable back-pressure regulator (0–300 bar) controlling the use of solvents under superheated conditions. The coil was heated in an oven to the desired temperature and solutions of the starting materials 1–8 were loaded into the reactor via a HPLC pump. Solvents were selected on the basis of the solubility of the starting materials. A schematic representation of the flow reactor setup is illustrated in Figure 1. Products 9–14 thus prepared were identified by means of HPLC–MS and NMR spectroscopic analysis. All physical and spectroscopic data of pyrimidinones 9–14 were identical with their literature data (Supporting Information File 1).
![[1860-5397-14-20-i1]](/bjoc/content/inline/1860-5397-14-20-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Flow synthesis for the preparation of fused pyrimidinones 9–14 by rDA reaction. Solvent and conditions (FR is the flow rate): (i) MeCN, toluene, FR = 0.5 mL min−1, 230–250 °C; (ii) MeOH, FR = 0.5 mL min−1, 120–150 °C; (iii) MeCN, FR = 0.5 mL min−1, 220–250 °C.
Scheme 1: Flow synthesis for the preparation of fused pyrimidinones 9–14 by rDA reaction. Solvent and conditi...
![[1860-5397-14-20-1]](/bjoc/content/figures/1860-5397-14-20-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Schematic outline of the continuous-flow reactor.
Figure 1: Schematic outline of the continuous-flow reactor.
The rDA reaction is basically a thermally-driven process. Consequently, by careful reaction parameter optimization, a balance should be found between the desired rDA cycloreversion reaction and the unwanted thermal degradation of the rDA product. The conversion and yield of a reaction under CF conditions is influenced directly by the residence time and reaction temperature, which are crucial determining factors in flow chemistry [18-20]. Thus, these two parameters were fine-tuned for all of the starting materials. The residence time was set by the use of coils with different lengths. The pressure and flow rate of the reactions were kept at constant values of 10 bars and 0.5 mL min−1, respectively. The full reaction parameter optimization is shown only for compound 1 in Table 1.
The tricyclic di-exo-2-(4-chlorophenyl)tetrahydro-5,8-methano-4(3H)-quinazoline (1) was dissolved in acetonitrile (MeCN) and first the effect of the temperature was investigated. The results show that with 10 min residence time the best conversion value (86%) was obtained at 230 °C (Table 1, entry 4). It should be noted that at higher temperature, a significant amount of degradation product was observed and a brown oil was isolated (Table 1, entries 5 and 6). To further improve the conversion, the residence time was increased by utilizing longer coils (Table 1, entries 7 and 8). It was found that complete conversion can be obtained at 15 min residence time and the desired rDA product was isolated with 92% yield (Table 1, entry 7, Table 2, entry 1). With longer residence times, again, degradation of the product was observed. Importantly, the complete reaction parameter optimization was carried out only in 105 min. The parameters of the optimized reaction conditions and related results are summarized in Table 2.
Table 2: Comparison between batch reactionsa and the CF process for the synthesis of pyrimidinones 9–14.
| starting material | product | batch reaction (lit.) | CF in the present work | |||
|---|---|---|---|---|---|---|
| method: yieldc [%] | solventb | temp [°C] | residence time [min] | yieldc [%] | ||
| 1 | 9 |
A: 85 [47]
B: 56 [46] |
MeCN | 230 | 15 | 92 |
| 2a | 9 |
B: 54 [46]
C: 85 [54] |
250 | 10 | 95 | |
| 2b | 9 |
A: 63 [49]
B: 58 [49] C: 72 [49] |
toluene | 230 | 30 | 93 |
| 3 | 10 | B: 70–80 [50] | MeOH | 130 | 10 | 95 |
| 4 | 11 | 150 | 10 | 97 | ||
| 5 | 12 | 120 | 10 | 95 | ||
| 6 | 13 | 130 | 10 | 94 | ||
| 7 | 14 |
B: 80 [45]
C: 89 [54] |
MeCN | 210 | 15 | 96 |
| 8a | 14 | B: 80 [45] | 220 | 10 | 96 | |
| 8b | 14 |
A: B: C: 0 [49]
(no rDA occurred) |
250 | 30 | 30d | |
| 8b | 15b | – | EtOH/H2O = 2:1 | 250 | 30 | 90 |
Batch reactiona: Method A: reflux, CB, 12 h; Method B: performed at their melting points; Method C: MW, solvent: DCB (2a), EtOH (7), solvent-free (8b). bSolvents were selected on the basis of solubilities. cIsolated yield. dAfter column chromatography.
In the case of di-endo-isomer 2a, higher temperature (250 °C) but a shorter residence time was satisfactory to isolate 9 in a yield of 95%. Furthermore, we proceeded to investigate the elimination of cyclohexadiene from compound 2b. Because of solubility reasons, the solvent was changed to toluene, which is known to be compatible with high-temperature conditions [51-53]. Retrodiene product 9 was afforded with full conversion and in an excellent yield of 93%, which is higher than the maximum yield (85%) reached in our previous batch work [54]. Importantly, this result was achieved with a residence time of 30 min.
Subsequently, tetracyclic methanopyrrolo-, methanopyrido- and methanoazepino[2,1-b]quinazolinones 3–6 were examined. Because of their excellent solubility, reactions were carried out in methanol (MeOH). Importantly, much milder reaction conditions gave satisfactory results. With the utilization of 120–150 °C and only 10 min residence time, full conversions and high yields (94–97%) were obtained. Lower yields were previously found (70–80%) using a batch process, even upon melting compounds 3–6 for 20 min [50].
The effect of the thioxo group on the rDA reaction was investigated too with compounds 7, 8a and 8b. In the case of 7, a yield of 96% was reached at full conversion at 210 °C in 15 min residence time. In the reaction of 8a, the di-endo isomer of 7, a slightly higher temperature was necessary, while an appropriate residence time of only 10 min was satisfactory to have 14 with 96% isolated yield.
On the basis of these encouraging results, we decided to further examine the scope and limitation of the rDA reaction with the use of our CF reactor. Di-endo-3-phenyl-2-thioxohexahydro-5,8-ethanoquinazolin-4(1H)-one (8b) was selected, since this compound did not lose cyclohexadiene to form monocyclic 14 under batch and microwave conditions [49]. A solution of 8b in MeCN was treated in the heated coil reactor at 250 °C, with a residence time of 30 min. Importantly, according to the HPLC–MS analysis, compound 8b underwent thermal decomposition but only a moderate conversion (36%) was detected and 14 was isolated by means of column chromatography with a yield of 30%. This result is due to the lack of the quasi-aromatic character of the leaving cyclohexadiene, and possibly also due to the temperature limitation of our system. Surprisingly, however, we could detect traces of di-endo-3-phenyl-4a,5,8,8a-tetrahydro-5,8-ethanoquinazolin-4(3H)-one (15b), resulting from desulfurisation of 8b (Scheme 2). This observation prompted us to investigate whether desulfurisation can occur under the flow reactor conditions. In the literature, a similar desulfurisation batch reaction was performed with nickel catalysis, in ethanol (EtOH)/water (2:1) solution [55-57]. Thus, thioxo derivative 8b was dissolved in this mixture, and the CF method was repeated. Desulfurisation of 8b, at 250 °C without adding any catalytic metal, provided tricyclic 15b in good yield (90%). Most probably, the reaction was catalyzed by nickel, a component of the 304 stainless steel reactor coil [58,59]. These results also underline the importance to select appropriate solvents and tubing [60,61] for thermally driven reactions. In support of our results, tricyclic 15b was also prepared alternately: the mixture of 3-aminobicyclo[2.2.2]oct-5-ene-carboxylic acid, triethylorthoformate, aniline and acetic acid was subjected to microwave irradiation at 120 W at 80 °C for 20 min. After completion of the reaction, as monitored by TLC, 20% methanolic solution in water was added to get precipitation. The solid was filtered off and washed with water to get di-endo-3-phenyl-4a,5,8,8a-tetrahydro-5,8-ethanoquinazolin-4(3H)-one (15b). All spectroscopic data of the alternately synthesized compound were the same as those obtained by the flow chemical method. The protocol for the synthesis of 15b and the 1H and 13C NMR spectra of 15b are shown in Supporting Information File 1 of this study.
![[1860-5397-14-20-i2]](/bjoc/content/inline/1860-5397-14-20-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of tricyclic ethanoquinazolin-4(3H)-one 15b; (i) MeCN, FR = 0.5 mL min−1, 220–250 °C; (ii) EtOH/H2O = 2:1, FR = 0.5 mL min−1, 250 °C; (iii) MeCN, FR = 0.5 mL min−1, 250 °C.
Scheme 2: Synthesis of tricyclic ethanoquinazolin-4(3H)-one 15b; (i) MeCN, FR = 0.5 mL min−1, 220–250 °C; (ii...
A further attempt was made to perform the rDA reaction with 15b at 250 °C with a residence time of 15 min in MeCN. However, the formation of 16 was not observed, that is the starting tetrahydroquinazolinone derivative 15b did not undergo a thermally driven rDA reaction (Scheme 2). Furthermore, by applying the same conditions on 14, no desulfurisation occurred and the formation of 16 was not detected either.
Conclusion
In the case of compounds 1–8, HPLC–MS measurements revealed full conversions to the desired pyrimidinones 9–14, whereas only a moderate conversion of 8b to 14 was observed. Mainly the retrodiene decomposition of compounds 1–8 occurred, since these latter possess the quasi-aromatic character, through the splitting-off of cyclopentadiene or cyclohexadiene. The stereochemistry (di-endo versus di-exo condensation) of the starting pyrimidinones 1, 2, 7 and 8 has no significant effect on the reaction yields. By using this safe, stable and scalable flow process, pyrimidinones 9–14 were afforded in high purity without the need for further purification steps. In addition, excellent yields and shorter reaction times are significant further advantages when compared to the corresponding batch processes. Moreover, the flow technology allowed the replacement of high-boiling and toxic solvents, which are commonly employed in batch process, e.g., CB or DCB, by less harmful, environmentally benign solvents such as toluene, MeCN, methanol, and ethanol.
In summary, we have developed a simple flow-based method for the preparation of pyrimidinone derivatives, precursors of a series of pharmacologically active materials, through the rDA reaction. The design of the reactor enabled accurate control of both residence time and reaction temperature. CF syntheses were performed under high-temperature conditions with varied solvents. The CF reactor set-up ensured enhanced safety and afforded yields higher than those for the batch and microwave processes. These could be achieved through careful reaction parameter optimization. It is particularly true for 8b, which was unreactive under batch conditions, in contrast to a yield of 30% in CF. We envisage that this method can be readily extended to the preparation of other synthetically important building blocks requiring harsh conditions in batch methods. A simple, efficient and scalable production was implemented with a short processing time, which might open up new horizons for a potential CF industrial synthesis of heterocycles.
Supporting Information
| Supporting Information File 1: Experimental procedures and analytical data. | ||
| Format: PDF | Size: 1.1 MB | Download |
References
-
Lapkin, A. A.; Plucinski, P. K. Engineering Factors for Efficient Flow rocesses in Chemical Industries. Chemical Reactions; Processes under Flow Conditions; Chapter 1, Vol. 5; The Royal Society of. Chemistry, 2010; pp 1–43.
Return to citation in text: [1] -
Jähnisch, K.; Hessel, V.; Löwe, H.; Baerns, M. Angew. Chem., Int. Ed. 2004, 43, 406–446. doi:10.1002/anie.200300577
Return to citation in text: [1] -
Wegner, J.; Ceylan, S.; Kirschning, A. Adv. Synth. Catal. 2012, 354, 17–57. doi:10.1002/adsc.201100584
Return to citation in text: [1] -
Rehm, T. H.; Hofmann, C.; Reinhard, D.; Kost, H.-J.; Löb, P.; Besold, M.; Welzel, K.; Barten, J.; Didenko, A.; Sevenard, D. V.; Lix, B.; Hillson, A. R.; Riegel, S. D. React. Chem. Eng. 2017, 2, 315–323. doi:10.1039/c7re00023e
Return to citation in text: [1] -
Roberge, D. M.; Zimmermann, B.; Rainone, F.; Gottsponer, M.; Eyholzer, M.; Kockmann, N. Org. Process Res. Dev. 2008, 12, 905–910. doi:10.1021/op8001273
Return to citation in text: [1] -
Hessel, V.; Kralisch, D.; Kockmann, N.; Noël, T.; Wang, Q. ChemSusChem 2013, 6, 746–789. doi:10.1002/cssc.201200766
Return to citation in text: [1] [2] -
Wegner, J.; Ceylan, S.; Kirschning, A. Chem. Commun. 2011, 47, 4583–4592. doi:10.1039/C0CC05060A
Return to citation in text: [1] [2] -
Wiles, C.; Watts, P. Green Chem. 2012, 14, 38–54. doi:10.1039/C1GC16022B
Return to citation in text: [1] [2] -
Newman, S. G.; Jensen, K. F. Green Chem. 2013, 15, 1456–1472. doi:10.1039/C3GC40374B
Return to citation in text: [1] [2] -
Wiles, C.; Watts, P. Green Chem. 2014, 16, 55–62. doi:10.1039/c3gc41797b
Return to citation in text: [1] [2] -
Müller, S. T. R.; Wirth, T. ChemSusChem 2015, 8, 245–250. doi:10.1002/cssc.201402874
Return to citation in text: [1] [2] -
Kockmann, N.; Thenée, P.; Fleischer-Trebes, C.; Laudadio, G.; Noël, T. React. Chem. Eng. 2017, 2, 258–280. doi:10.1039/c7re00021a
Return to citation in text: [1] [2] -
DeMello, A. J. Nature 2006, 442, 394–402. doi:10.1038/nature05062
Return to citation in text: [1] -
Noël, T.; Su, Y.; Hessel, V. Top. Organomet. Chem. 2016, 57, 1–41. doi:10.1007/3418_2015_152
Return to citation in text: [1] [2] -
Baxendale, I. R. J. Chem. Technol. Biotechnol. 2013, 88, 519–552. doi:10.1002/jctb.4012
Return to citation in text: [1] -
Hessel, V. Chem. Eng. Technol. 2009, 32, 1655–1681. doi:10.1002/ceat.200900474
Return to citation in text: [1] -
Illg, T.; Löb, P.; Hessel, V. Bioorg. Med. Chem. 2010, 18, 3707–3719. doi:10.1016/j.bmc.2010.03.073
Return to citation in text: [1] [2] -
Mándity, I. M.; Ötvös, S. B.; Szöllösi, G.; Fülöp, F. Chem. Rec. 2016, 16, 1018–1033. doi:10.1002/tcr.201500286
Return to citation in text: [1] [2] -
Hsieh, C.-T.; Ötvös, S. B.; Wu, Y.-C.; Mándity, I. M.; Chang, F.-R.; Fülöp, F. ChemPlusChem 2015, 80, 859–864. doi:10.1002/cplu.201402426
Return to citation in text: [1] [2] -
Mándity, I. M.; Ötvös, S. B.; Fülöp, F. ChemistryOpen 2015, 4, 212–223. doi:10.1002/open.201500018
Return to citation in text: [1] -
Movsisyan, M.; Delbeke, E. I. P.; Berton, J. K. E. T.; Battilocchio, C.; Ley, S. V.; Stevens, C. V. Chem. Soc. Rev. 2016, 45, 4892–4928. doi:10.1039/C5CS00902B
Return to citation in text: [1] -
Wollweber, H. Diels–Alder-Reaction; Georg Thieme Verlag: Stuttgart, 1972; pp 152–270.
Return to citation in text: [1] -
Ichihara, A. Synthesis 1987, 207–222. doi:10.1055/s-1987-27894
Return to citation in text: [1] -
Rickborn, B. The Retro–Diels–Alder Reaction Part I. C-C Dienophiles. Organic Reactions; John Wiley & Sons, Inc., 2004. doi:10.1002/0471264180.or052.01
Return to citation in text: [1] -
Klunder, A. J. H.; Zhu, J.; Zwanenburg, B. Chem. Rev. 1999, 99, 1163–1190. doi:10.1021/cr9803840
Return to citation in text: [1] -
Stájer, G.; Csende, F.; Fülöp, F. Curr. Org. Chem. 2003, 7, 1423–1432. doi:10.2174/1385272033486369
Return to citation in text: [1] [2] [3] -
González-Temprano, I.; Osante, I.; Lete, E.; Sotomayor, N. J. Org. Chem. 2004, 69, 3875–3885. doi:10.1021/jo049672o
Return to citation in text: [1] -
Suzuki, K.; Inomata, K.; Endo, Y. Org. Lett. 2004, 6, 409. doi:10.1021/ol036253p
Return to citation in text: [1] -
Nising, C. F.; Ohnemuller, U. K.; Bräse, S. Synthesis 2006, 2643–2645. doi:10.1055/s-2006-942484
Return to citation in text: [1] -
Citron, C. A.; Wickel, S. M.; Schulz, B.; Draeger, S.; Dickschat, J. S. Eur. J. Org. Chem. 2012, 6636–6646. doi:10.1002/ejoc.201200991
Return to citation in text: [1] -
Clay, D. R.; Rosenberg, A. G.; McIntosh, M. C. Tetrahedron: Asymmetry 2011, 22, 713–716. doi:10.1016/j.tetasy.2011.04.022
Return to citation in text: [1] -
Gallagher, T.; Sanchez, S.; Bateson, J. H.; O'Hanlon, P. J. Pure Appl. Chem. 2009, 77, 2033–2040. doi:10.1351/pac200577122033
Return to citation in text: [1] -
Eddolls, J. P.; Iqbal, M.; Robert, S. M.; Santoro, M. G. Tetrahedron 2004, 60, 2539–2550. doi:10.1016/j.tet.2004.01.047
Return to citation in text: [1] -
Iqbal, M.; Li, Y.; Evans, P. Tetrahedron 2004, 60, 2531–2538. doi:10.1016/j.tet.2004.01.048
Return to citation in text: [1] -
Arai, Y.; Kontani, T.; Koizumi, T. Chem. Lett. 1991, 2135–2138. doi:10.1246/cl.1991.2135
Return to citation in text: [1] [2] -
Hasbullah, S. A.; Jones, S. Tetrahedron: Asymmetry 2010, 21, 2719–2725. doi:10.1016/j.tetasy.2010.10.021
Return to citation in text: [1] -
Csende, F.; Stájer, G.; Fülöp, F. Comprehensive Organic Synthesis, 2nd ed.; Elsevier: Amsterdam, 2014; Vol. 5, pp 518–594.
Return to citation in text: [1] -
Stájer, G.; Miklós, F.; Kanizsai, I.; Csende, F.; Sillanpää, R.; Sohár, P. Eur. J. Org. Chem. 2004, 3701–3706. doi:10.1002/ejoc.200400247
Return to citation in text: [1] -
Fülöp, F.; Miklós, F.; Forró, E. Synlett 2008, 1687–1689. doi:10.1055/s-2008-1077793
Return to citation in text: [1] -
Miklós, F.; Stájer, G.; Fülöp, F. Lett. Org. Chem. 2006, 3, 915–916. doi:10.2174/157017806779468086
Return to citation in text: [1] -
Kummer, D. A.; Li, D.; Dion, A.; Myers, A. G. Chem. Sci. 2011, 2, 1710–1718. doi:10.1039/C1SC00303H
Return to citation in text: [1] -
Januszczyk, P.; Fogt, J.; Boryski, J.; Izawa, K.; Onishi, T.; Neyts, J.; De Clercq, E. Nucleosides, Nucleotides Nucleic Acids 2009, 28, 713–723. doi:10.1080/15257770903128870
Return to citation in text: [1] -
Bakavoli, M.; Bagherzadeh, G.; Vaseghifar, M.; Shiri, A.; Pordel, M.; Mashreghi, M.; Pordeli, P.; Araghi, M. Eur. J. Med. Chem. 2010, 45, 647–650. doi:10.1016/j.ejmech.2009.10.051
Return to citation in text: [1] -
Guo, C.; Linton, A.; Jalaie, M.; Kephart, S.; Ornelas, M.; Pairish, M.; Greasley, S.; Richardson, P.; Maegley, K.; Hickey, M.; Li, J.; Wu, X.; Ji, X.; Xie, Z. Bioorg. Med. Chem. Lett. 2013, 23, 3358–3363. doi:10.1016/j.bmcl.2013.03.090
Return to citation in text: [1] -
Stájer, G.; Szabó, A. E.; Pintye, J.; Bernáth, G.; Sohár, P. J. Chem. Soc., Perkin Trans. 1 1985, 2483–2487. doi:10.1039/p19850002483
Return to citation in text: [1] [2] [3] [4] [5] -
Stájer, G.; Szabó, A. E.; Fülöp, F.; Bernáth, G.; Sohár, P. Chem. Ber. 1987, 120, 259–264. doi:10.1002/cber.19871200302
Return to citation in text: [1] [2] [3] [4] -
Stájer, G.; Szabó, A. E.; Bernáth, G.; Sohár, P. Synthesis 1987, 290–292. doi:10.1055/s-1987-27922
Return to citation in text: [1] [2] [3] -
Csende, F.; Fülöp, F.; Stájer, G. Curr. Org. Synth. 2008, 5, 173–185. doi:10.2174/157017908784221576
Return to citation in text: [1] [2] -
Palkó, M.; Sohár, P.; Fülöp, F. Molecules 2011, 16, 7691–7705. doi:10.3390/molecules16097691
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Fülöp, F.; Palkó, M.; Bernáth, G.; Sohár, P. Synth. Commun. 1997, 27, 195–203. doi:10.1080/00397919708005019
Return to citation in text: [1] [2] [3] [4] -
Lamborelle, N.; Simon, J. F.; Luxen, A.; Monbaliu, J.-C. M. Org. Biomol. Chem. 2015, 13, 11602–11606. doi:10.1039/c5ob02036k
Return to citation in text: [1] -
Martin, R. E.; Lenz, M.; Alzieu, T.; Aebi, J. D.; Forzy, L. Tetrahedron Lett. 2013, 54, 6703–6707. doi:10.1016/j.tetlet.2013.09.069
Return to citation in text: [1] -
Patel, S. K.; Long, T. E. Tetrahedron Lett. 2009, 50, 5067–5070. doi:10.1016/j.tetlet.2009.06.082
Return to citation in text: [1] -
Miklós, F.; Stájer, G.; Fülöp, F. Lett. Org. Chem. 2006, 3, 915–916. doi:10.2174/157017806779468086
Return to citation in text: [1] [2] [3] -
Chakrabarty, M.; Sarkar, S.; Harigaya, Y. Synthesis 2003, 2292–2294. doi:10.1055/s-2003-42409
Return to citation in text: [1] -
Goff, D.; Zhang, J.; Singh, R.; Holland, S.; Heckrodt, T.; Ding, P.; Yu, J.; Litvak,, J. Bicyclic aryl and bicyclic heteroaryl substituted triazoles useful as Axl inhibitors. WO Patent WO2008083353A1, July 10, 2008.
Chem. Abstr. 2008, 149, 153089.
Return to citation in text: [1] -
Shao, D. Method for synthesizing 5,6-dihydropyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione. CN Patent CN104098562A, Oct 15, 2014.
Chem. Abstr. 2014, 161, 647237.
Return to citation in text: [1] -
Kawano, A.; Masuda, S.; Saito, M.; Tsuchiya, H.; Fujimoto, S. J. Electrochem. Soc. 2016, 163, C506–C513. doi:10.1149/2.0081609jes
Return to citation in text: [1] -
Oldfield, J. W.; Todd, B. Desalination 1999, 124, 75–84. doi:10.1016/S0011-9164(99)00090-9
Return to citation in text: [1] -
Bogdan, A. R.; Sach, N. W. Adv. Synth. Catal. 2009, 351, 849–854. doi:10.1002/adsc.200800758
Return to citation in text: [1] -
Bogdan, A. R.; James, J. Chem. – Eur. J. 2010, 16, 14506–14512. doi:10.1002/chem.201002215
Return to citation in text: [1]
| 46. | Stájer, G.; Szabó, A. E.; Fülöp, F.; Bernáth, G.; Sohár, P. Chem. Ber. 1987, 120, 259–264. doi:10.1002/cber.19871200302 |
| 54. | Miklós, F.; Stájer, G.; Fülöp, F. Lett. Org. Chem. 2006, 3, 915–916. doi:10.2174/157017806779468086 |
| 49. | Palkó, M.; Sohár, P.; Fülöp, F. Molecules 2011, 16, 7691–7705. doi:10.3390/molecules16097691 |
| 1. | Lapkin, A. A.; Plucinski, P. K. Engineering Factors for Efficient Flow rocesses in Chemical Industries. Chemical Reactions; Processes under Flow Conditions; Chapter 1, Vol. 5; The Royal Society of. Chemistry, 2010; pp 1–43. |
| 2. | Jähnisch, K.; Hessel, V.; Löwe, H.; Baerns, M. Angew. Chem., Int. Ed. 2004, 43, 406–446. doi:10.1002/anie.200300577 |
| 3. | Wegner, J.; Ceylan, S.; Kirschning, A. Adv. Synth. Catal. 2012, 354, 17–57. doi:10.1002/adsc.201100584 |
| 7. | Wegner, J.; Ceylan, S.; Kirschning, A. Chem. Commun. 2011, 47, 4583–4592. doi:10.1039/C0CC05060A |
| 8. | Wiles, C.; Watts, P. Green Chem. 2012, 14, 38–54. doi:10.1039/C1GC16022B |
| 9. | Newman, S. G.; Jensen, K. F. Green Chem. 2013, 15, 1456–1472. doi:10.1039/C3GC40374B |
| 10. | Wiles, C.; Watts, P. Green Chem. 2014, 16, 55–62. doi:10.1039/c3gc41797b |
| 11. | Müller, S. T. R.; Wirth, T. ChemSusChem 2015, 8, 245–250. doi:10.1002/cssc.201402874 |
| 12. | Kockmann, N.; Thenée, P.; Fleischer-Trebes, C.; Laudadio, G.; Noël, T. React. Chem. Eng. 2017, 2, 258–280. doi:10.1039/c7re00021a |
| 13. | DeMello, A. J. Nature 2006, 442, 394–402. doi:10.1038/nature05062 |
| 14. | Noël, T.; Su, Y.; Hessel, V. Top. Organomet. Chem. 2016, 57, 1–41. doi:10.1007/3418_2015_152 |
| 30. | Citron, C. A.; Wickel, S. M.; Schulz, B.; Draeger, S.; Dickschat, J. S. Eur. J. Org. Chem. 2012, 6636–6646. doi:10.1002/ejoc.201200991 |
| 31. | Clay, D. R.; Rosenberg, A. G.; McIntosh, M. C. Tetrahedron: Asymmetry 2011, 22, 713–716. doi:10.1016/j.tetasy.2011.04.022 |
| 49. | Palkó, M.; Sohár, P.; Fülöp, F. Molecules 2011, 16, 7691–7705. doi:10.3390/molecules16097691 |
| 7. | Wegner, J.; Ceylan, S.; Kirschning, A. Chem. Commun. 2011, 47, 4583–4592. doi:10.1039/C0CC05060A |
| 8. | Wiles, C.; Watts, P. Green Chem. 2012, 14, 38–54. doi:10.1039/C1GC16022B |
| 9. | Newman, S. G.; Jensen, K. F. Green Chem. 2013, 15, 1456–1472. doi:10.1039/C3GC40374B |
| 10. | Wiles, C.; Watts, P. Green Chem. 2014, 16, 55–62. doi:10.1039/c3gc41797b |
| 11. | Müller, S. T. R.; Wirth, T. ChemSusChem 2015, 8, 245–250. doi:10.1002/cssc.201402874 |
| 12. | Kockmann, N.; Thenée, P.; Fleischer-Trebes, C.; Laudadio, G.; Noël, T. React. Chem. Eng. 2017, 2, 258–280. doi:10.1039/c7re00021a |
| 32. | Gallagher, T.; Sanchez, S.; Bateson, J. H.; O'Hanlon, P. J. Pure Appl. Chem. 2009, 77, 2033–2040. doi:10.1351/pac200577122033 |
| 33. | Eddolls, J. P.; Iqbal, M.; Robert, S. M.; Santoro, M. G. Tetrahedron 2004, 60, 2539–2550. doi:10.1016/j.tet.2004.01.047 |
| 34. | Iqbal, M.; Li, Y.; Evans, P. Tetrahedron 2004, 60, 2531–2538. doi:10.1016/j.tet.2004.01.048 |
| 35. | Arai, Y.; Kontani, T.; Koizumi, T. Chem. Lett. 1991, 2135–2138. doi:10.1246/cl.1991.2135 |
| 51. | Lamborelle, N.; Simon, J. F.; Luxen, A.; Monbaliu, J.-C. M. Org. Biomol. Chem. 2015, 13, 11602–11606. doi:10.1039/c5ob02036k |
| 52. | Martin, R. E.; Lenz, M.; Alzieu, T.; Aebi, J. D.; Forzy, L. Tetrahedron Lett. 2013, 54, 6703–6707. doi:10.1016/j.tetlet.2013.09.069 |
| 53. | Patel, S. K.; Long, T. E. Tetrahedron Lett. 2009, 50, 5067–5070. doi:10.1016/j.tetlet.2009.06.082 |
| 6. | Hessel, V.; Kralisch, D.; Kockmann, N.; Noël, T.; Wang, Q. ChemSusChem 2013, 6, 746–789. doi:10.1002/cssc.201200766 |
| 28. | Suzuki, K.; Inomata, K.; Endo, Y. Org. Lett. 2004, 6, 409. doi:10.1021/ol036253p |
| 54. | Miklós, F.; Stájer, G.; Fülöp, F. Lett. Org. Chem. 2006, 3, 915–916. doi:10.2174/157017806779468086 |
| 4. | Rehm, T. H.; Hofmann, C.; Reinhard, D.; Kost, H.-J.; Löb, P.; Besold, M.; Welzel, K.; Barten, J.; Didenko, A.; Sevenard, D. V.; Lix, B.; Hillson, A. R.; Riegel, S. D. React. Chem. Eng. 2017, 2, 315–323. doi:10.1039/c7re00023e |
| 5. | Roberge, D. M.; Zimmermann, B.; Rainone, F.; Gottsponer, M.; Eyholzer, M.; Kockmann, N. Org. Process Res. Dev. 2008, 12, 905–910. doi:10.1021/op8001273 |
| 29. | Nising, C. F.; Ohnemuller, U. K.; Bräse, S. Synthesis 2006, 2643–2645. doi:10.1055/s-2006-942484 |
| 45. | Stájer, G.; Szabó, A. E.; Pintye, J.; Bernáth, G.; Sohár, P. J. Chem. Soc., Perkin Trans. 1 1985, 2483–2487. doi:10.1039/p19850002483 |
| 22. | Wollweber, H. Diels–Alder-Reaction; Georg Thieme Verlag: Stuttgart, 1972; pp 152–270. |
| 26. | Stájer, G.; Csende, F.; Fülöp, F. Curr. Org. Chem. 2003, 7, 1423–1432. doi:10.2174/1385272033486369 |
| 50. | Fülöp, F.; Palkó, M.; Bernáth, G.; Sohár, P. Synth. Commun. 1997, 27, 195–203. doi:10.1080/00397919708005019 |
| 21. | Movsisyan, M.; Delbeke, E. I. P.; Berton, J. K. E. T.; Battilocchio, C.; Ley, S. V.; Stevens, C. V. Chem. Soc. Rev. 2016, 45, 4892–4928. doi:10.1039/C5CS00902B |
| 27. | González-Temprano, I.; Osante, I.; Lete, E.; Sotomayor, N. J. Org. Chem. 2004, 69, 3875–3885. doi:10.1021/jo049672o |
| 45. | Stájer, G.; Szabó, A. E.; Pintye, J.; Bernáth, G.; Sohár, P. J. Chem. Soc., Perkin Trans. 1 1985, 2483–2487. doi:10.1039/p19850002483 |
| 17. | Illg, T.; Löb, P.; Hessel, V. Bioorg. Med. Chem. 2010, 18, 3707–3719. doi:10.1016/j.bmc.2010.03.073 |
| 18. | Mándity, I. M.; Ötvös, S. B.; Szöllösi, G.; Fülöp, F. Chem. Rec. 2016, 16, 1018–1033. doi:10.1002/tcr.201500286 |
| 19. | Hsieh, C.-T.; Ötvös, S. B.; Wu, Y.-C.; Mándity, I. M.; Chang, F.-R.; Fülöp, F. ChemPlusChem 2015, 80, 859–864. doi:10.1002/cplu.201402426 |
| 49. | Palkó, M.; Sohár, P.; Fülöp, F. Molecules 2011, 16, 7691–7705. doi:10.3390/molecules16097691 |
| 6. | Hessel, V.; Kralisch, D.; Kockmann, N.; Noël, T.; Wang, Q. ChemSusChem 2013, 6, 746–789. doi:10.1002/cssc.201200766 |
| 14. | Noël, T.; Su, Y.; Hessel, V. Top. Organomet. Chem. 2016, 57, 1–41. doi:10.1007/3418_2015_152 |
| 15. | Baxendale, I. R. J. Chem. Technol. Biotechnol. 2013, 88, 519–552. doi:10.1002/jctb.4012 |
| 16. | Hessel, V. Chem. Eng. Technol. 2009, 32, 1655–1681. doi:10.1002/ceat.200900474 |
| 17. | Illg, T.; Löb, P.; Hessel, V. Bioorg. Med. Chem. 2010, 18, 3707–3719. doi:10.1016/j.bmc.2010.03.073 |
| 23. | Ichihara, A. Synthesis 1987, 207–222. doi:10.1055/s-1987-27894 |
| 24. | Rickborn, B. The Retro–Diels–Alder Reaction Part I. C-C Dienophiles. Organic Reactions; John Wiley & Sons, Inc., 2004. doi:10.1002/0471264180.or052.01 |
| 25. | Klunder, A. J. H.; Zhu, J.; Zwanenburg, B. Chem. Rev. 1999, 99, 1163–1190. doi:10.1021/cr9803840 |
| 49. | Palkó, M.; Sohár, P.; Fülöp, F. Molecules 2011, 16, 7691–7705. doi:10.3390/molecules16097691 |
| 41. | Kummer, D. A.; Li, D.; Dion, A.; Myers, A. G. Chem. Sci. 2011, 2, 1710–1718. doi:10.1039/C1SC00303H |
| 35. | Arai, Y.; Kontani, T.; Koizumi, T. Chem. Lett. 1991, 2135–2138. doi:10.1246/cl.1991.2135 |
| 36. | Hasbullah, S. A.; Jones, S. Tetrahedron: Asymmetry 2010, 21, 2719–2725. doi:10.1016/j.tetasy.2010.10.021 |
| 54. | Miklós, F.; Stájer, G.; Fülöp, F. Lett. Org. Chem. 2006, 3, 915–916. doi:10.2174/157017806779468086 |
| 37. | Csende, F.; Stájer, G.; Fülöp, F. Comprehensive Organic Synthesis, 2nd ed.; Elsevier: Amsterdam, 2014; Vol. 5, pp 518–594. |
| 38. | Stájer, G.; Miklós, F.; Kanizsai, I.; Csende, F.; Sillanpää, R.; Sohár, P. Eur. J. Org. Chem. 2004, 3701–3706. doi:10.1002/ejoc.200400247 |
| 39. | Fülöp, F.; Miklós, F.; Forró, E. Synlett 2008, 1687–1689. doi:10.1055/s-2008-1077793 |
| 40. | Miklós, F.; Stájer, G.; Fülöp, F. Lett. Org. Chem. 2006, 3, 915–916. doi:10.2174/157017806779468086 |
| 50. | Fülöp, F.; Palkó, M.; Bernáth, G.; Sohár, P. Synth. Commun. 1997, 27, 195–203. doi:10.1080/00397919708005019 |
| 49. | Palkó, M.; Sohár, P.; Fülöp, F. Molecules 2011, 16, 7691–7705. doi:10.3390/molecules16097691 |
| 47. | Stájer, G.; Szabó, A. E.; Bernáth, G.; Sohár, P. Synthesis 1987, 290–292. doi:10.1055/s-1987-27922 |
| 46. | Stájer, G.; Szabó, A. E.; Fülöp, F.; Bernáth, G.; Sohár, P. Chem. Ber. 1987, 120, 259–264. doi:10.1002/cber.19871200302 |
| 45. | Stájer, G.; Szabó, A. E.; Pintye, J.; Bernáth, G.; Sohár, P. J. Chem. Soc., Perkin Trans. 1 1985, 2483–2487. doi:10.1039/p19850002483 |
| 49. | Palkó, M.; Sohár, P.; Fülöp, F. Molecules 2011, 16, 7691–7705. doi:10.3390/molecules16097691 |
| 18. | Mándity, I. M.; Ötvös, S. B.; Szöllösi, G.; Fülöp, F. Chem. Rec. 2016, 16, 1018–1033. doi:10.1002/tcr.201500286 |
| 19. | Hsieh, C.-T.; Ötvös, S. B.; Wu, Y.-C.; Mándity, I. M.; Chang, F.-R.; Fülöp, F. ChemPlusChem 2015, 80, 859–864. doi:10.1002/cplu.201402426 |
| 20. | Mándity, I. M.; Ötvös, S. B.; Fülöp, F. ChemistryOpen 2015, 4, 212–223. doi:10.1002/open.201500018 |
| 26. | Stájer, G.; Csende, F.; Fülöp, F. Curr. Org. Chem. 2003, 7, 1423–1432. doi:10.2174/1385272033486369 |
| 45. | Stájer, G.; Szabó, A. E.; Pintye, J.; Bernáth, G.; Sohár, P. J. Chem. Soc., Perkin Trans. 1 1985, 2483–2487. doi:10.1039/p19850002483 |
| 46. | Stájer, G.; Szabó, A. E.; Fülöp, F.; Bernáth, G.; Sohár, P. Chem. Ber. 1987, 120, 259–264. doi:10.1002/cber.19871200302 |
| 47. | Stájer, G.; Szabó, A. E.; Bernáth, G.; Sohár, P. Synthesis 1987, 290–292. doi:10.1055/s-1987-27922 |
| 48. | Csende, F.; Fülöp, F.; Stájer, G. Curr. Org. Synth. 2008, 5, 173–185. doi:10.2174/157017908784221576 |
| 49. | Palkó, M.; Sohár, P.; Fülöp, F. Molecules 2011, 16, 7691–7705. doi:10.3390/molecules16097691 |
| 60. | Bogdan, A. R.; Sach, N. W. Adv. Synth. Catal. 2009, 351, 849–854. doi:10.1002/adsc.200800758 |
| 61. | Bogdan, A. R.; James, J. Chem. – Eur. J. 2010, 16, 14506–14512. doi:10.1002/chem.201002215 |
| 50. | Fülöp, F.; Palkó, M.; Bernáth, G.; Sohár, P. Synth. Commun. 1997, 27, 195–203. doi:10.1080/00397919708005019 |
| 42. | Januszczyk, P.; Fogt, J.; Boryski, J.; Izawa, K.; Onishi, T.; Neyts, J.; De Clercq, E. Nucleosides, Nucleotides Nucleic Acids 2009, 28, 713–723. doi:10.1080/15257770903128870 |
| 43. | Bakavoli, M.; Bagherzadeh, G.; Vaseghifar, M.; Shiri, A.; Pordel, M.; Mashreghi, M.; Pordeli, P.; Araghi, M. Eur. J. Med. Chem. 2010, 45, 647–650. doi:10.1016/j.ejmech.2009.10.051 |
| 44. | Guo, C.; Linton, A.; Jalaie, M.; Kephart, S.; Ornelas, M.; Pairish, M.; Greasley, S.; Richardson, P.; Maegley, K.; Hickey, M.; Li, J.; Wu, X.; Ji, X.; Xie, Z. Bioorg. Med. Chem. Lett. 2013, 23, 3358–3363. doi:10.1016/j.bmcl.2013.03.090 |
| 55. | Chakrabarty, M.; Sarkar, S.; Harigaya, Y. Synthesis 2003, 2292–2294. doi:10.1055/s-2003-42409 |
| 56. |
Goff, D.; Zhang, J.; Singh, R.; Holland, S.; Heckrodt, T.; Ding, P.; Yu, J.; Litvak,, J. Bicyclic aryl and bicyclic heteroaryl substituted triazoles useful as Axl inhibitors. WO Patent WO2008083353A1, July 10, 2008.
Chem. Abstr. 2008, 149, 153089. |
| 57. |
Shao, D. Method for synthesizing 5,6-dihydropyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione. CN Patent CN104098562A, Oct 15, 2014.
Chem. Abstr. 2014, 161, 647237. |
| 26. | Stájer, G.; Csende, F.; Fülöp, F. Curr. Org. Chem. 2003, 7, 1423–1432. doi:10.2174/1385272033486369 |
| 45. | Stájer, G.; Szabó, A. E.; Pintye, J.; Bernáth, G.; Sohár, P. J. Chem. Soc., Perkin Trans. 1 1985, 2483–2487. doi:10.1039/p19850002483 |
| 46. | Stájer, G.; Szabó, A. E.; Fülöp, F.; Bernáth, G.; Sohár, P. Chem. Ber. 1987, 120, 259–264. doi:10.1002/cber.19871200302 |
| 47. | Stájer, G.; Szabó, A. E.; Bernáth, G.; Sohár, P. Synthesis 1987, 290–292. doi:10.1055/s-1987-27922 |
| 48. | Csende, F.; Fülöp, F.; Stájer, G. Curr. Org. Synth. 2008, 5, 173–185. doi:10.2174/157017908784221576 |
| 49. | Palkó, M.; Sohár, P.; Fülöp, F. Molecules 2011, 16, 7691–7705. doi:10.3390/molecules16097691 |
| 50. | Fülöp, F.; Palkó, M.; Bernáth, G.; Sohár, P. Synth. Commun. 1997, 27, 195–203. doi:10.1080/00397919708005019 |
| 58. | Kawano, A.; Masuda, S.; Saito, M.; Tsuchiya, H.; Fujimoto, S. J. Electrochem. Soc. 2016, 163, C506–C513. doi:10.1149/2.0081609jes |
| 59. | Oldfield, J. W.; Todd, B. Desalination 1999, 124, 75–84. doi:10.1016/S0011-9164(99)00090-9 |
© 2018 Nekkaa et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)