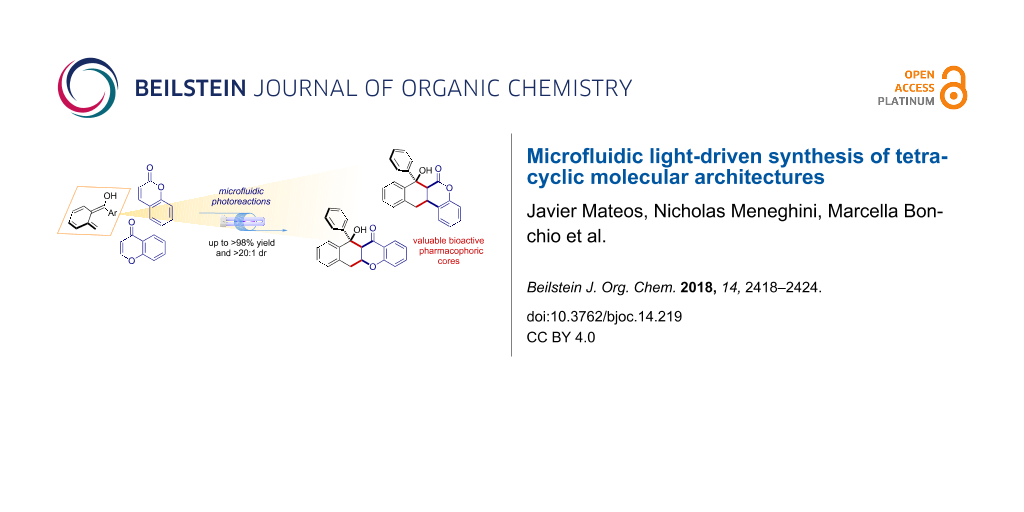
Abstract
Herein we report an effective synthetic method for the direct assembly of highly functionalized tetracyclic pharmacophoric cores. Coumarins and chromones undergo diastereoselective [4 + 2] cycloaddition reactions with light-generated photoenol intermediates. The reactions occur by aid of a microfluidic photoreactor (MFP) in high yield (up to >98%) and virtually complete diastereocontrol (>20:1 dr). The method is easily scaled-up to a parallel setup, furnishing 948 mg of product over a 14 h reaction time. Finally, a series of manipulations of the tetracyclic scaffold obtained gave access to valuable precursors of biologically active molecules.

Graphical Abstract
Introduction
In recent years synthetic photochemistry has become highly sophisticated [1]. The opportunity of using renewable energy sources to transform and functionalize organic molecules is receiving considerable interest from the scientific community [2]. Thus, innovative light-driven metal-free synthetic methods have been successfully developed [3]. More recently, the microfluidic photoreactor (MFP) technology has revealed to be a key technology applicable for diverse photochemical processes [4]. Microfluidic photoreactions allow an increased light penetration and surface-to-volume ratio together with a more uniform and effective irradiation of the reaction system [5], thus resulting in highly improved synthetic performances compared to the classical batch conditions. Recently, light-driven reactions of 2-methylbenzophenone (2-MBP) were reported to proceed smoothly under a MFP setup, furnishing highly diversified molecular scaffolds with enhanced yields and selectivities [6]. The chemistry is based on the ability of 2-MBP derivatives A of generating, upon light-irradiation, the highly reactive photoenol intermediate A' [7] and trapping of the latter by a competent electron-deficient reaction partner (Figure 1). The synthetic approach is not only restricted to electron-poor dienophiles such as maleimides B (see Figure 1a), but has also been implemented different reaction partners, allowing the light-promoted construction of biologically active natural products [8]. With this aim, electron-deficient chromophores, such as 3-coumarincarboxylates D, have been used as competent reaction partners of 2-MBPs A, furnishing 3-benzylated chromanones E through a Michael addition pathway (see Figure 1b) [6].
![[1860-5397-14-219-1]](/bjoc/content/figures/1860-5397-14-219-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: a) Light-driven reaction between 2-MBP A and maleimide B for the synthesis of C through a [4 + 2] cycloaddition manifold. b) Light-driven reaction between 2-MBP A and 3-substituted coumarin D for the synthesis of E through a Michael addition manifold. c) Light-driven reaction between 2-MBPs 1 and coumarin (2a) or chromone (3a) for the synthesis of privileged tetracyclic scaffolds 4 or 5.
Figure 1: a) Light-driven reaction between 2-MBP A and maleimide B for the synthesis of C through a [4 + 2] c...
Prompted by the interest of developing novel light-driven microfluidic methods for the construction of biologically relevant molecular scaffolds, we investigated the reaction between MBP 1 and 3-unsubstituted coumarin (2a) and chromone (3a, Figure 1c). It was anticipated that the successful development of these photoreactions would generate valuable privileged scaffolds, namely, naphthochromenones 4 and benzoxanthenes 5, through a diastereoselective light-driven [4 + 2] cycloaddition reaction. Interestingly, the tetracyclic scaffolds 4 and 5 are embodied in different biologically active molecules, with diverse pharmacological activities [9]. To the best of our knowledge, diastereoselective methods for the direct construction of naphthochromenone 4 are still missing. On the other hand, the reported construction of the benzoxanthene scaffold 5 relies on the use of harsh reaction conditions (e.g., 250 °C), leading to a mixture of regio- and diastereoisomers in moderate yields [10]. Hence, the development of an efficient synthetic method to access these privileged motifs still represents an open task in synthetic chemistry.
The method presented herein is suited for a broad range of coumarins 2 and chromones 3, used as precursors for the direct generation of the tetracyclic scaffold 4 and 5, with high synthetic performances (up to >98% yield) and complete diastereocontrol (>20:1). Additionally, manipulations of the naphthochromenone scaffold 4 give access to highly diversified molecular architectures, which are valuable intermediates in the synthesis of different biologically active molecules [11]. Noteworthy, the photoreactions presented herein do not proceed under conventional batch conditions, thus highlighting the importance of the MFP method enabling novel light-driven synthetic transformations.
Results and Discussion
The reaction between 2-methylbenzophenone (1a) and coumarin (2a) was initially screened in a MFP of 1000 μL volume, using 1.5 equiv of 1a and a residence time of 26.6 min (Table 1 and Table S3 in the Supporting Information File 1). Under these reaction conditions, product 4a formed in 57% yield as a single detectable diastereoisomer with a production of 0.077 mmol·h−1 (entry 1 in Table 1). For comparison, in entry 2 of Table 1 are reported the reaction conditions previously described for the synthesis of 4a [6]. Interestingly, a higher MFP volume resulted in a higher productivity: 0.077 mmol·h−1 vs 0.063 mmol·h−1 (entry 1 vs entry 2, Table 1). Reversing the reagents ratio, i.e., using a slight excess of coumarin (2a), turned out to be highly beneficial, giving the cyclized product 4a in 77% yield (Table 1, entry 3). Notably, the optimal reaction conditions for the light-driven [4 + 2] cycloaddition were achieved within a 1000 μL MFP with a residence time set at 35 min forming product 4a in quantitative yield, complete diastereocontrol and a productivity of 0.104 mmol·h−1 (Table 1, entry 4). On the contrary, when the same reaction was performed under batch conditions, the expected [4 + 2] cycloaddition product 4a was only formed in trace amounts along with extensive product decomposition (Table 1, entry 5). The enhanced reactivity under the MFP compared to the batch setup (Table 1, entry 4 vs entry 5) is attributed to the more efficient illumination and the shorter irradiation time within the MFP [6], thus successfully preventing the light-promoted product decomposition [12]. In fact, the irradiation for 8 h of an authentic sample of 4a resulted in the formation of a series of undefined decomposition products. Control experiments showed that in the absence of light irradiation, the cyclization product was not detected (Table 1, entry 6), confirming the photochemical nature of the present reaction.
Table 1: Light-driven reaction between 2-methylbenzophenone (1a) and coumarin (2a); selected optimization results.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-219-i2.svg?max-width=637&scale=1.0)
|
|||||
| entrya | residence time (min) | reactor volume (μL) | reagent ratio (1a:2a) | yield |
production
(mmol·h−1) |
| 1 | 26.6 | 1000 | 1.5:1 | 57 | 0.077 |
| 2b | 26.6 | 400 | 1.5:1 | 70c | 0.063 |
| 3 | 26.6 | 1000 | 1:3 | 77 | 0.104 |
| 4 | 35 | 1000 | 1:5 | >98%c | 0.104 |
| 5d,e | 480 | 1000 | 1:5 | – | – |
| 6f | 35 | 1000 | 1:5 | – | – |
aUnless otherwise noted, reaction conditions were as follows: a degassed solution of 1a and 2a in toluene (0.06 M) was irradiated for the indicated time at 25 ± 2 °C (see Supporting Information File 1 for details). All yields refer to NMR yields using trimethoxybenzene as the internal standard. The dr was inferred by 1H NMR analysis on the crude reaction mixture and in all the cases resulted >20:1. bReaction conditions as described in [6]. cIsolated yield. dReaction performed in batch. eExtensive decomposition of both starting reagents 1a and 2a was observed by 1H NMR analysis of the crude reaction mixture. fReaction performed under MFP setup in the absence of light. i.d. = internal diameter.
With the optimal reaction conditions in hand we next explored the generality and limitations of the photochemical transformation (Figure 2). First, different substitutions on the 2-MBP scaffold were evaluated. Electron-donating substituents on both aromatic rings gave excellent results, furnishing the corresponding naphthochromenones 4b and 4c as single detectable diastereoisomers (>20:1 dr), with yields spanning from 53% to 83% and short residence times (35 min). On the contrary, electron-withdrawing substituents resulted in inferior synthetic performances. Compounds 4d and 4e were isolated in 44% and 40%, respectively within 60 min. The optimized reaction conditions were also amenable to diverse coumarin scaffolds. Six and 7-substituted coumarins furnished the corresponding cyclic products 4f–h in moderate to excellent yields (41 to >98%) in a pure diastereoisomeric form. As a limitation of the present microfluidic photochemical method, thioxocoumarin 2e showed poor reactivity under the titled reaction conditions, producing only traces of the expected sulfur-containing adduct 4i. As a matter of fact, compound 2e showed a high tendency to undergo a light-promoted [2 + 2] dimerization reaction, thus preventing the envisaged [4 + 2] cycloaddition pathway [13].
![[1860-5397-14-219-2]](/bjoc/content/figures/1860-5397-14-219-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Generality and limits of the light-driven [4 + 2] cyclization reaction between 2-MBP 1a–g and coumarins 2a–c under MFP setup. *Residence time was 60 min.
Figure 2: Generality and limits of the light-driven [4 + 2] cyclization reaction between 2-MBP 1a–g and couma...
Next, the chromone scaffold 3a, which is a precursor of diverse classes of biologically active molecules [14], was evaluated under the developed MFP setup. Notably, the [4 + 2] cycloaddition product 5a formed in 72% yield and >20:1 dr, without the need of further condition adjustments. The relative syn configuration within 5a was inferred by 2D-NOESY experiments and confirmed by X-ray analysis of a suitable single crystal (Figure 3). Noteworthy, different 2-MBPs bearing electron-donating or electron-withdrawing groups underwent the light-driven [4 + 2] cycloaddition, affording the corresponding tetracyclic products 5b–f with high dr and in good yields spanning from 41% to 72% (Figure 3).
![[1860-5397-14-219-3]](/bjoc/content/figures/1860-5397-14-219-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Generality and limits of the light-driven [4 + 2] cyclization reaction between 2-MBP 1a–f and chromone (3a) under MFP setup.
Figure 3: Generality and limits of the light-driven [4 + 2] cyclization reaction between 2-MBP 1a–f and chrom...
In order to demonstrate the easy scalability of the present method (Scheme 1) two MFPs were used in a parallel setup producing 948 mg of 4a after 14 h with an overall productivity rate of 0.196 mmol·h−1 [15]. Subsequently, a series of manipulations were conducted on product 4a. Its treatment with a solution of sodium hydroxide in water promoted a quantitative lactone-opening/decarboxylation cascade sequence, yielding 2,4-dihydronaphthalene 6a in quantitative yield without the need of chromatographic purification. Interestingly, scaffold 6a is a valuable intermediate for the synthesis of biologically active natural compounds [16] and industrially relevant drugs [17] reminiscent of the bioactive tetralinolic pharmacophore core [18].
![[1860-5397-14-219-i1]](/bjoc/content/inline/1860-5397-14-219-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: MFP parallel setup for higher scale production of 4a (top) and different molecular scaffolds 6a–9a accessible after simple manipulation (Ar = o-OH-C6H4).
Scheme 1: MFP parallel setup for higher scale production of 4a (top) and different molecular scaffolds 6a–9a ...
Acidic treatment of 4a generated, after a simple extraction, the corresponding α,β-unsaturated compound 7a in 97% yield. A further manipulation involved the treatment of 4a with PhMgBr, converting the lactone moiety into the corresponding aromatic ketone. Product 8a formed in 71% yield without diastereoisomeric loss. Finally, LiAlH4 reduction of 4a furnished the bicyclic 1,3-diol 9a in quantitative yield, again without the need of chromatographic purification. Noteworthy, compounds 6a–9a embody different functionalities suitable for additional synthetic transformations.
Conclusion
In conclusion we have developed an effective light-driven microfluidic method for the synthesis of valuable tetracyclic molecular architectures using commercially available precursors and a common 365 nm bulb. The reaction does not proceed under conventional batch conditions, highlighting the essential role of the developed MFP. A wide series of naphthochromenones and benzoxanthenes were synthesized in high yields and excellent diastereoselectivity. Finally, the large-scale production and subsequent manipulations of product 4a demonstrated the high synthetic potential of the present MFP method, which is well-suited for the construction of diverse biologically active molecules.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data for products 4a–h, 5a–f and 6a–9a, NMR spectra, and CIF files for CCDC 1837120 and CCDC 1851516. | ||
| Format: PDF | Size: 4.2 MB | Download |
References
-
Ravelli, D.; Protti, S.; Fagnoni, M. Chem. Rev. 2016, 116, 9850–9913. doi:10.1021/acs.chemrev.5b00662
See for a review.
Return to citation in text: [1] -
Shaw, M. H.; Twilton, J.; MacMillan, D. W. C. J. Org. Chem. 2016, 81, 6898–6926. doi:10.1021/acs.joc.6b01449
Return to citation in text: [1] -
Zou, Y.-Q.; Hörmann, F. M.; Bach, T. Chem. Soc. Rev. 2018, 47, 278–290. doi:10.1039/C7CS00509A
Return to citation in text: [1] -
Cambié, D.; Bottecchia, C.; Straathof, N. J. W.; Hessel, V.; Noël, T. Chem. Rev. 2016, 116, 10276–10341. doi:10.1021/acs.chemrev.5b00707
Return to citation in text: [1] -
Su, Y.; Straathof, N. J. W.; Hessel, V.; Noël, T. Chem. – Eur. J. 2014, 20, 10562–10589. doi:10.1002/chem.201400283
Return to citation in text: [1] -
Mateos, J.; Cherubini-Celli, A.; Carofiglio, T.; Bonchio, M.; Marino, N.; Companyó, X.; Dell’Amico, L. Chem. Commun. 2018, 54, 6820–6823. doi:10.1039/C8CC01373J
Return to citation in text: [1] [2] [3] [4] [5] -
Sammes, P. G. Tetrahedron 1976, 32, 405–422. doi:10.1016/0040-4020(76)80055-5
Return to citation in text: [1] -
Nicolaou, K. C.; Gray, D.; Tae, J. Angew. Chem., Int. Ed. 2001, 40, 3675–3678. doi:10.1002/1521-3773(20011001)40:19<3675::AID-ANIE3675>3.0.CO;2-G
Return to citation in text: [1] -
Wright, P. M.; Seiple, I. B.; Myers, A. G. Angew. Chem., Int. Ed. 2014, 53, 8840–8869. doi:10.1002/anie.201310843
Return to citation in text: [1] -
Sandulache, A.; Silva, A. M. S.; Cavaleiro, J. A. S. Tetrahedron 2002, 58, 105–114. doi:10.1016/S0040-4020(01)01131-0
Return to citation in text: [1] -
Allred, T. K.; Manoni, F.; Harran, P. G. Chem. Rev. 2017, 117, 11994–12051. doi:10.1021/acs.chemrev.7b00126
Return to citation in text: [1] -
The batch reaction was performed using the same light source employed for the MFP setup and stopped at different reaction times. When the batch reaction was stopped at short reaction time (e.g., 2 h) the product 4a was only detected in low amount (<10%) along with recovery of the unreacted starting material 1a and 2a. At longer reaction time (e.g., 8 h) decomposition products were detected by 1H NMR analysis of the crude reaction mixture. See Supporting Information File 1 for details.
Return to citation in text: [1] -
1H NMR analysis of the crude reaction mixture collected after 30 min residence time under the MFP setup, showed a large amount of the [2 + 2] dimerization side product of 2e (57% conversion with respect to 2e). Other coumarins 2a–c showed inferior conversion to the corresponding dimerization side product (10–20%).
Return to citation in text: [1] -
Bauvois, B.; Puiffe, M.-L.; Bongui, J.-B.; Paillat, S.; Monneret, C.; Dauzonne, D. J. Med. Chem. 2003, 46, 3900–3910. doi:10.1021/jm021109f
Return to citation in text: [1] -
Su, Y.; Kuijpers, K.; Hessel, V.; Noël, T. React. Chem. Eng. 2016, 1, 73–81. doi:10.1039/C5RE00021A
Return to citation in text: [1] -
Lantaño, B.; Aguirre, J. M.; Drago, E. V.; de la Faba, D. J.; Pomilio, N.; Mufato, J. D. Magn. Reson. Chem. 2017, 55, 619–633. doi:10.1002/mrc.4564
Return to citation in text: [1] -
Sook, O.; Jang, B. S. Arch. Pharmacal Res. 1995, 18, 277–281.
Return to citation in text: [1] -
Hanaya, K.; Onodera, S.; Ikegami, Y.; Kudo, H.; Shimaya, K. J. Chem. Soc., Perkin Trans. 2 1981, 944–947. doi:10.1039/p29810000944
Return to citation in text: [1]
| 15. | Su, Y.; Kuijpers, K.; Hessel, V.; Noël, T. React. Chem. Eng. 2016, 1, 73–81. doi:10.1039/C5RE00021A |
| 13. | 1H NMR analysis of the crude reaction mixture collected after 30 min residence time under the MFP setup, showed a large amount of the [2 + 2] dimerization side product of 2e (57% conversion with respect to 2e). Other coumarins 2a–c showed inferior conversion to the corresponding dimerization side product (10–20%). |
| 14. | Bauvois, B.; Puiffe, M.-L.; Bongui, J.-B.; Paillat, S.; Monneret, C.; Dauzonne, D. J. Med. Chem. 2003, 46, 3900–3910. doi:10.1021/jm021109f |
| 1. |
Ravelli, D.; Protti, S.; Fagnoni, M. Chem. Rev. 2016, 116, 9850–9913. doi:10.1021/acs.chemrev.5b00662
See for a review. |
| 5. | Su, Y.; Straathof, N. J. W.; Hessel, V.; Noël, T. Chem. – Eur. J. 2014, 20, 10562–10589. doi:10.1002/chem.201400283 |
| 12. | The batch reaction was performed using the same light source employed for the MFP setup and stopped at different reaction times. When the batch reaction was stopped at short reaction time (e.g., 2 h) the product 4a was only detected in low amount (<10%) along with recovery of the unreacted starting material 1a and 2a. At longer reaction time (e.g., 8 h) decomposition products were detected by 1H NMR analysis of the crude reaction mixture. See Supporting Information File 1 for details. |
| 4. | Cambié, D.; Bottecchia, C.; Straathof, N. J. W.; Hessel, V.; Noël, T. Chem. Rev. 2016, 116, 10276–10341. doi:10.1021/acs.chemrev.5b00707 |
| 6. | Mateos, J.; Cherubini-Celli, A.; Carofiglio, T.; Bonchio, M.; Marino, N.; Companyó, X.; Dell’Amico, L. Chem. Commun. 2018, 54, 6820–6823. doi:10.1039/C8CC01373J |
| 3. | Zou, Y.-Q.; Hörmann, F. M.; Bach, T. Chem. Soc. Rev. 2018, 47, 278–290. doi:10.1039/C7CS00509A |
| 6. | Mateos, J.; Cherubini-Celli, A.; Carofiglio, T.; Bonchio, M.; Marino, N.; Companyó, X.; Dell’Amico, L. Chem. Commun. 2018, 54, 6820–6823. doi:10.1039/C8CC01373J |
| 2. | Shaw, M. H.; Twilton, J.; MacMillan, D. W. C. J. Org. Chem. 2016, 81, 6898–6926. doi:10.1021/acs.joc.6b01449 |
| 6. | Mateos, J.; Cherubini-Celli, A.; Carofiglio, T.; Bonchio, M.; Marino, N.; Companyó, X.; Dell’Amico, L. Chem. Commun. 2018, 54, 6820–6823. doi:10.1039/C8CC01373J |
| 6. | Mateos, J.; Cherubini-Celli, A.; Carofiglio, T.; Bonchio, M.; Marino, N.; Companyó, X.; Dell’Amico, L. Chem. Commun. 2018, 54, 6820–6823. doi:10.1039/C8CC01373J |
| 10. | Sandulache, A.; Silva, A. M. S.; Cavaleiro, J. A. S. Tetrahedron 2002, 58, 105–114. doi:10.1016/S0040-4020(01)01131-0 |
| 18. | Hanaya, K.; Onodera, S.; Ikegami, Y.; Kudo, H.; Shimaya, K. J. Chem. Soc., Perkin Trans. 2 1981, 944–947. doi:10.1039/p29810000944 |
| 8. | Nicolaou, K. C.; Gray, D.; Tae, J. Angew. Chem., Int. Ed. 2001, 40, 3675–3678. doi:10.1002/1521-3773(20011001)40:19<3675::AID-ANIE3675>3.0.CO;2-G |
| 11. | Allred, T. K.; Manoni, F.; Harran, P. G. Chem. Rev. 2017, 117, 11994–12051. doi:10.1021/acs.chemrev.7b00126 |
| 16. | Lantaño, B.; Aguirre, J. M.; Drago, E. V.; de la Faba, D. J.; Pomilio, N.; Mufato, J. D. Magn. Reson. Chem. 2017, 55, 619–633. doi:10.1002/mrc.4564 |
| 6. | Mateos, J.; Cherubini-Celli, A.; Carofiglio, T.; Bonchio, M.; Marino, N.; Companyó, X.; Dell’Amico, L. Chem. Commun. 2018, 54, 6820–6823. doi:10.1039/C8CC01373J |
| 9. | Wright, P. M.; Seiple, I. B.; Myers, A. G. Angew. Chem., Int. Ed. 2014, 53, 8840–8869. doi:10.1002/anie.201310843 |
© 2018 Mateos et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)