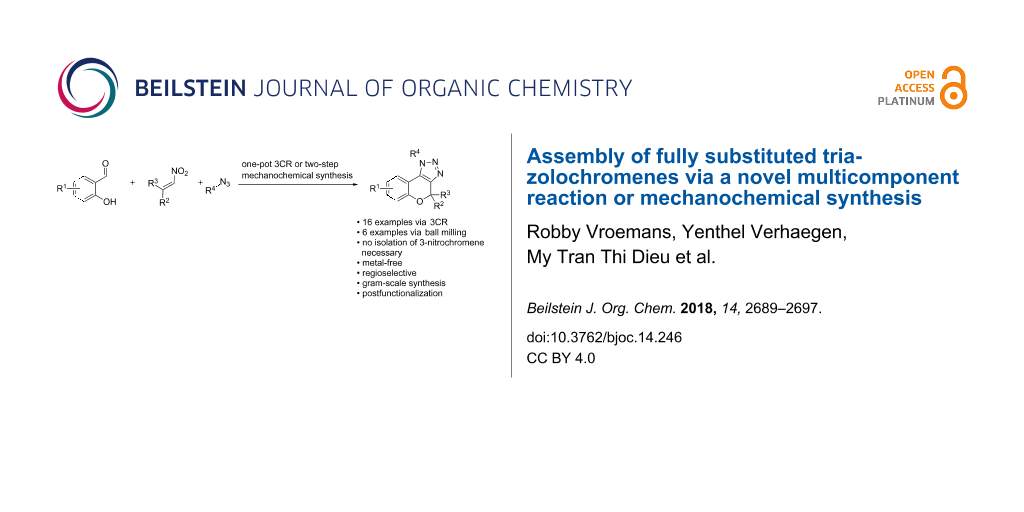
Abstract
A new metal-free one-pot three-component procedure towards fully substituted triazolochromenes has been developed, starting from commercially available materials. Salicylaldehydes and nitroalkenes were reacted under solvent-free conditions, followed by a 1,3-dipolar cycloaddition of the intermediate 3-nitro-2H-chromenes with organic azides in a one-pot two-step sequence. The triazolochromenes were formed with complete regioselectivity and new biologically relevant structures were synthesized via extension of the developed procedure and via postfunctionalization. The mechanochemical synthesis was carried out for several salicylaldehydes and gave a clear improvement in the yield of the corresponding triazolochromenes and consequently showed to be a viable alternative for solid salicylaldehydes.

Graphical Abstract
Introduction
Chromenes are important structural motifs and are omnipresent in nature and drugs for medicinal applications [1-4]. Vitamin E [5-8], arahypin-5 [9,10], THC and other cannabinoids [11-14] are only a few examples of biologically relevant chromenes. Hence, the search for new methodologies towards the rapid assembly of chromene analogs is of utmost importance for many researchers. In this regard, 3-nitrochromenes are easily available building blocks for chromene and chromane derivatives and are highly reactive due to the presence of the nitroalkene moiety, which enables them to undergo a high variety of reactions and functionalizations [15].
Combining the chromene core with the 1,2,3-triazole structural motif has led to some interesting new molecules [16-31]. Very recently, spiro-fused triazolochromenes were found to be active as antitubercular agents [32], indicating that the development of new triazolochromenes in a straightforward manner is still of major interest. Previously, NH-triazolochromenes were synthesized starting from 3-nitrochromenes with sodium azide [16-22], via intramolecular cyclization of a diazomethane group and a nitrile [23], or via our recently reported NH-triazole synthesis starting from 6-methoxyflavanone [24]. Furthermore, 1,4,5-trisubstituted 1,2,3-triazole annulated chromenes have been reported via an intramolecular arylation reaction of 1,2,3-triazoles [25-31]. Yet, the developed methodologies for trisubstituted triazolochromenes generally lack a substituent on the 2-position, except for a sporadic methyl group which drastically lowers the yield and often the use of transition metals is needed [28]. The additional substituents on the chromene core and 1,2,3-triazole offer a lot of possibilities for further derivatization and optimization towards biologically relevant structures such as flavonoid structures.
Our group developed a Knoevenagel-assisted three-component reaction of (protected) salicylaldehyde, ethyl nitroacetate and organic azides, in which the synthesis of both triazolocoumarin regioisomers was accomplished [33]. Interestingly, the expected regioisomer was not observed in the case of the in situ formed 3-nitrocoumarins. Hence, in our continued exploration towards novel multicomponent reactions for the assembly of triazole-fused (hetero)cycles [24,33-42], we opted to develop a new one-pot two-step three-component reaction starting from salicylaldehydes, nitroalkenes and organic azides, without isolation of the intermediate 3-nitrochromenes, in a regioselective manner and without the use of metals. Salicylaldehydes with a high melting point or low solubility proved difficult to convert to the intermediate 3-nitrochromene derivatives [15]. In this regard, applying mechanochemistry has been proven previously to be a viable alternative [43]. To the best of our knowledge, both the development of a metal-free sequential one-pot three-component reaction and the mechanochemically assisted 3-nitrochromene synthesis towards fully substituted triazolochromenes, without the isolation of the intermediate 3-nitrochromenes, have not been reported until now.
Results and Discussion
To prove the plausibility of the one-pot three-component reaction, we commenced our trials with the synthesis and isolation of 3-nitro-2H-chromene (3) as reported in the literature [15], followed by the 1,3-dipolar cycloaddition of the nitroalkene moiety with organic azides. We based the 1,3-dipolar cycloaddition reaction on the synthesis of NH-triazoles by Guan et al. using p-toluenesulfonic acid as the catalyst in DMF [19], but with benzyl azide (4a) instead of sodium azide (Scheme 1). Our initial test gave a promising result, since after a reaction time of five days for the cycloaddition step the desired product 5a was obtained in 67% yield, together with an oxidized ring opened side product 6 in 20% yield. The overall yield of 5a after two steps was 48%, considering that the 3-nitro-2H-chromene (3) was obtained in a yield of 71%.
![[1860-5397-14-246-i1]](/bjoc/content/inline/1860-5397-14-246-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Two-step reaction towards triazolochromene 5a and obtained oxidized side product 6.
Scheme 1: Two-step reaction towards triazolochromene 5a and obtained oxidized side product 6.
We continued our studies by verifying the obtained regiochemistry, in which we synthesized the different regioisomers 5a, 10 and 11 via different pathways (Scheme 2). The regiospecific syntheses [36] of compounds 5a and 11 were accomplished by triazolization of the corresponding flavanone 7 and 2-phenylchroman-3-one (8), respectively. As anticipated, these methods furnished both regioisomers in poor yields since the chromanones 7 and 8 are known to be unstable under the triazolization conditions [36]. Hence, no further attempts were made to improve these yields. Additionally, NH-triazole 9 could be alkylated using benzyl bromide and potassium carbonate in acetone providing a mixture of alkylated triazolochromenes 5a, 10 and 11. The polarity of the 2-alkylated triazolochromene 10 is significantly different than the other two which were obtained as an inseparable mixture of both regioisomers 5a and 11 in a 1:3 ratio. Comparing the 1H NMR spectra (see Supporting Information File 1, Figure S1 for NMR comparison), we can make unambiguous conclusions about the regiochemistry of the synthesized compounds 5a, 10 and 11 (Scheme 2). As the product contains a stereocenter, there is a possibility to see diastereotopic splitting of the benzylic protons. In the spectrum of the obtained product 5a starting from 3-nitro-2H-chromene and flavanone, this splitting is not observed (A2 pattern). The benzylic peak of the 2-alkylated product 10 shows an AB splitting pattern and the third regioisomer 11 shows a substantial AX splitting pattern. This striking difference can be rationalized in function of the proximity of the stereocenter to the diastereotopic protons. Further proof was provided by characterization of side product 6 [44], which is formed during the reaction by oxidation and ring-opening of triazolochromene 5a (Scheme 1). All these observations confirm the expected regioselectivity in the formation of triazolochromene 5a via 3-nitro-2H-chromene (3).
![[1860-5397-14-246-i2]](/bjoc/content/inline/1860-5397-14-246-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction pathways leading to the different regioisomers.
Scheme 2: Reaction pathways leading to the different regioisomers.
Next, the two-step synthesis was converted into a one-pot two-step synthesis, circumventing the need for isolating the intermediate 3-nitro-2H-chromene (3), which would greatly facilitate the purification of the overall reaction since the 3-nitro-2H-chromenes and their starting materials show similar retention factors. Since the triazolochromenes 5 are showing much lower retention factors, the one-pot synthesis would display a great improvement in the labor intensiveness both for the purification steps and reaction set-up. The reaction was further optimized using salicylaldehyde (1a), β-nitrostyrene (2a) and benzyl azide (3a) as model substrates (see Supporting Information File 1, pages S4–S8 for full description of the optimization study). The optimized conditions for the one-pot three-component reaction were determined to be 1 equivalent nitroalkene, 1.2 equivalents of salicylaldehyde and 0.1 equivalents of DABCO as catalyst in the first step at 40 °C, and 2 equivalents of benzyl azide, 2 equivalents of acetic acid, 0.3 equivalents of BHT as antioxidant, 4 Å MS and DMF under argon atmosphere at 120 °C in the second step. Crude NMR analysis of the reaction mixture under optimized conditions showed solely regioisomer 5a, which was obtained in 54% yield after chromatographic purification. Additionally, the optimized conditions gave improved yields compared to the two-step synthesis and circumvented the formation of oxidized side product 6. As variation of the substituents on the three different starting materials is necessary to obtain a diverse library, there is one main limitation to overcome. The first step of the reaction relies on the fluidity of salicylaldehyde (1a) to liquefy the reaction mixture. Salicylaldehyde analogs 1c–f are solids at 40 °C and hence, to overcome this problem, some slight modifications from the optimized conditions were done (see Supporting Information File 1, pages S6 and S7 for more detailed description of the performed experiments for compound 5a). Eventually, the use of two equivalents of triethylamine was needed but the overall yield of 5a was still lower as it only reached 38%.
With the obtained optimized conditions and proof of regioselectivity in hand, further investigation towards the generality of this three-component reaction was carried out by varying the substrate scope (Figure 1). We first studied a range of salicylaldehydes 1a–f, from which 5a and 5b were obtained in the best yields since salicylaldehydes 1a and 1b are liquids. As mentioned earlier, the altered conditions for solid salicylaldehydes result in general in a decrease in yield. Yet, we were able to diversify towards electron-rich triazolochromenes 5b and 5c, resulting in a drastic loss in yield for the more sterically hindered compound 5c. Furthermore, electron-deficient and halogenated analogs 5d–f were successfully synthesized. In a next series, we examined the substitution pattern on the nitroalkene part. Electron-rich substituents 3,4,5-trimethoxyphenyl and piperonyl were tolerated and furnished products 5g and 5h, respectively. 2,2-Dimethyl-substituted derivative 5i was prepared from 2-methyl-1-nitroprop-1-ene (2d) and interestingly, 1,4-bis((E)-2-nitrovinyl)benzene (2e) furnished bis-chromenotriazole 5p in 26% yield (Scheme 3). Finally, the scope with respect to organic azides was investigated by performing reactions with alkyl and aryl azides 4a–g. Electron-rich aliphatic azides produced products 5j–l in moderate yields. Additionally, electron-rich and electron-deficient aromatic azides were explored, resulting in slightly lower yields and elongated reaction times in the cycloaddition step compared to aliphatic azides. Unfortunately, this reaction has encountered some limitations towards certain substrates (not shown). In the case of a strongly electron-withdrawing substituent on the nitroalkene part for (E)-1-nitro-4-(2-nitrovinyl)benzene and sterically hindered 2-hydroxy-1-naphthaldehyde, only the oxidized product analogous to 6 was observed. 2-Hydroxy-4-nitrobenzaldehyde and 2,6-dihydroxybenzaldehyde were unreactive in the cycloaddition reaction.
![[1860-5397-14-246-1]](/bjoc/content/figures/1860-5397-14-246-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Scope with respect to various salicylaldehydes 1a–f, nitroalkenes 2a–d and organic azides 4a–g. aReaction conditions for two-step one-pot procedure: 1. 1 (1.2 equiv), 2 (1 equiv), DABCO (0.1 equiv), 1.5 h, 40 °C; 2. 4 (2 equiv), acetic acid (2 equiv), BHT (0.3 equiv), 4 Å MS (50 mg), DMF (0.1 mL), 24 h, 120 °C. bReaction conditions for two-step one-pot procedure with solid salicylaldehydes: 1. 1 (1.2 equiv), 2 (1 equiv), triethylamine (2 equiv), 1.5 h, 40 °C; 2. Same as for a. cReaction time for the second step: 27 h. dReaction time for the first step: 24 h; second step: 29 h. eReaction time for the first step: 2 h. fReaction time for the first step: 3 h. gReaction time for the first step: 38 h. hReaction time for the second step: 30 h. iReaction time for the second step: 45 h.
Figure 1: Scope with respect to various salicylaldehydes 1a–f, nitroalkenes 2a–d and organic azides 4a–g. aRe...
![[1860-5397-14-246-i3]](/bjoc/content/inline/1860-5397-14-246-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of bis-chromenotriazole 5p.
Scheme 3: Synthesis of bis-chromenotriazole 5p.
As previously mentioned, solid salicylaldehydes furnish triazolochromenes in diminished yields in the one-pot three-component reaction (Figure 1, compounds 5c–f). Hence, a mechanochemical two-step protocol was developed, since a report by Jia and Zhang et al. [43] previously showed that ball milling could be a convenient manner to produce 3-nitrochromenes. Because our first step is best performed solvent-free, we opted to try our own optimized solvent-free conditions for the in situ syntheses of 3-nitro-2H-chromenes, followed by the 1,3-dipolar cycloaddition in a reaction vial. Despite being a two-pot procedure, purification of the intermediate 3-nitro-2H-chromene is still circumvented. Hence, our initial trials were performed by using solid salicylaldehydes 1c–f (Figure 2), resulting in a significant increase in yield for triazolochromenes 5c–f compared to the one-pot procedure developed as described above (Figure 1). To compare the two methodologies, the two highest yielding liquid salicylaldehydes in the one-pot protocol, i.e., 1a and 1b, were reacted in the two-step mechanochemically assisted reaction, giving rise to slightly lowered yields for compounds 5a and 5b. Hence, the use of the ball milling procedure is advantageous when solid salicylaldehydes are used. Complementary to this the one-pot three-component reaction gave better results for liquid salicylaldehydes.
![[1860-5397-14-246-2]](/bjoc/content/figures/1860-5397-14-246-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Generality of products obtained via the two-pot mechanochemical procedure, varying the salicylaldehydes 1a–f. aReaction conditions for ball milling procedure, followed by 1,3-dipolar cycloaddition in a reaction vial: 1. 1 (1.2 equiv), 2a (1 equiv), DABCO (0.1 equiv), 15 min, 30 Hz; 2. 4a (2 equiv), acetic acid (2 equiv), BHT (0.3 equiv), 4 Å MS (50 mg), DMF (2 mL), 24 h, 120 °C. bReaction time for the first step: 2 h. The overall isolated yields are given for 2 steps.
Figure 2: Generality of products obtained via the two-pot mechanochemical procedure, varying the salicylaldeh...
In order to show the utility of the developed methodologies towards possible drug discovery, the scalability of the reactions was explored (Scheme 4). Both developed methodologies easily led to gram scale syntheses without significant loss in yield, i.e., 50% and 40% for the one-pot three-component reaction and mechanochemical procedure, respectively.
![[1860-5397-14-246-i4]](/bjoc/content/inline/1860-5397-14-246-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Scale-up of the one-pot three-component reaction and two-step ball milling procedure.
Scheme 4: Scale-up of the one-pot three-component reaction and two-step ball milling procedure.
Finally, the versatility of these novel methodologies was demonstrated by performing postfunctionalization strategies towards well-known biologically active analogs (Scheme 5) [3]. Pd-catalyzed reactions were effected on bromotriazolochromene 5e. The piperazin-1-ylchromenes have been identified to be potent inhibitors at the 5-HT1A receptor and at the 5-HT transporter [45,46]. Thus, Buchwald–Hartwig amination of 1-phenylpiperazine and 5e furnishes piperazin-1-ylchromene 12 in 64% yield. Furthermore, as highly methylated flavonoid derivatives [47] and 6-(3,5-dimethoxyphenyl)chromenes [48,49] have been demonstrated to be potent anti-seizure drugs and anticancer agents, respectively, a Suzuki–Miyaura reaction was performed yielding 13 in 51% yield. Since aldehydes are interesting and versatile functional moieties for further derivatization, e.g., used in the synthesis of heterocyclic scaffolds [33,34,50], several multicomponent reactions [40,51-53], etc., we wished to convert dimethyl acetal 5j into aldehyde appended triazolochromene 14 and at the same time examine the stability under strong acidic conditions. Aldehyde appended triazolochromene 14 was synthesized in 85% yield, providing the proof for their relative stability under acidic conditions. Finally, triazolium salt 15 was prepared from 5a in 60% yield and renders a polar triazolium annulated chromene.
![[1860-5397-14-246-i5]](/bjoc/content/inline/1860-5397-14-246-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Postfunctional transformations of triazolochromenes.
Scheme 5: Postfunctional transformations of triazolochromenes.
Conclusion
We developed a sequential one-pot three-component reaction to access a variety of novel triazolochromenes avoiding the purification of intermediate 3-nitro-2H-chromenes. The regiochemistry of the reaction was determined and proven, followed by a scope study using various salicylaldehydes, nitroalkenes and organic azides. Solid salicylaldehydes gave diminished yields in the one-pot three-component protocol, hence a two-step mechanochemical approach was developed offering higher yields and resulting in a complementary route for solid salicylaldehydes. The applicability of the newly developed protocols was shown by gram-scale syntheses and postfunctionalization reactions towards biologically relevant analogs. The biological data will be published in due course.
Supporting Information
Supporting information features the optimization studies, NMR comparison studies of the various regioisomers 5a, 10 and 11, experimental details and copies of 1H and 13C NMR spectra of compounds 5a–p and 10–15.
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 1.6 MB | Download |
Acknowledgements
We thank the KU Leuven for financial support. RV thanks the Fonds Wetenschappelijk Onderzoek – Vlaanderen (FWO) for a Ph.D. fellowship (1S13516N). Prof. Dr. Koen Binnemans is thanked for granting the permission to use the ball milling apparatus, Gerrit Van Haele and Karel Duerinckx are acknowledged for technical assistance of the used equipment. Mass spectrometry was made possible by the support of the Hercules Foundation of the Flemish Government (grant 20100225-7).
References
-
Patil, S. A.; Patil, R.; Pfeffer, L. M.; Miller, D. D. Future Med. Chem. 2013, 5, 1647–1660. doi:10.4155/fmc.13.126
Return to citation in text: [1] -
Pratap, R.; Ram, V. J. Chem. Rev. 2014, 114, 10476–10526. doi:10.1021/cr500075s
Return to citation in text: [1] -
Costa, M.; Dias, T. A.; Brito, A.; Proença, F. Eur. J. Med. Chem. 2016, 123, 487–507. doi:10.1016/j.ejmech.2016.07.057
Return to citation in text: [1] [2] -
Kumar, D.; Sharma, P.; Singh, H.; Nepali, K.; Gupta, G. K.; Jain, S. K.; Ntie-Kang, F. RSC Adv. 2017, 7, 36977–36999. doi:10.1039/C7RA05441F
Return to citation in text: [1] -
Brigelius-Flohé, R.; Traber, M. G. FASEB J. 1999, 13, 1145–1155. doi:10.1096/fasebj.13.10.1145
Return to citation in text: [1] -
Choe, E.; Min, D. B. Compr. Rev. Food Sci. Food Saf. 2009, 8, 345–358. doi:10.1111/j.1541-4337.2009.00085.x
Return to citation in text: [1] -
Lu, D.; Yang, Y.; Li, Y.; Sun, C. Curr. Pharm. Anal. 2015, 11, 66–78. doi:10.2174/1573412910666140630170055
Return to citation in text: [1] -
Péter, S.; Friedel, A.; Roos, F. F.; Wyss, A.; Eggersdorfer, M.; Hoffmann, K.; Weber, P. Int. J. Vitam. Nutr. Res. 2015, 85, 261–281. doi:10.1024/0300-9831/a000281
Return to citation in text: [1] -
Sobolev, V. S.; Neff, S. A.; Gloer, J. B. J. Agric. Food Chem. 2009, 57, 62–68. doi:10.1021/jf802891v
Return to citation in text: [1] -
Park, B. H.; Lee, H. J.; Lee, Y. R. J. Nat. Prod. 2011, 74, 644–649. doi:10.1021/np100696f
Return to citation in text: [1] -
Hampson, A. J.; Grimaldi, M.; Axelrod, J.; Wink, D. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 8268–8273. doi:10.1073/pnas.95.14.8268
Return to citation in text: [1] -
Ameri, A. Prog. Neurobiol. 1999, 58, 315–348. doi:10.1016/S0301-0082(98)00087-2
Return to citation in text: [1] -
Croxford, J. L. Drugs 2003, 17, 179–202. doi:10.2165/00023210-200317030-00004
Return to citation in text: [1] -
Reekie, T. A.; Scott, M. P.; Kassiou, M. Nat. Rev. Chem. 2017, 2, No. 0101. doi:10.1038/s41570-017-0101
Return to citation in text: [1] -
Korotaev, V. Y.; Sosnovskikh, V. Y.; Barkov, A. Y. Russ. Chem. Rev. 2013, 82, 1081–1116. doi:10.1070/RC2013v082n12ABEH004370
Return to citation in text: [1] [2] [3] -
Habib, P. M.; Raju, B. R.; Kavala, V.; Kuo, C.-W.; Yao, C.-F. Tetrahedron 2009, 65, 5799–5804. doi:10.1016/j.tet.2009.05.002
Return to citation in text: [1] [2] -
Das, B. C.; Mohapatra, S.; Campbell, P. D.; Nayak, S.; Mahalingam, S. M.; Evans, T. Tetrahedron Lett. 2010, 51, 2567–2570. doi:10.1016/j.tetlet.2010.02.143
Return to citation in text: [1] [2] -
Wang, T.; Hu, X.-C.; Huang, X.-J.; Li, X.-S.; Xie, J.-W. J. Braz. Chem. Soc. 2012, 23, 1119–1123. doi:10.1590/S0103-50532012000600017
Return to citation in text: [1] [2] -
Quan, X.-J.; Ren, Z.-H.; Wang, Y.-Y.; Guan, Z.-H. Org. Lett. 2014, 16, 5728–5731. doi:10.1021/ol5027975
Return to citation in text: [1] [2] [3] -
Schwendt, G.; Glasnov, T. Monatsh. Chem. 2017, 148, 69–75. doi:10.1007/s00706-016-1885-5
Return to citation in text: [1] [2] -
Korotaev, V. Y.; Kutyashev, I. B.; Barkov, A. Y.; Sosnovskikh, V. Y. Chem. Heterocycl. Compd. 2017, 53, 597–603. doi:10.1007/s10593-017-2097-6
Return to citation in text: [1] [2] -
Sharma, P.; Kumar, N. P.; Senwar, K. R.; Forero-Doria, O.; Nachtigall, F. M.; Santos, L. S.; Shankaraiah, N. J. Braz. Chem. Soc. 2017, 28, 589–597. doi:10.21577/0103-5053.20160203
Return to citation in text: [1] [2] -
Mani, N. S.; Fitzgerald, A. E. J. Org. Chem. 2014, 79, 8889–8894. doi:10.1021/jo5013288
Return to citation in text: [1] [2] -
Thomas, J.; Jana, S.; Liekens, S.; Dehaen, W. Chem. Commun. 2016, 52, 9236–9239. doi:10.1039/C6CC03744E
Return to citation in text: [1] [2] [3] -
Ackermann, L.; Jeyachandran, R.; Potukuchi, H. K.; Novák, P.; Büttner, L. Org. Lett. 2010, 12, 2056–2059. doi:10.1021/ol1005517
Return to citation in text: [1] [2] -
Reddy, M. N.; Swamy, K. C. K. Eur. J. Org. Chem. 2012, 2013–2022. doi:10.1002/ejoc.201101816
Return to citation in text: [1] [2] -
Schulman, J. M.; Friedman, A. A.; Panteleev, J.; Lautens, M. Chem. Commun. 2012, 48, 55–57. doi:10.1039/C1CC16110E
Return to citation in text: [1] [2] -
Jeyachandran, R.; Potukuchi, H. K.; Ackermann, L. Beilstein J. Org. Chem. 2012, 8, 1771–1777. doi:10.3762/bjoc.8.202
Return to citation in text: [1] [2] [3] -
Chen, C.-Y.; Yang, C.-H.; Hu, W.-P.; Vandavasi, J. K.; Chung, M.-I.; Wang, J.-J. RSC Adv. 2013, 3, 2710–2719. doi:10.1039/c2ra22799a
Return to citation in text: [1] [2] -
Bai, S.-T.; Xiong, D.-C.; Niu, Y.; Wu, Y.-F.; Ye, X.-S. Tetrahedron 2015, 71, 4909–4919. doi:10.1016/j.tet.2015.05.108
Return to citation in text: [1] [2] -
Mondal, B.; Roy, B. Tetrahedron Lett. 2015, 56, 6123–6127. doi:10.1016/j.tetlet.2015.09.096
Return to citation in text: [1] [2] -
Ashok, D.; Chiranjeevi, P.; Kumar, A. V.; Sarasija, M.; Krishna, V. S.; Sriram, D.; Balasubramanian, S. RSC Adv. 2018, 8, 16997–17007. doi:10.1039/C8RA03197E
Return to citation in text: [1] -
Thomas, J.; John, J.; Parekh, N.; Dehaen, W. Angew. Chem., Int. Ed. 2014, 53, 10155–10159. doi:10.1002/anie.201403453
Return to citation in text: [1] [2] [3] -
John, J.; Thomas, J.; Parekh, N.; Dehaen, W. Eur. J. Org. Chem. 2015, 4922–4930. doi:10.1002/ejoc.201500459
Return to citation in text: [1] [2] -
John, J.; Thomas, J.; Dehaen, W. Chem. Commun. 2015, 51, 10797–10806. doi:10.1039/C5CC02319J
Return to citation in text: [1] -
Thomas, J.; Jana, S.; John, J.; Liekens, S.; Dehaen, W. Chem. Commun. 2016, 52, 2885–2888. doi:10.1039/C5CC08347H
Return to citation in text: [1] [2] [3] -
Thomas, J.; Goyvaerts, V.; Liekens, S.; Dehaen, W. Chem. – Eur. J. 2016, 22, 9966–9970. doi:10.1002/chem.201601928
Return to citation in text: [1] -
Jana, S.; Thomas, J.; Dehaen, W. J. Org. Chem. 2016, 81, 12426–12432. doi:10.1021/acs.joc.6b02607
Return to citation in text: [1] -
Opsomer, T.; Thomas, J.; Dehaen, W. Synthesis 2017, 49, 4191–4198. doi:10.1055/s-0036-1588856
Return to citation in text: [1] -
Vroemans, R.; Bamba, F.; Winters, J.; Thomas, J.; Jacobs, J.; Van Meervelt, L.; John, J.; Dehaen, W. Beilstein J. Org. Chem. 2018, 14, 626–633. doi:10.3762/bjoc.14.49
Return to citation in text: [1] [2] -
Silveira-Dorta, G.; Jana, S.; Borkova, L.; Thomas, J.; Dehaen, W. Org. Biomol. Chem. 2018, 16, 3168–3176. doi:10.1039/C8OB00533H
Return to citation in text: [1] -
Jalani, H. B.; Karagöz, A. Ç.; Tsogoeva, S. B. Synthesis 2017, 49, 29–41. doi:10.1055/s-0036-1588904
Return to citation in text: [1] -
Liu, S.-X.; Jia, C.-M.; Yao, B.-Y.; Chen, X.-L.; Zhang, Q. Synthesis 2016, 48, 407–412. doi:10.1055/s-0035-1560964
Return to citation in text: [1] [2] -
Gangaprasad, D.; Raj, J. P.; Kiranmye, T.; Karthikeyan, K.; Elangovan, J. Eur. J. Org. Chem. 2016, 5642–5646. doi:10.1002/ejoc.201601121
Return to citation in text: [1] -
Heinrich, T.; Böttcher, H.; Gericke, R.; Bartoszyk, G. D.; Anzali, S.; Seyfried, C. A.; Greiner, H. E.; van Amsterdam, C. J. Med. Chem. 2004, 47, 4684–4692. doi:10.1021/jm040793q
Return to citation in text: [1] -
Heinrich, T.; Böttcher, H.; Schiemann, K.; Hölzemann, G.; Schwarz, M.; Bartoszyk, G. D.; van Amsterdam, C.; Greiner, H. E.; Seyfried, C. A. Bioorg. Med. Chem. 2004, 12, 4943–4952. doi:10.1016/j.bmc.2004.07.014
Return to citation in text: [1] -
Copmans, D.; Orellana-Paucar, A. M.; Steurs, G.; Zhang, Y.; Ny, A.; Foubert, K.; Exarchou, V.; Siekierska, A.; Kim, Y.; De Borggraeve, W.; Dehaen, W.; Pieters, L.; de Witte, P. A. M. Neurochem. Int. 2018, 112, 124–133. doi:10.1016/j.neuint.2017.11.011
Return to citation in text: [1] -
Das, S. G.; Doshi, J. M.; Tian, D.; Addo, S. N.; Srinivasan, B.; Hermanson, D. L.; Xing, C. J. Med. Chem. 2009, 52, 5937–5949. doi:10.1021/jm9005059
Return to citation in text: [1] -
Das, S. G.; Srinivasan, B.; Hermanson, D. L.; Bleeker, N. P.; Doshi, J. M.; Tang, R.; Beck, W. T.; Xing, C. J. Med. Chem. 2011, 54, 5937–5948. doi:10.1021/jm200764t
Return to citation in text: [1] -
Ellis, G. P. In The Chemistry of Heterocyclic Compounds, Synthesis of Fused Heterocycles; Taylor, E. C., Ed.; John Wiley & Sons Ltd., 1987; Vol. 47. doi:10.1002/9780470187302
Return to citation in text: [1] -
Zhu, J.; Bienaymé, H. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005. doi:10.1002/3527605118
Return to citation in text: [1] -
Brauch, S.; van Berkel, S. S.; Westermann, B. Chem. Soc. Rev. 2013, 42, 4948–4962. doi:10.1039/c3cs35505e
Return to citation in text: [1] -
Váradi, A.; Palmer, T. C.; Dardashti, R. N.; Majumdar, S. Molecules 2016, 21, 19–40. doi:10.3390/molecules21010019
Return to citation in text: [1]
| 40. | Vroemans, R.; Bamba, F.; Winters, J.; Thomas, J.; Jacobs, J.; Van Meervelt, L.; John, J.; Dehaen, W. Beilstein J. Org. Chem. 2018, 14, 626–633. doi:10.3762/bjoc.14.49 |
| 51. | Zhu, J.; Bienaymé, H. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005. doi:10.1002/3527605118 |
| 52. | Brauch, S.; van Berkel, S. S.; Westermann, B. Chem. Soc. Rev. 2013, 42, 4948–4962. doi:10.1039/c3cs35505e |
| 53. | Váradi, A.; Palmer, T. C.; Dardashti, R. N.; Majumdar, S. Molecules 2016, 21, 19–40. doi:10.3390/molecules21010019 |
| 1. | Patil, S. A.; Patil, R.; Pfeffer, L. M.; Miller, D. D. Future Med. Chem. 2013, 5, 1647–1660. doi:10.4155/fmc.13.126 |
| 2. | Pratap, R.; Ram, V. J. Chem. Rev. 2014, 114, 10476–10526. doi:10.1021/cr500075s |
| 3. | Costa, M.; Dias, T. A.; Brito, A.; Proença, F. Eur. J. Med. Chem. 2016, 123, 487–507. doi:10.1016/j.ejmech.2016.07.057 |
| 4. | Kumar, D.; Sharma, P.; Singh, H.; Nepali, K.; Gupta, G. K.; Jain, S. K.; Ntie-Kang, F. RSC Adv. 2017, 7, 36977–36999. doi:10.1039/C7RA05441F |
| 15. | Korotaev, V. Y.; Sosnovskikh, V. Y.; Barkov, A. Y. Russ. Chem. Rev. 2013, 82, 1081–1116. doi:10.1070/RC2013v082n12ABEH004370 |
| 15. | Korotaev, V. Y.; Sosnovskikh, V. Y.; Barkov, A. Y. Russ. Chem. Rev. 2013, 82, 1081–1116. doi:10.1070/RC2013v082n12ABEH004370 |
| 11. | Hampson, A. J.; Grimaldi, M.; Axelrod, J.; Wink, D. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 8268–8273. doi:10.1073/pnas.95.14.8268 |
| 12. | Ameri, A. Prog. Neurobiol. 1999, 58, 315–348. doi:10.1016/S0301-0082(98)00087-2 |
| 13. | Croxford, J. L. Drugs 2003, 17, 179–202. doi:10.2165/00023210-200317030-00004 |
| 14. | Reekie, T. A.; Scott, M. P.; Kassiou, M. Nat. Rev. Chem. 2017, 2, No. 0101. doi:10.1038/s41570-017-0101 |
| 43. | Liu, S.-X.; Jia, C.-M.; Yao, B.-Y.; Chen, X.-L.; Zhang, Q. Synthesis 2016, 48, 407–412. doi:10.1055/s-0035-1560964 |
| 9. | Sobolev, V. S.; Neff, S. A.; Gloer, J. B. J. Agric. Food Chem. 2009, 57, 62–68. doi:10.1021/jf802891v |
| 10. | Park, B. H.; Lee, H. J.; Lee, Y. R. J. Nat. Prod. 2011, 74, 644–649. doi:10.1021/np100696f |
| 33. | Thomas, J.; John, J.; Parekh, N.; Dehaen, W. Angew. Chem., Int. Ed. 2014, 53, 10155–10159. doi:10.1002/anie.201403453 |
| 5. | Brigelius-Flohé, R.; Traber, M. G. FASEB J. 1999, 13, 1145–1155. doi:10.1096/fasebj.13.10.1145 |
| 6. | Choe, E.; Min, D. B. Compr. Rev. Food Sci. Food Saf. 2009, 8, 345–358. doi:10.1111/j.1541-4337.2009.00085.x |
| 7. | Lu, D.; Yang, Y.; Li, Y.; Sun, C. Curr. Pharm. Anal. 2015, 11, 66–78. doi:10.2174/1573412910666140630170055 |
| 8. | Péter, S.; Friedel, A.; Roos, F. F.; Wyss, A.; Eggersdorfer, M.; Hoffmann, K.; Weber, P. Int. J. Vitam. Nutr. Res. 2015, 85, 261–281. doi:10.1024/0300-9831/a000281 |
| 24. | Thomas, J.; Jana, S.; Liekens, S.; Dehaen, W. Chem. Commun. 2016, 52, 9236–9239. doi:10.1039/C6CC03744E |
| 33. | Thomas, J.; John, J.; Parekh, N.; Dehaen, W. Angew. Chem., Int. Ed. 2014, 53, 10155–10159. doi:10.1002/anie.201403453 |
| 34. | John, J.; Thomas, J.; Parekh, N.; Dehaen, W. Eur. J. Org. Chem. 2015, 4922–4930. doi:10.1002/ejoc.201500459 |
| 35. | John, J.; Thomas, J.; Dehaen, W. Chem. Commun. 2015, 51, 10797–10806. doi:10.1039/C5CC02319J |
| 36. | Thomas, J.; Jana, S.; John, J.; Liekens, S.; Dehaen, W. Chem. Commun. 2016, 52, 2885–2888. doi:10.1039/C5CC08347H |
| 37. | Thomas, J.; Goyvaerts, V.; Liekens, S.; Dehaen, W. Chem. – Eur. J. 2016, 22, 9966–9970. doi:10.1002/chem.201601928 |
| 38. | Jana, S.; Thomas, J.; Dehaen, W. J. Org. Chem. 2016, 81, 12426–12432. doi:10.1021/acs.joc.6b02607 |
| 39. | Opsomer, T.; Thomas, J.; Dehaen, W. Synthesis 2017, 49, 4191–4198. doi:10.1055/s-0036-1588856 |
| 40. | Vroemans, R.; Bamba, F.; Winters, J.; Thomas, J.; Jacobs, J.; Van Meervelt, L.; John, J.; Dehaen, W. Beilstein J. Org. Chem. 2018, 14, 626–633. doi:10.3762/bjoc.14.49 |
| 41. | Silveira-Dorta, G.; Jana, S.; Borkova, L.; Thomas, J.; Dehaen, W. Org. Biomol. Chem. 2018, 16, 3168–3176. doi:10.1039/C8OB00533H |
| 42. | Jalani, H. B.; Karagöz, A. Ç.; Tsogoeva, S. B. Synthesis 2017, 49, 29–41. doi:10.1055/s-0036-1588904 |
| 23. | Mani, N. S.; Fitzgerald, A. E. J. Org. Chem. 2014, 79, 8889–8894. doi:10.1021/jo5013288 |
| 25. | Ackermann, L.; Jeyachandran, R.; Potukuchi, H. K.; Novák, P.; Büttner, L. Org. Lett. 2010, 12, 2056–2059. doi:10.1021/ol1005517 |
| 26. | Reddy, M. N.; Swamy, K. C. K. Eur. J. Org. Chem. 2012, 2013–2022. doi:10.1002/ejoc.201101816 |
| 27. | Schulman, J. M.; Friedman, A. A.; Panteleev, J.; Lautens, M. Chem. Commun. 2012, 48, 55–57. doi:10.1039/C1CC16110E |
| 28. | Jeyachandran, R.; Potukuchi, H. K.; Ackermann, L. Beilstein J. Org. Chem. 2012, 8, 1771–1777. doi:10.3762/bjoc.8.202 |
| 29. | Chen, C.-Y.; Yang, C.-H.; Hu, W.-P.; Vandavasi, J. K.; Chung, M.-I.; Wang, J.-J. RSC Adv. 2013, 3, 2710–2719. doi:10.1039/c2ra22799a |
| 30. | Bai, S.-T.; Xiong, D.-C.; Niu, Y.; Wu, Y.-F.; Ye, X.-S. Tetrahedron 2015, 71, 4909–4919. doi:10.1016/j.tet.2015.05.108 |
| 31. | Mondal, B.; Roy, B. Tetrahedron Lett. 2015, 56, 6123–6127. doi:10.1016/j.tetlet.2015.09.096 |
| 16. | Habib, P. M.; Raju, B. R.; Kavala, V.; Kuo, C.-W.; Yao, C.-F. Tetrahedron 2009, 65, 5799–5804. doi:10.1016/j.tet.2009.05.002 |
| 17. | Das, B. C.; Mohapatra, S.; Campbell, P. D.; Nayak, S.; Mahalingam, S. M.; Evans, T. Tetrahedron Lett. 2010, 51, 2567–2570. doi:10.1016/j.tetlet.2010.02.143 |
| 18. | Wang, T.; Hu, X.-C.; Huang, X.-J.; Li, X.-S.; Xie, J.-W. J. Braz. Chem. Soc. 2012, 23, 1119–1123. doi:10.1590/S0103-50532012000600017 |
| 19. | Quan, X.-J.; Ren, Z.-H.; Wang, Y.-Y.; Guan, Z.-H. Org. Lett. 2014, 16, 5728–5731. doi:10.1021/ol5027975 |
| 20. | Schwendt, G.; Glasnov, T. Monatsh. Chem. 2017, 148, 69–75. doi:10.1007/s00706-016-1885-5 |
| 21. | Korotaev, V. Y.; Kutyashev, I. B.; Barkov, A. Y.; Sosnovskikh, V. Y. Chem. Heterocycl. Compd. 2017, 53, 597–603. doi:10.1007/s10593-017-2097-6 |
| 22. | Sharma, P.; Kumar, N. P.; Senwar, K. R.; Forero-Doria, O.; Nachtigall, F. M.; Santos, L. S.; Shankaraiah, N. J. Braz. Chem. Soc. 2017, 28, 589–597. doi:10.21577/0103-5053.20160203 |
| 28. | Jeyachandran, R.; Potukuchi, H. K.; Ackermann, L. Beilstein J. Org. Chem. 2012, 8, 1771–1777. doi:10.3762/bjoc.8.202 |
| 32. | Ashok, D.; Chiranjeevi, P.; Kumar, A. V.; Sarasija, M.; Krishna, V. S.; Sriram, D.; Balasubramanian, S. RSC Adv. 2018, 8, 16997–17007. doi:10.1039/C8RA03197E |
| 16. | Habib, P. M.; Raju, B. R.; Kavala, V.; Kuo, C.-W.; Yao, C.-F. Tetrahedron 2009, 65, 5799–5804. doi:10.1016/j.tet.2009.05.002 |
| 17. | Das, B. C.; Mohapatra, S.; Campbell, P. D.; Nayak, S.; Mahalingam, S. M.; Evans, T. Tetrahedron Lett. 2010, 51, 2567–2570. doi:10.1016/j.tetlet.2010.02.143 |
| 18. | Wang, T.; Hu, X.-C.; Huang, X.-J.; Li, X.-S.; Xie, J.-W. J. Braz. Chem. Soc. 2012, 23, 1119–1123. doi:10.1590/S0103-50532012000600017 |
| 19. | Quan, X.-J.; Ren, Z.-H.; Wang, Y.-Y.; Guan, Z.-H. Org. Lett. 2014, 16, 5728–5731. doi:10.1021/ol5027975 |
| 20. | Schwendt, G.; Glasnov, T. Monatsh. Chem. 2017, 148, 69–75. doi:10.1007/s00706-016-1885-5 |
| 21. | Korotaev, V. Y.; Kutyashev, I. B.; Barkov, A. Y.; Sosnovskikh, V. Y. Chem. Heterocycl. Compd. 2017, 53, 597–603. doi:10.1007/s10593-017-2097-6 |
| 22. | Sharma, P.; Kumar, N. P.; Senwar, K. R.; Forero-Doria, O.; Nachtigall, F. M.; Santos, L. S.; Shankaraiah, N. J. Braz. Chem. Soc. 2017, 28, 589–597. doi:10.21577/0103-5053.20160203 |
| 23. | Mani, N. S.; Fitzgerald, A. E. J. Org. Chem. 2014, 79, 8889–8894. doi:10.1021/jo5013288 |
| 24. | Thomas, J.; Jana, S.; Liekens, S.; Dehaen, W. Chem. Commun. 2016, 52, 9236–9239. doi:10.1039/C6CC03744E |
| 25. | Ackermann, L.; Jeyachandran, R.; Potukuchi, H. K.; Novák, P.; Büttner, L. Org. Lett. 2010, 12, 2056–2059. doi:10.1021/ol1005517 |
| 26. | Reddy, M. N.; Swamy, K. C. K. Eur. J. Org. Chem. 2012, 2013–2022. doi:10.1002/ejoc.201101816 |
| 27. | Schulman, J. M.; Friedman, A. A.; Panteleev, J.; Lautens, M. Chem. Commun. 2012, 48, 55–57. doi:10.1039/C1CC16110E |
| 28. | Jeyachandran, R.; Potukuchi, H. K.; Ackermann, L. Beilstein J. Org. Chem. 2012, 8, 1771–1777. doi:10.3762/bjoc.8.202 |
| 29. | Chen, C.-Y.; Yang, C.-H.; Hu, W.-P.; Vandavasi, J. K.; Chung, M.-I.; Wang, J.-J. RSC Adv. 2013, 3, 2710–2719. doi:10.1039/c2ra22799a |
| 30. | Bai, S.-T.; Xiong, D.-C.; Niu, Y.; Wu, Y.-F.; Ye, X.-S. Tetrahedron 2015, 71, 4909–4919. doi:10.1016/j.tet.2015.05.108 |
| 31. | Mondal, B.; Roy, B. Tetrahedron Lett. 2015, 56, 6123–6127. doi:10.1016/j.tetlet.2015.09.096 |
| 24. | Thomas, J.; Jana, S.; Liekens, S.; Dehaen, W. Chem. Commun. 2016, 52, 9236–9239. doi:10.1039/C6CC03744E |
| 36. | Thomas, J.; Jana, S.; John, J.; Liekens, S.; Dehaen, W. Chem. Commun. 2016, 52, 2885–2888. doi:10.1039/C5CC08347H |
| 15. | Korotaev, V. Y.; Sosnovskikh, V. Y.; Barkov, A. Y. Russ. Chem. Rev. 2013, 82, 1081–1116. doi:10.1070/RC2013v082n12ABEH004370 |
| 19. | Quan, X.-J.; Ren, Z.-H.; Wang, Y.-Y.; Guan, Z.-H. Org. Lett. 2014, 16, 5728–5731. doi:10.1021/ol5027975 |
| 48. | Das, S. G.; Doshi, J. M.; Tian, D.; Addo, S. N.; Srinivasan, B.; Hermanson, D. L.; Xing, C. J. Med. Chem. 2009, 52, 5937–5949. doi:10.1021/jm9005059 |
| 49. | Das, S. G.; Srinivasan, B.; Hermanson, D. L.; Bleeker, N. P.; Doshi, J. M.; Tang, R.; Beck, W. T.; Xing, C. J. Med. Chem. 2011, 54, 5937–5948. doi:10.1021/jm200764t |
| 33. | Thomas, J.; John, J.; Parekh, N.; Dehaen, W. Angew. Chem., Int. Ed. 2014, 53, 10155–10159. doi:10.1002/anie.201403453 |
| 34. | John, J.; Thomas, J.; Parekh, N.; Dehaen, W. Eur. J. Org. Chem. 2015, 4922–4930. doi:10.1002/ejoc.201500459 |
| 50. | Ellis, G. P. In The Chemistry of Heterocyclic Compounds, Synthesis of Fused Heterocycles; Taylor, E. C., Ed.; John Wiley & Sons Ltd., 1987; Vol. 47. doi:10.1002/9780470187302 |
| 45. | Heinrich, T.; Böttcher, H.; Gericke, R.; Bartoszyk, G. D.; Anzali, S.; Seyfried, C. A.; Greiner, H. E.; van Amsterdam, C. J. Med. Chem. 2004, 47, 4684–4692. doi:10.1021/jm040793q |
| 46. | Heinrich, T.; Böttcher, H.; Schiemann, K.; Hölzemann, G.; Schwarz, M.; Bartoszyk, G. D.; van Amsterdam, C.; Greiner, H. E.; Seyfried, C. A. Bioorg. Med. Chem. 2004, 12, 4943–4952. doi:10.1016/j.bmc.2004.07.014 |
| 47. | Copmans, D.; Orellana-Paucar, A. M.; Steurs, G.; Zhang, Y.; Ny, A.; Foubert, K.; Exarchou, V.; Siekierska, A.; Kim, Y.; De Borggraeve, W.; Dehaen, W.; Pieters, L.; de Witte, P. A. M. Neurochem. Int. 2018, 112, 124–133. doi:10.1016/j.neuint.2017.11.011 |
| 43. | Liu, S.-X.; Jia, C.-M.; Yao, B.-Y.; Chen, X.-L.; Zhang, Q. Synthesis 2016, 48, 407–412. doi:10.1055/s-0035-1560964 |
| 3. | Costa, M.; Dias, T. A.; Brito, A.; Proença, F. Eur. J. Med. Chem. 2016, 123, 487–507. doi:10.1016/j.ejmech.2016.07.057 |
| 36. | Thomas, J.; Jana, S.; John, J.; Liekens, S.; Dehaen, W. Chem. Commun. 2016, 52, 2885–2888. doi:10.1039/C5CC08347H |
| 44. | Gangaprasad, D.; Raj, J. P.; Kiranmye, T.; Karthikeyan, K.; Elangovan, J. Eur. J. Org. Chem. 2016, 5642–5646. doi:10.1002/ejoc.201601121 |
© 2018 Vroemans et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)