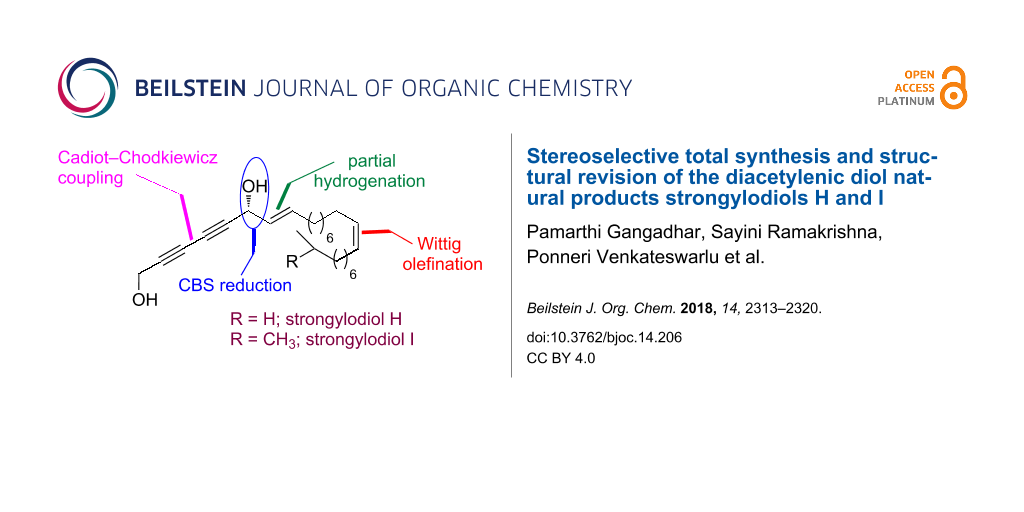
Abstract
The stereoselective total synthesis of strongylodiol H and I has been accomplished. The synthetic procedure comprised the stereoselective reduction of a ketone functionality in an ene–yne–one employing CBS as a catalyst and a Cadiot–Chodkiewicz coupling reaction as the key reaction steps. A common aldehyde intermediate has been used for the synthesis of both strongylodiols.

Graphical Abstract
Introduction
Diacetylenic polyol compounds [1,2] originated from marine sources continue to attract significant interest owing to their structural architectures and impressive biological properties that include antibacterial [3], anticancer [4-6], antiviral [7] and neuritogenic activities [8]. Watanabe et al. [9,10] have isolated strongylodiols A–J (1–10, Figure 1) [11] from the Okinawan marine sponge of the genus Petrosia (Strongylophora). The structural elucidation was accomplished by spectroscopic analyses and chemical reactions. It was found that these strongylodiols were present as an enantiomeric mixture with different ratios after analysis of their corresponding MNA ((R)- and (S)-methoxy(2-naphthyl)acetic acid) derivatives. Though petrosiols A, D and E along with strongylodiols C and D were found to display neuronal differentiation of PC12 cells in a dose-dependent manner and induce neuronal outgrowth, strongylodiols C and D also displayed cytotoxicity at higher concentrations [11]. There have been few contributions on the total synthesis of strongylodiols [12,13] employing alkynylation of an unsaturated aliphatic aldehyde catalyzed by Trost’s pro-phenol ligand [12,14], β-elimination of epoxy chloride [15], Noyori’s asymmetric reduction of ynones [16], diyne addition to long chain aliphatic aldehydes in the presence of N-methylephedrine [17] or an amino alcohol–zinc complex [18] and the Cadiot–Chodkiewicz cross-coupling reaction as key steps [14-16].
![[1860-5397-14-206-1]](/bjoc/content/figures/1860-5397-14-206-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Proposed structures of a selection of diacetylenic polyol natural products.
Figure 1: Proposed structures of a selection of diacetylenic polyol natural products.
In continuation to our research interest on the synthesis of acetylenic compounds [19-21], recently we have accomplished the total synthesis of the diacetylenic polyol natural products petrosiols A, D, E (11,12,13) [22,23] and strongylodiols A–D (1–4) [24]. Herein we describe the total synthesis of strongylodiols H and I (9 and 10).
Results and Discussion
Retrosynthesis for strongylodiol H and strongylodiol I
The retrosynthetic analysis for strongylodiols H and I is delineated in Scheme 1. We envisaged that the target molecules strongylodiol H (9) and strongylodiol I (10) can be synthesized by a Wittig reaction of a common intermediate aldehyde 14 with triphenylphosphonium Wittig salts 15 and 16, respectively, followed by the desilylation (TBDPS removal) to yield the title products. The intermediate aldehyde 14 can be synthesized from ketone 17 in a four-step sequence by a stereoselective keto reduction, TBDPS protection, TBS deprotection and an oxidation reaction. The ketone 17 can be easily synthesized by a coupling reaction of 18 with 19 followed by an oxidation reaction. While compound 19 can be obtained from 20 in 5 steps, compound 18 is easily accessible from readily available propargyl alcohol. Compound 20 in turn can be synthesized from commercially available 1,8-octanediol in a 3-step sequence (Scheme 1).
![[1860-5397-14-206-i1]](/bjoc/content/inline/1860-5397-14-206-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthesis of strongylodiols H and I.
Scheme 1: Retrosynthesis of strongylodiols H and I.
Synthesis of strongylodiols H and I
The synthesis began with the C-alkylation reaction of propargyl alcohol with tert-butyl((8-iodooctyl)oxy)dimethylsilane [14] (21, prepared from 1,8-octanediol in two steps) in the presence of n-BuLi to provide alkynol 20 in 72% yield. The trans-selective reduction [25] of alkyne 20 was easily achieved with sodium bis(2-methoxyethoxy)aluminum hydride (Red-Al) as a hydride-transfer reagent to furnish the corresponding (E)-allylic alcohol 23. A Swern oxidation of 23 provided the corresponding aldehyde which was subjected to an addition reaction with lithium(trimethylsilyl)acetylide to get the coupled product 24. The latter compound on further treatment with K2CO3 in MeOH [26] furnished the desilylated propargylic alcohol 19 (Scheme 2).
![[1860-5397-14-206-i2]](/bjoc/content/inline/1860-5397-14-206-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of alkyne 19 and iodo intermediate 21. Reagents and conditions: (a) n-BuLi, THF, −78 °C to rt, 21, 12 h, 72%; (b) Red-Al, anh. Et2O, −20 °C, 8 h, 95%; (c) (i) DMSO, CH2Cl2, (COCl)2, −78 °C, (ii) TMS-C≡CH, n-BuLi, THF, −78 °C to rt, 3 h, 89%; (d) K2CO3, MeOH, 0 °C to rt, 1 h, 92%; (e) TBSCl, imidazole, 0 °C to rt, 1 h, 98%; (f) I2, TPP, imidazole, 0 °C to rt, 1 h, 92%.
Scheme 2: Synthesis of alkyne 19 and iodo intermediate 21. Reagents and conditions: (a) n-BuLi, THF, −78 °C t...
The copper(I)-catalyzed Cadiot–Chodkiewicz [27] cross-coupling reaction between bromoalkyne 18 [28] and terminal alkyne 19 provided the corresponding diynes 25 and 25a in a 1:1 ratio. Though we had an option to proceed further with the mixture of 25 and 25a affording both enantiomers that could be separated later, we focused our attention towards the synthesis of the required chiral compound. Thus, the mixture of 25 and 25a was oxidized under Dess–Martin conditions to give the prochiral ene–yne–one 17 in 87% yield. The ketone 17 was then subjected to a stereoselective asymmetric reduction [23,29-31] in the presence of (S)-CBS as the catalyst to yield the chiral propargylic alcohol 25 with 92% ee (Scheme 3) [32].
![[1860-5397-14-206-i3]](/bjoc/content/inline/1860-5397-14-206-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Stereoselective synthesis of (R)-25. Reagents and conditions: (a) CuCl, NH2OH·HCl, 30% n-BuNH2, Et2O, 1 h, 68%; (b) DMP, CH2Cl2, 0 °C to rt, 1 h, 87%; (c) (S)-CBS catalyst, BH3·DMS, THF, −50 °C, 16 h, 85%.
Scheme 3: Stereoselective synthesis of (R)-25. Reagents and conditions: (a) CuCl, NH2OH·HCl, 30% n-BuNH2, Et2...
After derivatization and determination through Mosher’s ester analysis [33-35], the absolute configuration of the newly generated secondary hydroxy group-bearing carbon center (C6) was determined as R configuration (see Supporting Information File 1). The NMR analysis of the Mosher’s esters showed a positive chemical shift difference ∆δ for the protons at C7 and C8 indicating the R configuration at C6 in compound 25 (see Figure 2).
![[1860-5397-14-206-2]](/bjoc/content/figures/1860-5397-14-206-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Absolute configuration analysis of alcohol 25. ∆δ = δS − δR for the (R)- and (S)-MTPA ester of alcohol 25.
Figure 2: Absolute configuration analysis of alcohol 25. ∆δ = δS − δR for the (R)- and (S)-MTPA ester of alco...
Having determined the stereochemistry at the newly generated carbon center in intermediate 25, we proceeded further to extend the chain at the right hand side. Towards this, the free secondary hydroxy group in 25 was masked as its corresponding TBDPS ether 26 and then treated with PPTS in MeOH [36] to afford the disilylated primary alcohol 27 in 95% yield. Treatment of alcohol 27 with IBX [37] furnished the corresponding aldehyde 14 which was subjected to Wittig olefination reaction with triphenylphosphonium salt of n-nonyl bromide 15 in presence of n-BuLi to produce the corresponding Z-olefin 28 exclusively in 83% yield. Di-desilylation of compound 28 was easily achieved with n-tetrabutylammonium fluoride to furnish the target molecule strongylodiol H 9 in 85% yield (Scheme 4).
![[1860-5397-14-206-i4]](/bjoc/content/inline/1860-5397-14-206-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of strongylodiol H (9). Reagents and conditions: (a) TBDPSCl, imidazole, CH2Cl2, 0 °C to rt, 2 h, 95%; (b) PPTS, MeOH, 0 °C to rt, 2 h, 95%; (c) IBX, THF/DMSO, 0 °C to rt, 1 h, 97%; (d) 15, n-BuLi, THF, −78 °C to rt, 2 h, 83%; (e) TBAF, THF, 0 °C to rt, 2 h, 85%.
Scheme 4: Synthesis of strongylodiol H (9). Reagents and conditions: (a) TBDPSCl, imidazole, CH2Cl2, 0 °C to ...
Though the 1H NMR and 13C NMR spectra of the synthesized product were in full agreement with that of the reported natural product [10], the specific rotation of our synthetic product was determined as [α]D25 = +42.2 (c 0.81, CHCl3), and for the natural product it was found to be [α]D25 = −43.8 (c 0.35, CHCl3) [10]. Having identical spectral data (see Table 1 for comparative 1H and 13C NMR data for the synthetic and natural strongylodiol H), but with the opposite sign of rotation, and based on the outcome of our synthesis, we unambiguously revise the structure of the natural product as 9a which is the enantiomer of the proposed structure 9 (Figure 3).
Table 1: Comparison of 1H and 13C NMR data of strongylodiol H (isolated natural product vs synthetic).
| position |
1H NMR for isolated compound [10]
(CDCl3, 500 MHz) |
1H NMR for synthesized
compound (CDCl3, 400 MHz) |
13C NMR for isolated compound [10] (CDCl3,
125 MHz) |
13C NMR for synthesized compound (CDCl3, 100 MHz) |
|---|---|---|---|---|
| 1 | 4.36 (d, 6.3) | 4.35 s | 51.5 | 51.42 |
| 2 | – | – | 77.9 | 77.96 |
| 3 | – | – | 69.8 | 69.78 |
| 4 | – | – | 69.8 | 69.75 |
| 5 | – | – | 78.7 | 78.66 |
| 6 | 4.89 (brt, 6.2) | 4.89 (d, 5.8) | 63.3 | 63.29 |
| 7 | 5.57 (dd, 6.2, 15.3) | 5.57 (dd, 6.2, 15.3) | 127.7 | 127.65 |
| 8 | 5.89 (td, 6.7, 15.3) | 5.93–5.85 m | 135.2 | 135.19 |
| 9 | 2.06 (q, 6.7) | 2.14–1.97 m | 31.9 | 31.96 |
| 10 | 1.39 m | 1.49–1.21 m | 28.8 | 28.76 |
| 11–13 | 1.24–1.31 m | – | – | |
| 14 | 1.34 m | 28.7 | 28.76 | |
| 15 | 2.01 (q, 6.1) | 2.14–1.97 m | 27.18 | 27.19 |
| 16 | 5.35 m | 5.37–5.32 m | 129.8 | 129.79 |
| 17 | 130.0 | 129.95 | ||
| 18 | 2.01 (q, 6.1) | 2.14–1.97 m | 27.21 | 27.19 |
| 19 | 2.14 (t, 6.7) | 29.8 | 29.72 | |
| 20–22 | 1.24–1.31 m | 1.49–1.21 m | – | – |
| 23 | 1.24–1.31 m | 32.0 | 31.96 | |
| 24 | 1.24–1.31 m | 22.7 | 22.67 | |
| 25 | 0.88 (t, 6.7) | 0.88 (t, 6.7) | 14.1 | 14.10 |
![[1860-5397-14-206-3]](/bjoc/content/figures/1860-5397-14-206-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Previously proposed and revised structure of strongylodiol H (9).
Figure 3: Previously proposed and revised structure of strongylodiol H (9).
Further, to reconfirm the structural revision, we synthesized the other enantiomer of strongylodiol H. Towards this we proceeded for the stereoselective reduction of prochiral ketone 17 with (R)-CBS as the catalyst furnishing the chiral propargylic alcohol 25a with 92% ee (Scheme 5).
![[1860-5397-14-206-i5]](/bjoc/content/inline/1860-5397-14-206-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of compound 25a. Reagents and conditions: (a) (R)-CBS catalyst, BH3·DMS, −50 °C, 16 h, 86%.
Scheme 5: Synthesis of compound 25a. Reagents and conditions: (a) (R)-CBS catalyst, BH3·DMS, −50 °C, 16 h, 86...
After securing the absolute configuration at C6 in compound 25a by employing the Mosher’s ester analysis indicating S-configuration (see Supporting Information File 1), compound 25a was subjected to TBDPS protection followed by TBS deprotection to give the primary alcohol which was subjected to oxidation to yield aldehyde 14a (Scheme 6). The latter was then subjected to a Wittig reaction with 15 followed by silyl deprotection as performed for 14 to furnish 9a (see Supporting Information File 1 for experimental details). The sign of the optical rotation for 9a {[α]D25 = −40.2 (c 0.72, CHCl3)} was found to be in accordance with that reported for the isolated natural product {[α]D25 = −43.8 (c 0.35, CHCl3)} [10]. Thus, the total synthesis of the natural product (−)-strongylodiol H has been accomplished successfully.
![[1860-5397-14-206-i6]](/bjoc/content/inline/1860-5397-14-206-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of strongylodiol I (10a). Reagents and conditions: (a) (i) TBDPSCl, imidazole, CH2Cl2, 0 °C to rt, 1 h, 96%; (ii) PPTS, MeOH, 0 °C to rt, 2 h, 87%; (iii) IBX, DMSO/THF 1:1, 0 °C to rt, 1 h, 98%; (b) 16, n-BuLi, THF, −78 °C to rt, 2 h, 81%; (c) TBAF, THF, 0 °C to rt, 2 h, 82%.
Scheme 6: Synthesis of strongylodiol I (10a). Reagents and conditions: (a) (i) TBDPSCl, imidazole, CH2Cl2, 0 ...
As the proposed structure for strongylodiol H was revised, we next turned our attention to the total synthesis of strongylodiol I, having an additional methyl group attached to carbon atom C24. Towards this, aldehyde 14a was subjected to a Wittig reaction with the triphenylphosphonium salt of 1-iodo-8-methylnonane 16, prepared earlier in our group [24], to furnish the corresponding Z-olefin 29 exclusively in 81% yield. Exposure of 29 to n-tetrabutylammonium fluoride provided the natural product (S)-strongylodiol I (10a) in 82% yield (Scheme 6). The analytical data and specific rotation of the synthetic product were found to be comparable with the previously reported data of the isolated product {[α]D25 = −30.2 (c 1.7, CHCl3)}; Lit. {[α]D25 = −33.4 (c 0.35, CHCl3)} [10] (see Table 2 for comparative 1H and 13C NMR data for the synthetic and natural strongylodiol I). Thus, our synthesis has led us to revise the proposed structure of strongylodiol I as 10a. Based on these findings we believe, that the structures of other strongylodiols E, F, G and J may have to be revised accordingly as they contain only one similar chiral center as observed in strongylodiols H and I.
Table 2: Comparison of 1H and 13C NMR data of strongylodiol I (isolated natural product vs synthetic).
| position |
1H NMR for isolated compound [10] (CDCl3,
500 MHz) |
1H NMR for synthesized compound (CDCl3,
500 MHz) |
13C NMR for isolated compound [10] (CDCl3, 125 MHz) | 13C NMR for synthesized compound (CDCl3, 125 MHz) |
|---|---|---|---|---|
| 1 | 4.35 (d, 5.8) | 4.34 s | 51.5 | 51.42 |
| 2 | – | – | 77.9 | 77.95 |
| 3 | – | – | 69.80 | 69.76 |
| 4 | – | – | 68.83 | 69.76 |
| 5 | – | – | 78.7 | 78.66 |
| 6 | 4.89 (brt, 6.0) | 4.89 (d, 5.9) | 63.3 | 63.29 |
| 7 | 5.51 (brdd, 6.0, 15.3) | 5.57 (dd, 6.2, 15.3) | 127.7 | 127.66 |
| 8 | 5.90 (td, 6.8, 15.3) | 5.93–5.85 m | 135.2 | 135.19 |
| 9 | 2.06 (q, 6.8) | 2.10–1.97 m | 32.0 | 31.96 |
| 10 | 1.39 m | 1.57–1.11 m | 28.8 | 28.75 |
| 11–13 | 1.24–1.30 m | – | – | |
| 14 | 1.34 m | 29.74 | 28.73 | |
| 15 | 2.02 (q, 6.1) | 2.10–1.97 m | 27.19 | 27.18 |
| 16 | 5.34 m | 5.38–5.31 m | 129.8 | 129.79 |
| 17 | 130.0 | 129.95 | ||
| 18 | 2.02 (q, 6.0) | 2.10–1.97 m | 27.22 | 27.20 |
| 19 | 1.34 m | 1.57–1.11 m | 29.77 | 29.79 |
| 20–21 | 1.24–1.30 m | – | – | |
| 22 | 1.24–1.31 m | 27.4 | 27.36 | |
| 23 | 1.15 m | 39.0 | 39.02 | |
| 24 | 1.52 m | 28.0 | 27.94 | |
| 25–26 | 0.86 (d, 6.6) | 0.86 (d, 6.7) | 22.7 | 22.64 |
Conclusion
In conclusion, we have accomplished the first enantioselective total synthesis of the two diacetylenic diol natural products strongylodiol H and strongylodiol I and of an enantiomer of strongylodiol H. Our synthesis assisted us to revise the structure of both natural products strongylodiol H and I. The synthetic procedure involved the alkylation of the commercially available propargyl alcohol, a Cadiot–Chodkiewicz cross coupling, and CBS catalyzed reduction of the intermediate ene–yne–one as the key steps. The natural products strongylodiol H and I were obtained in 16.2%, 15.4% yields, respectively, from the commercially available propargyl alcohol involving 13 linear steps, respectively. Investigations towards exploring the current strategy for accessing other natural products and their analogues are currently underway.
References
-
Shi Shun, A. L. K.; Tykwinski, R. R. Angew. Chem., Int. Ed. 2006, 45, 1034–1057. doi:10.1002/anie.200502071
Return to citation in text: [1] -
Minto, R. E.; Blacklock, B. J. Prog. Lipid Res. 2008, 47, 233–306. doi:10.1016/j.plipres.2008.02.002
Return to citation in text: [1] -
Ankisetty, S.; Slattery, M. Mar. Drugs 2012, 10, 1037–1043. doi:10.3390/md10051037
Return to citation in text: [1] -
Lee, C.-Y.; Yun, J. H.; Kang, K.; Nho, C.-W.; Shin, D. Bioorg. Med. Chem. Lett. 2015, 25, 4020–4023. doi:10.1016/j.bmcl.2015.05.098
Return to citation in text: [1] -
Guo, L.; Song, L.; Wang, Z.; Zhao, W.; Mao, W.; Yin, M. Chem.-Biol. Interact. 2009, 181, 138–143. doi:10.1016/j.cbi.2009.04.015
Return to citation in text: [1] -
Matsunaga, H.; Saita, T.; Nagumo, F.; Mori, M.; Katano, M. Cancer Chemother. Pharmacol. 1995, 35, 291–296. doi:10.1007/BF00689447
Return to citation in text: [1] -
Zhao, Y.; Geng, C.-A.; Sun, C.-L.; Ma, Y.-B.; Huang, X.-Y.; Cao, T.-W.; He, K.; Wang, H.; Zhang, X.-M.; Chen, J.-J. Fitoterapia 2014, 95, 187–193. doi:10.1016/j.fitote.2014.03.017
Return to citation in text: [1] -
Aoki, S.; Matsui, K.; Wei, H.; Murakami, N.; Kobayashi, M. Tetrahedron 2002, 58, 5417–5422. doi:10.1016/S0040-4020(02)00519-7
Return to citation in text: [1] -
Watanabe, K.; Tsuda, Y.; Yamane, Y.; Takahashi, H.; Iguchi, K.; Naoki, H.; Fujita, T.; Van Soest, R. W. M. Tetrahedron Lett. 2000, 41, 9271–9276. doi:10.1016/S0040-4039(00)01692-0
Return to citation in text: [1] -
Watanabe, K.; Tsuda, Y.; Hamada, M.; Omori, M.; Mori, G.; Iguchi, K.; Naoki, H.; Fujita, T.; Van Soest, R. W. M. J. Nat. Prod. 2005, 68, 1001–1005. doi:10.1021/np040233u
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Horikawa, K.; Yagyu, T.; Yoshioka, Y.; Fujiwara, T.; Kanamoto, A.; Okamoto, T.; Ojika, M. Tetrahedron 2013, 69, 101–106. doi:10.1016/j.tet.2012.10.063
Return to citation in text: [1] [2] -
Trost, B. M.; Chan, V. S.; Yamamoto, D. J. Am. Chem. Soc. 2010, 132, 5186–5192. doi:10.1021/ja910656b
Return to citation in text: [1] [2] -
Gupta, N.; Shallu; Kad, G. L.; Singh, J. Nat. Prod. Res. 2014, 28, 424–430. doi:10.1080/14786419.2013.867855
Return to citation in text: [1] -
Liu, F.; Zhong, J.; Li, S.; Li, M.; Wu, L.; Wang, Q.; Mao, J.; Liu, S.; Zheng, B.; Wang, M.; Bian, Q. J. Nat. Prod. 2016, 79, 244–247. doi:10.1021/acs.jnatprod.5b00713
Return to citation in text: [1] [2] [3] -
Yadav, J. S.; Mishra, R. K. Tetrahedron Lett. 2002, 43, 1739–1741. doi:10.1016/S0040-4039(02)00066-7
Return to citation in text: [1] [2] -
Kirkham, J. E. D.; Courtney, T. D. L.; Lee, V.; Baldwin, J. E. Tetrahedron 2005, 61, 7219–7232. doi:10.1016/j.tet.2005.05.034
Return to citation in text: [1] [2] -
Reber, S.; Knopfel, T. F.; Carreira, E. M. Tetrahedron 2003, 59, 6813–6817. doi:10.1016/S0040-4020(03)00905-0
Return to citation in text: [1] -
Zheng, B.; L, S.-N.; Mao, J.-Y.; Wang, B.; Bian, Q.-H.; Liu, S.-Z.; Zhong, J.-C.; Guo, H.-C.; Wang, M. Chem. – Eur. J. 2012, 18, 9208–9211. doi:10.1002/chem.201200728
Return to citation in text: [1] -
Yadav, J. S.; Boyapelly, K.; Sathish Reddy, A.; Srihari, P.; Vangala, J. R.; Kalivendi, S. V. J. Org. Chem. 2011, 76, 2568–2576. doi:10.1021/jo102445h
Return to citation in text: [1] -
Srihari, P.; Sathish Reddy, A.; Yadav, J. S.; Yedlapudi, D.; Kalivendi, S. V. Tetrahedron Lett. 2013, 54, 5616–5618. doi:10.1016/j.tetlet.2013.08.020
Return to citation in text: [1] -
Sathish Reddy, A.; Gangadhar, P.; Srihari, P. Tetrahedron: Asymmetry 2013, 24, 1524–1530. doi:10.1016/j.tetasy.2013.09.017
Return to citation in text: [1] -
Sathish Reddy, A.; Srihari, P. Tetrahedron Lett. 2013, 54, 6370–6372. doi:10.1016/j.tetlet.2013.09.058
Return to citation in text: [1] -
Gangadhar, P.; Sathish Reddy, A.; Srihari, P. Tetrahedron 2016, 72, 5807–5817. doi:10.1016/j.tet.2016.08.009
Return to citation in text: [1] [2] -
Gangadhar, P.; Rajesh, P.; Srihari, P. ChemistrySelect 2017, 2, 4106–4110. doi:10.1002/slct.201700898
Return to citation in text: [1] [2] -
Zhou, S.-Z.; Anné, S.; Vandewalle, M. Tetrahedron Lett. 1996, 37, 7637–7640. doi:10.1016/0040-4039(96)01700-5
Return to citation in text: [1] -
Moilanen, S. B.; Tan, D. S. Org. Biomol. Chem. 2005, 3, 798–803. doi:10.1039/b417429a
Return to citation in text: [1] -
Chodkiewicz, W.; Cadiot, P. C. R. Hebd. Seances Acad. Sci. 1955, 241, 1055–1057.
Return to citation in text: [1] -
Yoshida, M.; Sugimoto, K.; Ihara, M. Org. Lett. 2004, 6, 1979–1982. doi:10.1021/ol049438k
Return to citation in text: [1] -
Corey, E. J.; Bakshi, R. K.; Shibata, S. J. Am. Chem. Soc. 1987, 109, 5551–5553. doi:10.1021/ja00252a056
Return to citation in text: [1] -
Corey, E. J.; Bakshi, R. K. Tetrahedron Lett. 1990, 31, 611–614. doi:10.1016/S0040-4039(00)94581-7
Return to citation in text: [1] -
Trost, B. M.; Quintard, A. Org. Lett. 2012, 14, 4698–4700. doi:10.1021/ol302074h
Return to citation in text: [1] -
The percentage of (R)- and (S)-isomer was analysed after derivatizing the alcohol as the corresponding (R)- or (S)-MTPA ester (Mosher’s ester) followed by LCMS analysis.
Return to citation in text: [1] -
Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. J. Am. Chem. Soc. 1991, 113, 4092–4096. doi:10.1021/ja00011a006
Return to citation in text: [1] -
Freire, F.; Seco, J. M.; Quiñoá, E.; Riguera, R. J. Org. Chem. 2005, 70, 3778–3790. doi:10.1021/jo048643a
Return to citation in text: [1] -
Hoye, T. R.; Jeffrey, C. S.; Shao, F. Nat. Protoc. 2007, 2, 2451–2458. doi:10.1038/nprot.2007.354
Return to citation in text: [1] -
Valli, M.; Bruno, P.; Sbarbada, D.; Porta, A.; Vidari, G.; Zanoni, G. J. Org. Chem. 2013, 78, 5556–5567. doi:10.1021/jo4004647
Return to citation in text: [1] -
Frigerio, M.; Santagostino, M. Tetrahedron Lett. 1994, 35, 8019–8022. doi:10.1016/0040-4039(94)80038-3
Return to citation in text: [1]
| 37. | Frigerio, M.; Santagostino, M. Tetrahedron Lett. 1994, 35, 8019–8022. doi:10.1016/0040-4039(94)80038-3 |
| 10. | Watanabe, K.; Tsuda, Y.; Hamada, M.; Omori, M.; Mori, G.; Iguchi, K.; Naoki, H.; Fujita, T.; Van Soest, R. W. M. J. Nat. Prod. 2005, 68, 1001–1005. doi:10.1021/np040233u |
| 10. | Watanabe, K.; Tsuda, Y.; Hamada, M.; Omori, M.; Mori, G.; Iguchi, K.; Naoki, H.; Fujita, T.; Van Soest, R. W. M. J. Nat. Prod. 2005, 68, 1001–1005. doi:10.1021/np040233u |
| 1. | Shi Shun, A. L. K.; Tykwinski, R. R. Angew. Chem., Int. Ed. 2006, 45, 1034–1057. doi:10.1002/anie.200502071 |
| 2. | Minto, R. E.; Blacklock, B. J. Prog. Lipid Res. 2008, 47, 233–306. doi:10.1016/j.plipres.2008.02.002 |
| 8. | Aoki, S.; Matsui, K.; Wei, H.; Murakami, N.; Kobayashi, M. Tetrahedron 2002, 58, 5417–5422. doi:10.1016/S0040-4020(02)00519-7 |
| 14. | Liu, F.; Zhong, J.; Li, S.; Li, M.; Wu, L.; Wang, Q.; Mao, J.; Liu, S.; Zheng, B.; Wang, M.; Bian, Q. J. Nat. Prod. 2016, 79, 244–247. doi:10.1021/acs.jnatprod.5b00713 |
| 15. | Yadav, J. S.; Mishra, R. K. Tetrahedron Lett. 2002, 43, 1739–1741. doi:10.1016/S0040-4039(02)00066-7 |
| 16. | Kirkham, J. E. D.; Courtney, T. D. L.; Lee, V.; Baldwin, J. E. Tetrahedron 2005, 61, 7219–7232. doi:10.1016/j.tet.2005.05.034 |
| 10. | Watanabe, K.; Tsuda, Y.; Hamada, M.; Omori, M.; Mori, G.; Iguchi, K.; Naoki, H.; Fujita, T.; Van Soest, R. W. M. J. Nat. Prod. 2005, 68, 1001–1005. doi:10.1021/np040233u |
| 7. | Zhao, Y.; Geng, C.-A.; Sun, C.-L.; Ma, Y.-B.; Huang, X.-Y.; Cao, T.-W.; He, K.; Wang, H.; Zhang, X.-M.; Chen, J.-J. Fitoterapia 2014, 95, 187–193. doi:10.1016/j.fitote.2014.03.017 |
| 19. | Yadav, J. S.; Boyapelly, K.; Sathish Reddy, A.; Srihari, P.; Vangala, J. R.; Kalivendi, S. V. J. Org. Chem. 2011, 76, 2568–2576. doi:10.1021/jo102445h |
| 20. | Srihari, P.; Sathish Reddy, A.; Yadav, J. S.; Yedlapudi, D.; Kalivendi, S. V. Tetrahedron Lett. 2013, 54, 5616–5618. doi:10.1016/j.tetlet.2013.08.020 |
| 21. | Sathish Reddy, A.; Gangadhar, P.; Srihari, P. Tetrahedron: Asymmetry 2013, 24, 1524–1530. doi:10.1016/j.tetasy.2013.09.017 |
| 4. | Lee, C.-Y.; Yun, J. H.; Kang, K.; Nho, C.-W.; Shin, D. Bioorg. Med. Chem. Lett. 2015, 25, 4020–4023. doi:10.1016/j.bmcl.2015.05.098 |
| 5. | Guo, L.; Song, L.; Wang, Z.; Zhao, W.; Mao, W.; Yin, M. Chem.-Biol. Interact. 2009, 181, 138–143. doi:10.1016/j.cbi.2009.04.015 |
| 6. | Matsunaga, H.; Saita, T.; Nagumo, F.; Mori, M.; Katano, M. Cancer Chemother. Pharmacol. 1995, 35, 291–296. doi:10.1007/BF00689447 |
| 17. | Reber, S.; Knopfel, T. F.; Carreira, E. M. Tetrahedron 2003, 59, 6813–6817. doi:10.1016/S0040-4020(03)00905-0 |
| 10. | Watanabe, K.; Tsuda, Y.; Hamada, M.; Omori, M.; Mori, G.; Iguchi, K.; Naoki, H.; Fujita, T.; Van Soest, R. W. M. J. Nat. Prod. 2005, 68, 1001–1005. doi:10.1021/np040233u |
| 3. | Ankisetty, S.; Slattery, M. Mar. Drugs 2012, 10, 1037–1043. doi:10.3390/md10051037 |
| 18. | Zheng, B.; L, S.-N.; Mao, J.-Y.; Wang, B.; Bian, Q.-H.; Liu, S.-Z.; Zhong, J.-C.; Guo, H.-C.; Wang, M. Chem. – Eur. J. 2012, 18, 9208–9211. doi:10.1002/chem.201200728 |
| 10. | Watanabe, K.; Tsuda, Y.; Hamada, M.; Omori, M.; Mori, G.; Iguchi, K.; Naoki, H.; Fujita, T.; Van Soest, R. W. M. J. Nat. Prod. 2005, 68, 1001–1005. doi:10.1021/np040233u |
| 12. | Trost, B. M.; Chan, V. S.; Yamamoto, D. J. Am. Chem. Soc. 2010, 132, 5186–5192. doi:10.1021/ja910656b |
| 13. | Gupta, N.; Shallu; Kad, G. L.; Singh, J. Nat. Prod. Res. 2014, 28, 424–430. doi:10.1080/14786419.2013.867855 |
| 15. | Yadav, J. S.; Mishra, R. K. Tetrahedron Lett. 2002, 43, 1739–1741. doi:10.1016/S0040-4039(02)00066-7 |
| 10. | Watanabe, K.; Tsuda, Y.; Hamada, M.; Omori, M.; Mori, G.; Iguchi, K.; Naoki, H.; Fujita, T.; Van Soest, R. W. M. J. Nat. Prod. 2005, 68, 1001–1005. doi:10.1021/np040233u |
| 11. | Horikawa, K.; Yagyu, T.; Yoshioka, Y.; Fujiwara, T.; Kanamoto, A.; Okamoto, T.; Ojika, M. Tetrahedron 2013, 69, 101–106. doi:10.1016/j.tet.2012.10.063 |
| 16. | Kirkham, J. E. D.; Courtney, T. D. L.; Lee, V.; Baldwin, J. E. Tetrahedron 2005, 61, 7219–7232. doi:10.1016/j.tet.2005.05.034 |
| 24. | Gangadhar, P.; Rajesh, P.; Srihari, P. ChemistrySelect 2017, 2, 4106–4110. doi:10.1002/slct.201700898 |
| 11. | Horikawa, K.; Yagyu, T.; Yoshioka, Y.; Fujiwara, T.; Kanamoto, A.; Okamoto, T.; Ojika, M. Tetrahedron 2013, 69, 101–106. doi:10.1016/j.tet.2012.10.063 |
| 10. | Watanabe, K.; Tsuda, Y.; Hamada, M.; Omori, M.; Mori, G.; Iguchi, K.; Naoki, H.; Fujita, T.; Van Soest, R. W. M. J. Nat. Prod. 2005, 68, 1001–1005. doi:10.1021/np040233u |
| 9. | Watanabe, K.; Tsuda, Y.; Yamane, Y.; Takahashi, H.; Iguchi, K.; Naoki, H.; Fujita, T.; Van Soest, R. W. M. Tetrahedron Lett. 2000, 41, 9271–9276. doi:10.1016/S0040-4039(00)01692-0 |
| 10. | Watanabe, K.; Tsuda, Y.; Hamada, M.; Omori, M.; Mori, G.; Iguchi, K.; Naoki, H.; Fujita, T.; Van Soest, R. W. M. J. Nat. Prod. 2005, 68, 1001–1005. doi:10.1021/np040233u |
| 12. | Trost, B. M.; Chan, V. S.; Yamamoto, D. J. Am. Chem. Soc. 2010, 132, 5186–5192. doi:10.1021/ja910656b |
| 14. | Liu, F.; Zhong, J.; Li, S.; Li, M.; Wu, L.; Wang, Q.; Mao, J.; Liu, S.; Zheng, B.; Wang, M.; Bian, Q. J. Nat. Prod. 2016, 79, 244–247. doi:10.1021/acs.jnatprod.5b00713 |
| 10. | Watanabe, K.; Tsuda, Y.; Hamada, M.; Omori, M.; Mori, G.; Iguchi, K.; Naoki, H.; Fujita, T.; Van Soest, R. W. M. J. Nat. Prod. 2005, 68, 1001–1005. doi:10.1021/np040233u |
| 14. | Liu, F.; Zhong, J.; Li, S.; Li, M.; Wu, L.; Wang, Q.; Mao, J.; Liu, S.; Zheng, B.; Wang, M.; Bian, Q. J. Nat. Prod. 2016, 79, 244–247. doi:10.1021/acs.jnatprod.5b00713 |
| 22. | Sathish Reddy, A.; Srihari, P. Tetrahedron Lett. 2013, 54, 6370–6372. doi:10.1016/j.tetlet.2013.09.058 |
| 23. | Gangadhar, P.; Sathish Reddy, A.; Srihari, P. Tetrahedron 2016, 72, 5807–5817. doi:10.1016/j.tet.2016.08.009 |
| 24. | Gangadhar, P.; Rajesh, P.; Srihari, P. ChemistrySelect 2017, 2, 4106–4110. doi:10.1002/slct.201700898 |
| 33. | Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. J. Am. Chem. Soc. 1991, 113, 4092–4096. doi:10.1021/ja00011a006 |
| 34. | Freire, F.; Seco, J. M.; Quiñoá, E.; Riguera, R. J. Org. Chem. 2005, 70, 3778–3790. doi:10.1021/jo048643a |
| 35. | Hoye, T. R.; Jeffrey, C. S.; Shao, F. Nat. Protoc. 2007, 2, 2451–2458. doi:10.1038/nprot.2007.354 |
| 36. | Valli, M.; Bruno, P.; Sbarbada, D.; Porta, A.; Vidari, G.; Zanoni, G. J. Org. Chem. 2013, 78, 5556–5567. doi:10.1021/jo4004647 |
| 23. | Gangadhar, P.; Sathish Reddy, A.; Srihari, P. Tetrahedron 2016, 72, 5807–5817. doi:10.1016/j.tet.2016.08.009 |
| 29. | Corey, E. J.; Bakshi, R. K.; Shibata, S. J. Am. Chem. Soc. 1987, 109, 5551–5553. doi:10.1021/ja00252a056 |
| 30. | Corey, E. J.; Bakshi, R. K. Tetrahedron Lett. 1990, 31, 611–614. doi:10.1016/S0040-4039(00)94581-7 |
| 31. | Trost, B. M.; Quintard, A. Org. Lett. 2012, 14, 4698–4700. doi:10.1021/ol302074h |
| 32. | The percentage of (R)- and (S)-isomer was analysed after derivatizing the alcohol as the corresponding (R)- or (S)-MTPA ester (Mosher’s ester) followed by LCMS analysis. |
| 27. | Chodkiewicz, W.; Cadiot, P. C. R. Hebd. Seances Acad. Sci. 1955, 241, 1055–1057. |
| 28. | Yoshida, M.; Sugimoto, K.; Ihara, M. Org. Lett. 2004, 6, 1979–1982. doi:10.1021/ol049438k |
| 25. | Zhou, S.-Z.; Anné, S.; Vandewalle, M. Tetrahedron Lett. 1996, 37, 7637–7640. doi:10.1016/0040-4039(96)01700-5 |
| 26. | Moilanen, S. B.; Tan, D. S. Org. Biomol. Chem. 2005, 3, 798–803. doi:10.1039/b417429a |
© 2018 Gangadhar et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)