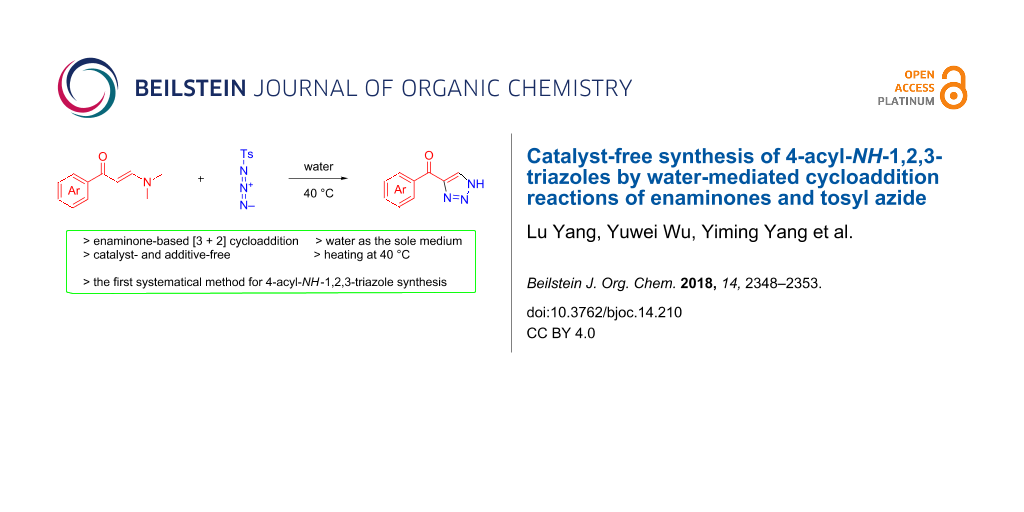
Abstract
The synthesis of 4-acyl-NH-1,2,3-triazoles has been accomplished with high efficiency through the cycloaddition reactions between N,N-dimethylenaminones and tosyl azide. This method is featured with extraordinary sustainability by employing water as the sole medium, free of any catalyst or additive, authentically mild conditions (40 °C stirring) as well as practical scalability.

Graphical Abstract
Introduction
Discovering sustainable chemical syntheses constitutes one central issue of modern organic chemistry. A large number of strategies and concepts promoting sustainable syntheses have been conceived over the past decades. Methods employing water as reaction medium are amongst the most promising ones by avoiding the application of volatile organic solvents during the reaction process [1-3]. Besides acting as a safer and environmentally benign alternative to organic solvents, on water reactions are known for their accelerated reaction rates and improved synthetic selectivity [4-6]. Being inspired by these commonly recognized green features, flourishing advances in the research of water-mediated or promoted organic syntheses, including those reactions involving valuable C–C [7-11], C–heteroatom [12-16], heteroatom–heteroatom [17,18] bond formation as well as divergent cascade reactions [19-23], are presently taking place to guide the progress of sustainable organic synthesis.
1,2,3-Triazole is a heterocyclic moiety showing exceptionally broad and important applications as privileged structure in the discovery of biologically functional scaffolds, organic materials preparation, as directing group in transition-metal-catalyzed transformations and as key building block in the synthesis of numerous organic compounds [24-28]. The amazingly rapid and broad permeation of 1,2,3-triazoles to multidisciplinary areas can majorly be attributed to the occurrence of robust synthetic methods toward this heterocycle. The copper-catalyzed click [3 + 2] cycloaddition of azides and alkynes [29-32], for example, has served enormously to the advances in both the preparation and application of 1,2,3-triazoles. In addition, the discovery of other metal-catalyzed alkyne–azide cycloadditions (MAAC) providing 1,2,3-triazoles with diverse substitution patterns triggers the continuous development of these metal-catalyzed cycloaddition strategies [33-35]. Alongside the vast progress happened in MAAC-based 1,2,3-triazole synthesis, the past decade has witnessed the emergence of another powerful cycloaddition tool for the 1,2,3-triazole synthesis: the metal-free cycloaddition of azides with activated dipolarophiles. As synthetic tools being able to provide 1,2,3-triazoles using an organocatalyst or other non-metal catalysts, this method shows distinctive advantages in enabling the production of 1,2,3-triazoles free of any heavy metal contamination [36-38].
Generally, the cycloaddition of azides with activated dipolarophiles such as strained cyclic alkynes, enamines, enolates, electron-deficient olefins, ylides, iminium cations and alkyne anions, etc., have been identified as reliable approaches to access 1,2,3-triazole scaffolds with multiple substitution patterns [39-44]. In addition, the azide-free annulation has evolved also as another sustainable strategy for the synthesis of many 1,2,3-triazoles in the past decade [45-50]. More notably, besides occurring as active intermediate in the enamine-mediated cycloaddition for 1,2,3-triazole construction, enamines with good stability and easy availability such as enaminones have exhibited also conspicuously versatile application in the metal-free synthesis of divergent 1,2,3-triazoles by directly acting as starting materials [51-54]. In 2016, Dehaen and co-workers [55] reported the synthesis of N-substituted 1,2,3-triazoles via the reactions of organoazides and the in situ prepared N,N-dimethylenaminones by 150 °C microwave irradiation and subsequent heating in toluene at 100 °C, providing an effective protocol of enaminone-based 1,2,3-triazole synthesis. Interestingly, our continuous adventure in enaminone-based organic transformations has led us to the discovery that the cycloaddition of N,N-dimethylenaminones and tosyl azide efficiently affords NH-1,2,3-triazoles with water as the only medium, and not any catalyst or additive is required. Considering the featured functions of NH-1,2,3-triazoles [56-61] as well as the urgent desire in finding more sustainable methods enabling 1,2,3-triazole synthesis, we report herein our results in the water-mediated, catalyst-free synthesis of NH-1,2,3-triazoles through the cycloaddition of enaminone and sulfonyl azide with mild heating (40 °C) and simple operation.
Results and Discussion
To start the work, the reaction of enaminone 1a and tosyl azide (2) was tentatively run in water by heating at 60 °C in the presence of t-BuONa, which provided NH-1,2,3-triazole product 3a with 52% yield together with N,N-dimethyl tosyl amide as byproduct (entry 1, Table 1). Varying the additive to AcOH didn’t lead to an improved result (entry 2, Table 1). To our delight, the parallel entry without using any catalyst or additive afforded 3a with identically good yield (entry 3, Table 1). With this encouraging result, we then carried out a systematic screen of the reaction parameters using water as the fixed reaction medium. First, a slight increase in the loading of tosyl azide was able to evidently enhance the yield of 3a (entries 4 and 5, Table 1). Furthermore, the examination on the impact of the reaction temperature led to the observation of an excellent product yield by running the reaction at 40 °C (entries 6–8, Table 1). The variation on the volume of the water, on the other hand, gave no better reaction results (entries 9 and 10, Table 1). Finally, a control experiment employing EtOH as the reaction medium gave 3a with evidently lower yield than the equivalent reaction using water (entry 11, Table 1).
Table 1: Screen and optimization of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-210-i3.svg?max-width=637&scale=1.0)
|
|||
| entry | T (°C) | additive | yield (%)b |
| 1 | 60 | t-BuONa | 52 |
| 2 | 60 | AcOH | 45 |
| 3 | 60 | – | 52 |
| 4c | 60 | – | 75 |
| 5d | 60 | – | 76 |
| 6c | 80 | – | 80 |
| 7c | 100 | – | 72 |
| 8c | 40 | – | 89 |
| 9c,e | 40 | – | 83 |
| 10c,f | 40 | – | 83 |
| 11c,g | 40 | – | 22 |
aGeneral conditions: enaminone 1a (0.2 mmol), tosyl azide (2, 0.2 mmol), additive (1 equiv) were stirred for 20 h in water (2.0 mL). bYield of isolated product based on 1a. c0.3 mmol 2. d0.24 mmol 2. eH2O (3 mL) was used. fH2O (1 mL) was used. gEtOH was used as alternative reaction medium.
To examine the scope of this water-mediated 1,2,3-triazole synthesis, a broad range of enaminones 1 was then employed to react with tosyl azide under the optimal conditions. According to the acquired results (Figure 1), satisfactory tolerance of this water-mediated, catalyst-free protocol was verified by the smooth synthesis of the 4-acyl-NH-1,2,3-triazoles 3a–t containing versatile substructures (Figure 1). Besides the successful reactions employing enaminones independently containing electron-withdrawing and donating groups in the phenyl ring (H, alkyl, alkoxyl, halogen, CF3 and cyano, etc.), the substitution in ortho- (3n, 3o, Figure 1) and meta-position of the phenyl ring (3k–m, Figure 1) were also readily compatible with the synthesis. More notably, those enaminones functionalized with disubstituted phenyls (3p–r, Table 2) as well as heteroaryl-based enaminones (3s and 3t, Figure 1) also participated in the reaction to provide the divergently functionalized NH-1,2,3-triazoles. The products were generally furnished with good to excellent yield, and the variation of product yields was found to associate with both the electron property and the sites of the substituent in the aryl ring of 1. However, when methyl-functionalized enaminone, N,N-dimethyl nitroenaminone, N,N-dimethyl cyanoenaminone, or pyridine-3-yl-functionalized enaminone was individually utilized, the expected reaction did not take place (3u–x, Figure 1). Moreover, it is notable that no N-sulfonyl-1,2,3-triazole was isolated from any of the above experiments, indicating the excellent chemoselectivity of the present synthetic method.
![[1860-5397-14-210-1]](/bjoc/content/figures/1860-5397-14-210-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Scope of the water-mediated synthesis of 4-acyl-NH-1,2,3-triazoles. General conditions: enaminone 1 (0.2 mmol), tosyl azide 2 (0.3 mmol) and water (2 mL), stirred at 40 °C for 20 h (yields of isolated products are based on 1).
Figure 1: Scope of the water-mediated synthesis of 4-acyl-NH-1,2,3-triazoles. General conditions: enaminone 1...
In order to illustrate the potential application of this authentically green synthetic method, a gram scale synthesis of product 3a was conducted starting from enaminone 1a and tosyl azide (2). As expected, this entry turned out to be highly efficient affording product 3a with excellent yield (Scheme 1). In addition, the appearance of the reaction mixture before and after the reaction indicated the reaction as a heterogeneous “on water” process (Scheme 1).
![[1860-5397-14-210-i1]](/bjoc/content/inline/1860-5397-14-210-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The gram scale synthesis of 3a: (a) before reaction; (b) completed reaction; (c) the purified product 3a.
Scheme 1: The gram scale synthesis of 3a: (a) before reaction; (b) completed reaction; (c) the purified produ...
Based on the known works employing organic solvents for similar synthesis and the present results [55], a possible mechanism for the reaction is proposed (Scheme 2). The reaction starts from the cycloaddition of enaminones 1 and tosyl azide (2) to provide 1,2,3-triazoline 4 which couples to water by strong hydrogen bond effect [51]. The presence of the hydrogen bonds may promote the elimination of the amino group and the acidic C–H bond at the α-position of the acyl group, which affords N-tosyl-1,2,3-triazole 5. Under the present reaction conditions, the intermediate 5 can undergo aminolysis and/or hydrolysis to provide the target products 3. The participation of water throughout the reaction also explains the high efficiency of the method using water as reaction medium.
![[1860-5397-14-210-i2]](/bjoc/content/inline/1860-5397-14-210-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The proposed reaction mechanism.
Scheme 2: The proposed reaction mechanism.
Conclusion
In summary, by means of the cycloaddition reactions between tertiary enaminones and tosyl azide employing water the sole reaction medium, a series of 4-acyl-NH-1,2,3-triazoles has been efficiently synthesized under catalyst-free and very mild heating conditions, thus providing the first water-mediated metal-free method toward the synthesis of 4-acyl-NH-1,2,3-triazoles. The present method benefits from unique sustainability not only due to the metal/additive-free cycloaddition reaction, but also by applying the completely green reaction medium water and mild reaction temperature.
Supporting Information
| Supporting Information File 1: General experimental information, experimental details of the synthesis of products 3, full characterization data as well as 1H/13C NMR spectra of all products. | ||
| Format: PDF | Size: 1.4 MB | Download |
Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (no. 21562025), Natural Science Foundation of Jiangxi Province (20161ACB21010) and an Open Project of the Key Laboratory of Rheumatic Diseases of Traditional Chinese Medicine in Zhejiang Province.
References
-
Lipshutz, B. H.; Ghorai, S.; Cortes-Clerget, M. Chem. – Eur. J. 2018, 24, 6672. doi:10.1002/chem.201705499
Return to citation in text: [1] -
Simon, M.-O.; Li, C.-J. Chem. Soc. Rev. 2012, 41, 1415. doi:10.1039/C1CS15222J
Return to citation in text: [1] -
Anastas, P.; Eghbali, N. Chem. Soc. Rev. 2010, 39, 301. doi:10.1039/B918763B
Return to citation in text: [1] -
Gawande, M. B.; Bonifácio, V. D. B.; Luque, R.; Branco, P. S.; Varma, R. S. Chem. Soc. Rev. 2013, 42, 5522. doi:10.1039/c3cs60025d
Return to citation in text: [1] -
Butler, R. N.; Goyne, A. G. Chem. Rev. 2010, 110, 6302. doi:10.1021/cr100162c
Return to citation in text: [1] -
Chanda, A.; Fokin, V. V. Chem. Rev. 2009, 109, 725. doi:10.1021/cr800448q
Return to citation in text: [1] -
Guo, W.; Wu, B.; Zhou, X.; Chen, P.; Wang, X.; Zhou, Y.-G.; Liu, Y.; Li, C. Angew. Chem., Int. Ed. 2015, 54, 4522. doi:10.1002/anie.201409894
Return to citation in text: [1] -
Li, Y.; Huang, Y.; Gui, Y.; Sun, J.; Li, J.; Zha, Z.; Wang, Z. Org. Lett. 2017, 19, 6416. doi:10.1021/acs.orglett.7b03299
Return to citation in text: [1] -
Zhang, F.-Z.; Tian, Y.; Li, G.-X.; Qu, J. J. Org. Chem. 2015, 80, 1107. doi:10.1021/jo502636d
Return to citation in text: [1] -
Álvarez, M.; Gava, R.; Rodríguez, M. R.; Rull, S. G.; Pérez, P. J. ACS Catal. 2017, 7, 3707. doi:10.1021/acscatal.6b03669
Return to citation in text: [1] -
Zhang, N.; Yang, D.; Wei, W.; Yuan, L.; Nie, F.; Tian, L.; Wang, H. J. Org. Chem. 2015, 80, 3258. doi:10.1021/jo502642n
Return to citation in text: [1] -
Xie, L.-Y.; Li, Y.-J.; Qu, J.; Duan, Y.; Hu, J.; Liu, K.-J.; Cao, Z.; He, W.-M. Green Chem. 2017, 19, 5642. doi:10.1039/C7GC02304A
Return to citation in text: [1] -
Xiao, F.; Chen, S.; Tian, J.; Huang, H.; Liu, Y.; Deng, G.-J. Green Chem. 2016, 18, 1538. doi:10.1039/C5GC02292D
Return to citation in text: [1] -
Liu, K.-J.; Fu, Y.-L.; Xie, L.-Y.; Wu, C.; He, W.-B.; Peng, S.; Wang, Z.; Bao, W.-H.; Cao, Z.; Xu, X.; He, W.-M. ACS Sustainable Chem. Eng. 2018, 6, 4916. doi:10.1021/acssuschemeng.7b04400
Return to citation in text: [1] -
Wu, C.; Xin, X.; Fu, Z.-M.; Xie, L.-Y.; Liu, K.-J.; Wang, Z.; Li, W.; Yuan, Z.-H.; He, W.-M. Green Chem. 2017, 19, 1983. doi:10.1039/C7GC00283A
Return to citation in text: [1] -
Li, W.; Yin, G.; Huang, L.; Xiao, Y.; Fu, Z.; Xin, X.; Liu, F.; Li, Z.; He, W. Green Chem. 2016, 18, 4879. doi:10.1039/C6GC01196A
Return to citation in text: [1] -
Tang, L.; Yang, Y.; Wen, L.; Yang, X.; Wang, Z. Green Chem. 2016, 18, 1224. doi:10.1039/C5GC02755A
Return to citation in text: [1] -
Lin, Y.-m.; Lu, G.-p.; Wang, G.-x.; Yi, W.-b. J. Org. Chem. 2017, 82, 382. doi:10.1021/acs.joc.6b02459
Return to citation in text: [1] -
Köhling, S.; Exner, M. P.; Nojoumi, S.; Schiller, J.; Budisa, N.; Rademann, J. Angew. Chem., Int. Ed. 2016, 55, 15510. doi:10.1002/anie.201607228
Return to citation in text: [1] -
Chen, D.; Feng, Q.; Yang, Y.; Cai, X.-M.; Wang, F.; Huang, S. Chem. Sci. 2017, 8, 1601. doi:10.1039/C6SC04504A
Return to citation in text: [1] -
Yang, J.; Mei, F.; Fu, S.; Gu, Y. Green Chem. 2018, 20, 1367. doi:10.1039/C7GC03644B
Return to citation in text: [1] -
Liu, J.; Lei, M.; Hu, L. Green Chem. 2012, 14, 2534. doi:10.1039/c2gc35745c
Return to citation in text: [1] -
Reddy, G. T.; Kumar, G.; Reddy, N. C. G. Adv. Synth. Catal. 2018, 360, 995. doi:10.1002/adsc.201701063
Return to citation in text: [1] -
Kolb, H. C.; Sharpless, K. B. Drug Discovery Today 2003, 8, 1128. doi:10.1016/S1359-6446(03)02933-7
Return to citation in text: [1] -
Nandivada, H.; Jiang, X.; Lahann, J. Adv. Mater. 2007, 19, 2197. doi:10.1002/adma.200602739
Return to citation in text: [1] -
Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Chem. Rev. 2013, 113, 4905. doi:10.1021/cr200409f
Return to citation in text: [1] -
Chen, Z.; Liu, Z.; Gao, G.; Li, H.; Ren, H. Adv. Synth. Catal. 2017, 359, 202. doi:10.1002/adsc.201600918
Return to citation in text: [1] -
Xie, L.-Y.; Qu, J.; Peng, S.; Liu, K.-J.; Wang, Z.; Ding, M.-H.; Wang, Y.; Cao, Z.; He, W.-M. Green Chem. 2018, 20, 760. doi:10.1039/C7GC03106H
Return to citation in text: [1] -
Huisgen, R. Angew. Chem., Int. Ed. Engl. 1963, 2, 565. doi:10.1002/anie.196305651
Return to citation in text: [1] -
Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004. doi:10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5
Return to citation in text: [1] -
Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057. doi:10.1021/jo011148j
Return to citation in text: [1] -
Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302. doi:10.1039/b904091a
Return to citation in text: [1] -
Zhang, L.; Chen, X.; Xue, P.; Sun, H. H. Y.; Williams, I. D.; Sharpless, K. B.; Fokin, V. V.; Jia, G. J. Am. Chem. Soc. 2005, 127, 15998. doi:10.1021/ja054114s
Return to citation in text: [1] -
Destito, P.; Couceiro, J. R.; Faustino, H.; López, F.; Mascareñas, J. L. Angew. Chem., Int. Ed. 2017, 56, 10766. doi:10.1002/anie.201705006
Return to citation in text: [1] -
Johansson, J. R.; Lincoln, P.; Nordén, B.; Kann, N. J. Org. Chem. 2011, 76, 2355. doi:10.1021/jo200134a
Return to citation in text: [1] -
Ramasastry, S. S. V. Angew. Chem., Int. Ed. 2014, 53, 14310. doi:10.1002/anie.201409410
Return to citation in text: [1] -
Lima, C. G. S.; Ali, A.; van Berkel, S. S.; Westermann, B.; Paixão, M. W. Chem. Commun. 2015, 51, 10784. doi:10.1039/C5CC04114G
Return to citation in text: [1] -
Thomas, J.; Jana, S.; John, J.; Liekens, S.; Dehaen, W. Chem. Commun. 2016, 52, 2885. doi:10.1039/C5CC08347H
Return to citation in text: [1] -
Ramachary, D. B.; Shashank, A. B.; Karthik, S. Angew. Chem., Int. Ed. 2014, 53, 10420. doi:10.1002/anie.201406721
Return to citation in text: [1] -
Li, W.; Wang, J. Angew. Chem., Int. Ed. 2014, 53, 14186. doi:10.1002/anie.201408265
Return to citation in text: [1] -
Agard, N. J.; Preschner, J. A.; Bertozzi, C. R. J. Am. Chem. Soc. 2004, 126, 15046. doi:10.1021/ja044996f
Return to citation in text: [1] -
Ramachary, D. B.; Ramakumar, K.; Narayana, V. V. Chem. – Eur. J. 2008, 14, 9143. doi:10.1002/chem.200801325
Return to citation in text: [1] -
Belkheira, M.; Abed, D. E.; Pons, J.-M.; Bressy, C. Chem. – Eur. J. 2011, 17, 12917. doi:10.1002/chem.201102046
Return to citation in text: [1] -
Kwok, S. W.; Fotsing, J. R.; Fraser, R. J.; Rodionov, V. O.; Fokin, V. V. Org. Lett. 2010, 12, 4217. doi:10.1021/ol101568d
Return to citation in text: [1] -
Wan, J.-P.; Hu, D.; Liu, Y.; Sheng, S. ChemCatChem 2015, 7, 901. doi:10.1002/cctc.201500001
Return to citation in text: [1] -
Chen, Z.; Cao, G.; Song, J.; Ren, H. Chin. J. Chem. 2017, 35, 1797. doi:10.1002/cjoc.201700459
Return to citation in text: [1] -
van Berkel, S. S.; Brauch, S.; Gabriel, L.; Henze, M.; Stark, S.; Vasilev, D.; Wessjohann, L. A.; Abbas, M.; Westermann, B. Angew. Chem., Int. Ed. 2012, 51, 5343. doi:10.1002/anie.201108850
Return to citation in text: [1] -
Chen, Z.; Yan, Q.; Liu, Z.; Zhang, Y. Chem. – Eur. J. 2014, 20, 17635. doi:10.1002/chem.201405057
Return to citation in text: [1] -
Cai, Z.-J.; Lu, X.-M.; Zi, Y.; Yang, C.; Shen, L.-J.; Li, J.; Wang, S.-Y.; Ji, S.-J. Org. Lett. 2014, 16, 5108. doi:10.1021/ol502431b
Return to citation in text: [1] -
Wan, J.-P.; Cao, S.; Liu, Y. J. Org. Chem. 2015, 80, 9028. doi:10.1021/acs.joc.5b01121
Return to citation in text: [1] -
Bakulev, V. A.; Beryozkina, T.; Thomas, J.; Dehaen, W. Eur. J. Org. Chem. 2018, 262. doi:10.1002/ejoc.201701031
and references cited therein.
Return to citation in text: [1] [2] -
Cheng, G.; Zeng, X.; Shen, J.; Wang, X.; Cui, X. Angew. Chem., Int. Ed. 2013, 52, 13265. doi:10.1002/anie.201307499
Return to citation in text: [1] -
Wan, J.-P.; Cao, S.; Liu, Y. Org. Lett. 2016, 18, 6034. doi:10.1021/acs.orglett.6b02975
Return to citation in text: [1] -
Efimov, I.; Bakulev, V.; Beliaev, N.; Beryozkina, T.; Knippschild, U.; Leban, J.; Zhi-Jin, F.; Eltsov, O.; Slepukhin, P.; Ezhikova, M.; Dehaen, W. Eur. J. Org. Chem. 2014, 3684. doi:10.1002/ejoc.201402130
Return to citation in text: [1] -
Thomas, J.; Goyvaerts, V.; Liekens, S.; Dehaen, W. Chem. – Eur. J. 2016, 22, 9966. doi:10.1002/chem.201601928
Return to citation in text: [1] [2] -
Thomas, J.; Jana, S.; Liekens, S.; Dehaen, W. Chem. Commun. 2016, 52, 9236. doi:10.1039/C6CC03744E
Return to citation in text: [1] -
Cohrt, A. E.; Jensen, J. F.; Nielsen, T. E. Org. Lett. 2010, 12, 5414. doi:10.1021/ol102209p
Return to citation in text: [1] -
Hu, Q.; Liu, Y.; Deng, X.; Li, Y.; Chen, Y. Adv. Synth. Catal. 2016, 358, 1689. doi:10.1002/adsc.201600098
Return to citation in text: [1] -
Qvortrup, K.; Nielsen, T. E. Chem. Commun. 2011, 47, 3278. doi:10.1039/c0cc05274d
Return to citation in text: [1] -
Deng, X.; Lei, X.; Nie, G.; Jia, L.; Li, Y.; Chen, Y. J. Org. Chem. 2017, 82, 6163. doi:10.1021/acs.joc.7b00752
Return to citation in text: [1] -
Bakulev, V. A.; Beryozkina, T. A. Chem. Heterocycl. Compd. 2016, 52, 4. doi:10.1007/s10593-016-1821-y
Return to citation in text: [1]
| 51. |
Bakulev, V. A.; Beryozkina, T.; Thomas, J.; Dehaen, W. Eur. J. Org. Chem. 2018, 262. doi:10.1002/ejoc.201701031
and references cited therein. |
| 1. | Lipshutz, B. H.; Ghorai, S.; Cortes-Clerget, M. Chem. – Eur. J. 2018, 24, 6672. doi:10.1002/chem.201705499 |
| 2. | Simon, M.-O.; Li, C.-J. Chem. Soc. Rev. 2012, 41, 1415. doi:10.1039/C1CS15222J |
| 3. | Anastas, P.; Eghbali, N. Chem. Soc. Rev. 2010, 39, 301. doi:10.1039/B918763B |
| 17. | Tang, L.; Yang, Y.; Wen, L.; Yang, X.; Wang, Z. Green Chem. 2016, 18, 1224. doi:10.1039/C5GC02755A |
| 18. | Lin, Y.-m.; Lu, G.-p.; Wang, G.-x.; Yi, W.-b. J. Org. Chem. 2017, 82, 382. doi:10.1021/acs.joc.6b02459 |
| 56. | Thomas, J.; Jana, S.; Liekens, S.; Dehaen, W. Chem. Commun. 2016, 52, 9236. doi:10.1039/C6CC03744E |
| 57. | Cohrt, A. E.; Jensen, J. F.; Nielsen, T. E. Org. Lett. 2010, 12, 5414. doi:10.1021/ol102209p |
| 58. | Hu, Q.; Liu, Y.; Deng, X.; Li, Y.; Chen, Y. Adv. Synth. Catal. 2016, 358, 1689. doi:10.1002/adsc.201600098 |
| 59. | Qvortrup, K.; Nielsen, T. E. Chem. Commun. 2011, 47, 3278. doi:10.1039/c0cc05274d |
| 60. | Deng, X.; Lei, X.; Nie, G.; Jia, L.; Li, Y.; Chen, Y. J. Org. Chem. 2017, 82, 6163. doi:10.1021/acs.joc.7b00752 |
| 61. | Bakulev, V. A.; Beryozkina, T. A. Chem. Heterocycl. Compd. 2016, 52, 4. doi:10.1007/s10593-016-1821-y |
| 12. | Xie, L.-Y.; Li, Y.-J.; Qu, J.; Duan, Y.; Hu, J.; Liu, K.-J.; Cao, Z.; He, W.-M. Green Chem. 2017, 19, 5642. doi:10.1039/C7GC02304A |
| 13. | Xiao, F.; Chen, S.; Tian, J.; Huang, H.; Liu, Y.; Deng, G.-J. Green Chem. 2016, 18, 1538. doi:10.1039/C5GC02292D |
| 14. | Liu, K.-J.; Fu, Y.-L.; Xie, L.-Y.; Wu, C.; He, W.-B.; Peng, S.; Wang, Z.; Bao, W.-H.; Cao, Z.; Xu, X.; He, W.-M. ACS Sustainable Chem. Eng. 2018, 6, 4916. doi:10.1021/acssuschemeng.7b04400 |
| 15. | Wu, C.; Xin, X.; Fu, Z.-M.; Xie, L.-Y.; Liu, K.-J.; Wang, Z.; Li, W.; Yuan, Z.-H.; He, W.-M. Green Chem. 2017, 19, 1983. doi:10.1039/C7GC00283A |
| 16. | Li, W.; Yin, G.; Huang, L.; Xiao, Y.; Fu, Z.; Xin, X.; Liu, F.; Li, Z.; He, W. Green Chem. 2016, 18, 4879. doi:10.1039/C6GC01196A |
| 55. | Thomas, J.; Goyvaerts, V.; Liekens, S.; Dehaen, W. Chem. – Eur. J. 2016, 22, 9966. doi:10.1002/chem.201601928 |
| 7. | Guo, W.; Wu, B.; Zhou, X.; Chen, P.; Wang, X.; Zhou, Y.-G.; Liu, Y.; Li, C. Angew. Chem., Int. Ed. 2015, 54, 4522. doi:10.1002/anie.201409894 |
| 8. | Li, Y.; Huang, Y.; Gui, Y.; Sun, J.; Li, J.; Zha, Z.; Wang, Z. Org. Lett. 2017, 19, 6416. doi:10.1021/acs.orglett.7b03299 |
| 9. | Zhang, F.-Z.; Tian, Y.; Li, G.-X.; Qu, J. J. Org. Chem. 2015, 80, 1107. doi:10.1021/jo502636d |
| 10. | Álvarez, M.; Gava, R.; Rodríguez, M. R.; Rull, S. G.; Pérez, P. J. ACS Catal. 2017, 7, 3707. doi:10.1021/acscatal.6b03669 |
| 11. | Zhang, N.; Yang, D.; Wei, W.; Yuan, L.; Nie, F.; Tian, L.; Wang, H. J. Org. Chem. 2015, 80, 3258. doi:10.1021/jo502642n |
| 51. |
Bakulev, V. A.; Beryozkina, T.; Thomas, J.; Dehaen, W. Eur. J. Org. Chem. 2018, 262. doi:10.1002/ejoc.201701031
and references cited therein. |
| 52. | Cheng, G.; Zeng, X.; Shen, J.; Wang, X.; Cui, X. Angew. Chem., Int. Ed. 2013, 52, 13265. doi:10.1002/anie.201307499 |
| 53. | Wan, J.-P.; Cao, S.; Liu, Y. Org. Lett. 2016, 18, 6034. doi:10.1021/acs.orglett.6b02975 |
| 54. | Efimov, I.; Bakulev, V.; Beliaev, N.; Beryozkina, T.; Knippschild, U.; Leban, J.; Zhi-Jin, F.; Eltsov, O.; Slepukhin, P.; Ezhikova, M.; Dehaen, W. Eur. J. Org. Chem. 2014, 3684. doi:10.1002/ejoc.201402130 |
| 4. | Gawande, M. B.; Bonifácio, V. D. B.; Luque, R.; Branco, P. S.; Varma, R. S. Chem. Soc. Rev. 2013, 42, 5522. doi:10.1039/c3cs60025d |
| 5. | Butler, R. N.; Goyne, A. G. Chem. Rev. 2010, 110, 6302. doi:10.1021/cr100162c |
| 6. | Chanda, A.; Fokin, V. V. Chem. Rev. 2009, 109, 725. doi:10.1021/cr800448q |
| 55. | Thomas, J.; Goyvaerts, V.; Liekens, S.; Dehaen, W. Chem. – Eur. J. 2016, 22, 9966. doi:10.1002/chem.201601928 |
| 33. | Zhang, L.; Chen, X.; Xue, P.; Sun, H. H. Y.; Williams, I. D.; Sharpless, K. B.; Fokin, V. V.; Jia, G. J. Am. Chem. Soc. 2005, 127, 15998. doi:10.1021/ja054114s |
| 34. | Destito, P.; Couceiro, J. R.; Faustino, H.; López, F.; Mascareñas, J. L. Angew. Chem., Int. Ed. 2017, 56, 10766. doi:10.1002/anie.201705006 |
| 35. | Johansson, J. R.; Lincoln, P.; Nordén, B.; Kann, N. J. Org. Chem. 2011, 76, 2355. doi:10.1021/jo200134a |
| 39. | Ramachary, D. B.; Shashank, A. B.; Karthik, S. Angew. Chem., Int. Ed. 2014, 53, 10420. doi:10.1002/anie.201406721 |
| 40. | Li, W.; Wang, J. Angew. Chem., Int. Ed. 2014, 53, 14186. doi:10.1002/anie.201408265 |
| 41. | Agard, N. J.; Preschner, J. A.; Bertozzi, C. R. J. Am. Chem. Soc. 2004, 126, 15046. doi:10.1021/ja044996f |
| 42. | Ramachary, D. B.; Ramakumar, K.; Narayana, V. V. Chem. – Eur. J. 2008, 14, 9143. doi:10.1002/chem.200801325 |
| 43. | Belkheira, M.; Abed, D. E.; Pons, J.-M.; Bressy, C. Chem. – Eur. J. 2011, 17, 12917. doi:10.1002/chem.201102046 |
| 44. | Kwok, S. W.; Fotsing, J. R.; Fraser, R. J.; Rodionov, V. O.; Fokin, V. V. Org. Lett. 2010, 12, 4217. doi:10.1021/ol101568d |
| 29. | Huisgen, R. Angew. Chem., Int. Ed. Engl. 1963, 2, 565. doi:10.1002/anie.196305651 |
| 30. | Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004. doi:10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5 |
| 31. | Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057. doi:10.1021/jo011148j |
| 32. | Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302. doi:10.1039/b904091a |
| 45. | Wan, J.-P.; Hu, D.; Liu, Y.; Sheng, S. ChemCatChem 2015, 7, 901. doi:10.1002/cctc.201500001 |
| 46. | Chen, Z.; Cao, G.; Song, J.; Ren, H. Chin. J. Chem. 2017, 35, 1797. doi:10.1002/cjoc.201700459 |
| 47. | van Berkel, S. S.; Brauch, S.; Gabriel, L.; Henze, M.; Stark, S.; Vasilev, D.; Wessjohann, L. A.; Abbas, M.; Westermann, B. Angew. Chem., Int. Ed. 2012, 51, 5343. doi:10.1002/anie.201108850 |
| 48. | Chen, Z.; Yan, Q.; Liu, Z.; Zhang, Y. Chem. – Eur. J. 2014, 20, 17635. doi:10.1002/chem.201405057 |
| 49. | Cai, Z.-J.; Lu, X.-M.; Zi, Y.; Yang, C.; Shen, L.-J.; Li, J.; Wang, S.-Y.; Ji, S.-J. Org. Lett. 2014, 16, 5108. doi:10.1021/ol502431b |
| 50. | Wan, J.-P.; Cao, S.; Liu, Y. J. Org. Chem. 2015, 80, 9028. doi:10.1021/acs.joc.5b01121 |
| 24. | Kolb, H. C.; Sharpless, K. B. Drug Discovery Today 2003, 8, 1128. doi:10.1016/S1359-6446(03)02933-7 |
| 25. | Nandivada, H.; Jiang, X.; Lahann, J. Adv. Mater. 2007, 19, 2197. doi:10.1002/adma.200602739 |
| 26. | Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Chem. Rev. 2013, 113, 4905. doi:10.1021/cr200409f |
| 27. | Chen, Z.; Liu, Z.; Gao, G.; Li, H.; Ren, H. Adv. Synth. Catal. 2017, 359, 202. doi:10.1002/adsc.201600918 |
| 28. | Xie, L.-Y.; Qu, J.; Peng, S.; Liu, K.-J.; Wang, Z.; Ding, M.-H.; Wang, Y.; Cao, Z.; He, W.-M. Green Chem. 2018, 20, 760. doi:10.1039/C7GC03106H |
| 19. | Köhling, S.; Exner, M. P.; Nojoumi, S.; Schiller, J.; Budisa, N.; Rademann, J. Angew. Chem., Int. Ed. 2016, 55, 15510. doi:10.1002/anie.201607228 |
| 20. | Chen, D.; Feng, Q.; Yang, Y.; Cai, X.-M.; Wang, F.; Huang, S. Chem. Sci. 2017, 8, 1601. doi:10.1039/C6SC04504A |
| 21. | Yang, J.; Mei, F.; Fu, S.; Gu, Y. Green Chem. 2018, 20, 1367. doi:10.1039/C7GC03644B |
| 22. | Liu, J.; Lei, M.; Hu, L. Green Chem. 2012, 14, 2534. doi:10.1039/c2gc35745c |
| 23. | Reddy, G. T.; Kumar, G.; Reddy, N. C. G. Adv. Synth. Catal. 2018, 360, 995. doi:10.1002/adsc.201701063 |
| 36. | Ramasastry, S. S. V. Angew. Chem., Int. Ed. 2014, 53, 14310. doi:10.1002/anie.201409410 |
| 37. | Lima, C. G. S.; Ali, A.; van Berkel, S. S.; Westermann, B.; Paixão, M. W. Chem. Commun. 2015, 51, 10784. doi:10.1039/C5CC04114G |
| 38. | Thomas, J.; Jana, S.; John, J.; Liekens, S.; Dehaen, W. Chem. Commun. 2016, 52, 2885. doi:10.1039/C5CC08347H |
© 2018 Yang et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)