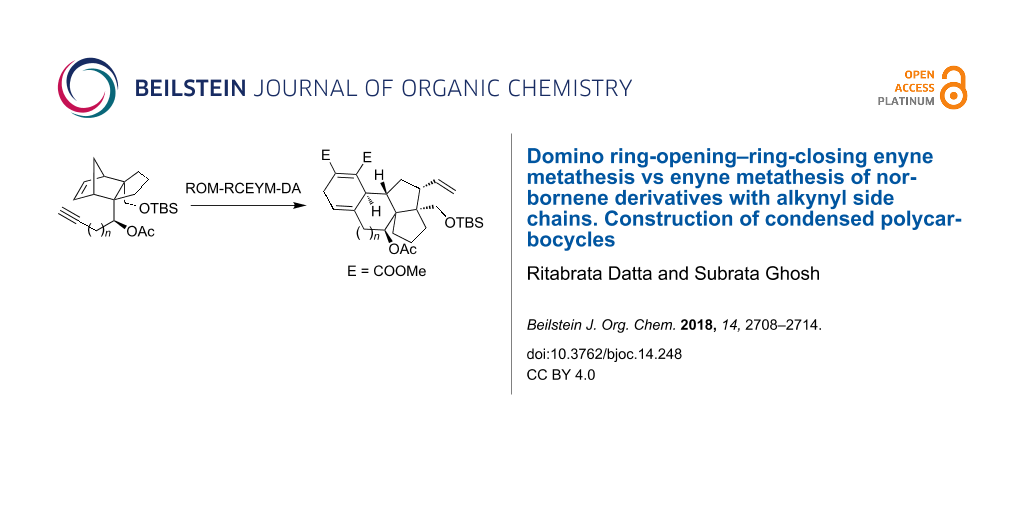
Abstract
The metathesis of norbornene derivatives with alkynyl side-chain with Grubbs’ ruthenium alkylidine as catalyst has been investigated with the objective of constructing condensed polycyclic structures. This investigation demonstrated that the generally observed domino reaction course involving a ring-opening metathesis of the norbornene unit and a ring-closing enyne metathesis is influenced to a great extent by the nature of the functional group and the substrate structure and may follow a different reaction course than what is usually observed. In cases where ROM–RCEYM occurred, the resulting 1,3-diene reacts in situ with the dienophile to provide condensed tetracyclic systems.

Graphical Abstract
Introduction
The metathesis of norbornene derivatives having an alkene side-chain on the norbornene nucleus with Grubbs’ ruthenium catalysts has been extensively investigated. Generally the reaction proceeds through a domino process involving a ring opening of the norbornene nucleus and ring closing with the alkene side chains to produce ring rearrangement products (path 1, Scheme 1) [1-4]. This protocol has been employed by several groups [5-22] as well as by our group [23-33] for the synthesis of a variety of complex ring systems such as condensed, bridged and spirocycles difficult to obtain otherwise. On the contrary, the domino process involving a ring-opening metathesis (ROM) followed by a ring-closing enyne metathesis (RCEYM) [34-37] of norbornene derivatives with a suitably located alkynyl side-chain on the nucleus (path 2, Scheme 1) to form carbocycles has been less explored. The greatest advantage of this protocol lies in its potential in increasing the molecular complexity through Diels–Alder reaction of the resulting ring system. Domino metathesis of oxa- and aza-norbornenes with alkyne side chains [38-40] as well as norbornene derivatives having ether linked alkynes [41,42] in combination with Diels–Alder reaction of the resulting 1,3-dienes have been investigated to construct polycycles with heteroatoms. In spite of the great potential little attention has been paid [43] for exploring its application in the synthesis of complex carbocyclic ring systems, backbones of innumerable natural products.
![[1860-5397-14-248-i1]](/bjoc/content/inline/1860-5397-14-248-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Metathesis of norbornene derivatives.
Scheme 1: Metathesis of norbornene derivatives.
We undertook a program for the synthesis of condensed polycarbocyclic scaffolds using a metathesis of norbornene derivatives with suitably located alkynyl side-chains as the key step. The structurally unique sesterterpenes retigeranic acid A (1a) and retigeranic acid B (1b, Figure 1) are representative examples of such complex polycyclic structures [44-47]. We speculated that domino ROM–RCEYM of the norbornene derivative 2 would provide the tricyclic 1,3-diene 3 which on Diels–Alder reaction with a dienophile would enable access to condensed polycyclic structures 4 (Scheme 2). Thus an appropriately chosen norbornene derivative and a dienophile may provide the B/C/D/E ring system of retigeranic acids. Herein we describe the results of metathesis of norbornene derivatives 2 with alkynyl side-chains.
![[1860-5397-14-248-1]](/bjoc/content/figures/1860-5397-14-248-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of retigeranic acids A (1a) and B (1b).
Figure 1: Structures of retigeranic acids A (1a) and B (1b).
![[1860-5397-14-248-i2]](/bjoc/content/inline/1860-5397-14-248-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
Initially Grubbs’ 1st generation catalyst (G-I) was used for metathesis of norbornene derivatives 2. In case G-I failed to accomplish metathesis in the desired direction, 2nd generation catalyst (G-II) was used. The norbornene derivative 7a was first chosen for investigating ROM–RCEYM. Compound 7a was prepared in the following way (Scheme 3). Reaction of the known lactol 5 [33] with propargyl magnesium bromide afforded the diol 6 in 88% yield (For detailed experimental procedures and characterization data see Supporting Information File 1). The stereochemical orientation of the secondary hydroxy group was determined through X-ray crystal structure of a compound derived from it in a subsequent step. The primary hydroxy group in the diol 6 was then selectively protected to provide the silyl ether 7a in 92% yield. Two different paths can be invoked for metathesis of compound 7a. Metathesis initiation may occur by attack of the ruthenium alkylidene at the alkyne unit to produce the more substituted vinyl alkylidine intermediate 8a which may undergo concomitant ROM–RCM with the norbornene nucleus to provide the triene 9a (path 1).
![[1860-5397-14-248-i3]](/bjoc/content/inline/1860-5397-14-248-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Metathesis of norbornene derivatives 7a and 7b.
Scheme 3: Metathesis of norbornene derivatives 7a and 7b.
Alternatively the metathesis initiation may occur initially at the norbornene double bond to provide the ring-opened ruthenium alkylidine intermediate 10 (path 2). The latter then undergoes RCEYM to provide the tricycle 9a. With this background a solution of the compound 7a in toluene under ethylene atmosphere was heated at 65 °C with Grubbs 1st generation catalyst (G-I). Compound 7a was found to be inert even after a prolonged reaction time. However, with G-II as the catalyst the metathesis went smoothly. Without isolation, the metathesis product was treated in situ with dimethyl acetylenedicarboxylate (DMAD). In case the Diels–Alder reaction would take place through the triene 9a the tetracyclic structure 12a would be formed. However, 13C NMR spectra of the product revealed the presence of eight methylene carbon signals at δ 28.6, 28.9, 30.9, 33.5, 36.7, 41.1, 45.7 and 68.8, one more aliphatic methylene unit than what the structure 12a requires (see Supporting Information File 1). This indicates that the metathesis product is not 9a. The structure of the metathesis product was finally settled by X-ray crystal structure (Figure 2) [48] (see Supporting Information File 2) of the 3,5-dinitrobenzoate derivative 13, mp 171–172 °C, prepared in two steps (51%) from the metathesis product on reaction with 3,5-dinitrobenzoyl chloride (DNBC) followed by acid-induced desilylation. Thus compound 7b on metathesis produced exclusively triene 11 and accordingly the structure of the Diels–Alder adduct is 14. The formation of triene 11 could be attributed to cross metathesis of the ruthenium alkylidene 8a with ethylene. No product arising out of ROM of norbornene derivative 7a was formed. It is worth mentioning that Spandl et al. [43] reported the metathesis of norbornene derivatives with an alkynyl side chain affording the major product arising from domino ROM–RCEYM while the enyne metathesis product was observed only in very low yield.
![[1860-5397-14-248-2]](/bjoc/content/figures/1860-5397-14-248-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ORTEP of compound 13 (ellipsoids at 30% probability).
Figure 2: ORTEP of compound 13 (ellipsoids at 30% probability).
In order to realize our objective and to find out if the free hydroxy group has any influence on the outcome of the metathesis, the hydroxy group in compound 7a was protected to provide the acetate derivative 7b. The metathesis of compound 7b with G-I as the catalyst proceeded smoothly and the resulting product without isolation was allowed to react with DMAD to produce the tetracycle 12b in overall 66% yield. The structure of compound 12b was established through analysis of its NMR spectra. Isolation of 12b dictated that metathesis of 7b proceeded through the formation of the triene 9b. Stereochemical assignment to the adduct follows from addition of the dienophile from the least hindered face (opposite to CH2OTBS group) of the diene. Thus unlike metathesis of 7a, metathesis of its acetate analogue 7b occurred through a domino ROM–RCEYM process. Addition of the Ru-carbene 10 arising from ring opening of norbornene unit in 7b could add to the acetylenic unit of another molecule of 7b leading to copolymerization. However, this process generally does not take place under such low molar concentration of the substrate [38-43]. We also did not isolate any copolymerization product. This may be attributed to the much faster rate of addition of the Ru-carbene 10 to the yne unit intramolecularly resulting in ring closure rather than intermolecular addition to an acetylenic unit of another molecule of 7b. It may be noted that changing the functional group from hydroxy to acetate the metathesis followed a different reaction course.
In order to construct a polycyclic structure analogous to the B/C/D/E ring of retigeranic acids, the norbornene derivative 16 was chosen. Addition of lithium (trimethylsilyl)acetylide to the lactol 5 followed by desilylation by using methanolic K2CO3 afforded diol 15 (Scheme 4). The primary hydroxy group in compound 15 was selectively protected to produce the silyl ether 16 in 95% yield. The attempted metathesis of compound 16 with G-I or G-II catalyst under the conditions used for the metathesis of 7a led to a complete recovery of 16. Since metathesis of the acetate derivative 7b proceeded smoothly in the desired direction, we chose to use the acetate 17 for metathesis. The acetate 17 also remained inert when subjected to metathesis conditions with G-I as well as with G-II. Neither ring opening of the norbornene nucleus nor cross metathesis of the alkyne with ethylene did occur. To have an understanding about the inertness of 17 towards metathesis we decided to prepare the ring-opened product 18 using an alternative path. The double bond in the norbornene nucleus in compound 17 was cleaved in the traditional way by treatment with OsO4/NaIO4 and the resulting dialdehyde on Wittig reaction provided the diene 18 in 66% yield in two steps. Amazingly when compound 18 was treated with G-I or G-II as catalyst, the metathesis was found to take place. After disappearance of the starting material (TLC), the reaction mixture was allowed to react with DMAD. The product obtained in 76% yield was assigned the structure 20 based on spectral data. Isolation of 20 indicates that metathesis of 18 proceeded through RCEYM to produce the triene 19. The latter then after in situ Diels–Alder reaction with DMAD delivered the product 20. The tetracyclic compound 20 represents the B/C/D/E tetracyclic core structure of retigeranic acids.
![[1860-5397-14-248-i4]](/bjoc/content/inline/1860-5397-14-248-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Metathesis of the norbornene derivative 17.
Scheme 4: Metathesis of the norbornene derivative 17.
Based on the above observations a mechanistic rationale regarding the metathesis of norbornene derivatives with an alkynyl side chain may be postulated (Figure 3). Possibly the metathesis is initiated at the acetylenic unit to form the ruthenium alkylidine such as 8. In case of 8a the ruthenium alkylidine is stabilized by formation of the chelate 21 (R = H) which prohibits intramolecular addition of the ruthenium alkylidine to form ruthena cyclobutane 22. The alkylidine 21 then undergoes cross metathesis with ethylene to form the product 11. The ruthenium alkylidine 8b possibly fails to form chelate 21 (R = Ac) due to the electron deficient nature of the OAc group. It forms intramolecularly the ruthena cyclobutane 22 which undergoes ring opening to give rise to the triene 9b. That the metathesis does not proceed through path 2 (Scheme 3) involving ROM–RCM is indicated by failure of the norbornene derivative 17 to undergo ROM. Steric shielding of the acetylenic unit in 17 inhibits metathesis initiation at the acetylenic unit. The norbornene derivative 17 just remains inert under metathesis conditions. Thus metathesis in these examples proceeds through path 1 (Scheme 3).
![[1860-5397-14-248-3]](/bjoc/content/figures/1860-5397-14-248-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Probable metathesis intermediates.
Figure 3: Probable metathesis intermediates.
Conclusion
In conclusion we have developed a protocol for the synthesis of condensed polycycles from metathesis of norbornene derivatives with alkynyl side-chain. This investigation demonstrated that domino metathesis of norbornene derivatives with alkynyl side-chain requires metathesis initiation at the acetylene unit. Further, the nature of functional groups as well as the substrate structure play a significant role in determining the metathesis reaction course.
Experimental
General experimental methods are similar as described in [49]
Synthesis of triene 11. A solution of the silyl ether 7a (120 mg, 0.35 mmol) in degassed toluene (7 mL) with Grubbs’ catalyst G-II (30 mg, 0.035 mmol) was heated at 65 °C for 6 h under a positive pressure of ethylene atmosphere. After completion (TLC) of the reaction toluene was removed under vacuo. The residual mass was purified by column chromatography (7% EA/PE) to afford diene 11 (89 mg, 69%) as an oil; 1H NMR (500 MHz) δ 6.42 (dd, J = 11, 17.5 Hz, 1H), 6.12 (s, 2H), 5.28 (d, J = 17.5 Hz, 1H), 5.15 (d, J = 29 Hz, 2H), 5.04 (d, J = 11 Hz, 1H), 3.98 (s, 1H), 3.63 (d, J = 10 Hz, 1H), 3.56 (s, 2H), 2.53 (s, 1H), 2.45–2.42 (m, 1H), 2.36–2.22 (m, 3H), 1.95–1.92 (m, 1H), 1.82–1.74 (m, 3H), 1.47–1.38 (m, 2H), 1.33–1.28 (m, 1H), 0.89 (s, 9H), 0.07 (s, 6H); 13C NMR (125 MHz) δ 144.3, 139.8, 136.4, 136.1, 117.0, 113.1, 71.9, 68.6, 63.4, 61.2, 52.2, 52.0, 45.8, 36.8, 36.0, 33.8, 28.8, 26.0 (× 3), 18.4, −5.4, −5.6; HRMS–ESI m/z: [M + Na]+ calcd for C23H38O2SiNa 397.2539; found, 397.2537.
Diels–Alder reaction of diene 11. Synthesis of adduct 14. A mixture of the diene 11 (40 mg, 0.11 mmol) and dimethyl acetylenedicarboxylate (0.02 mL, 0.16 mmol) in toluene (5 mL) was heated at 65 °C for 2 h. The solvent was removed under reduced pressure and was purified by column chromatography (12% EA/PE) to afford the Diels–Alder adduct 14 (42 mg, 76%) as an oil; 1H NMR (500 MHz) δ 6.12–6.11 (m, 2H), 5.52 (s, 1H), 4.26 (s, 1H), 3.89–3.88 (m, 1H), 3.78 (s, 3H), 3.76 (s, 3H), 3.60–3.52 (m, 3H), 3.05–2.97 (m, 3H), 2.45 (s, 1H), 2.32 (s, 1H), 2.18–2.17 (m, 3H), 1.93–1.89 (m, 1H), 1.83–1.78 (m, 2H), 1.75–1.68 (m, 2H), 1.48–1.41 (m, 1H), 1.39–1.33 (m, 2H), 0.88 (s, 9H), 0.08 (s, 6H); 13C NMR (75 MHz) δ 169.0, 168.7, 136.4, 135.8, 133.6, 132.4, 132.2, 117.9, 72.0, 68.8, 63.1, 61.2, 52.3 (× 2), 52.1 (× 2), 45.7, 41.1, 36.8, 33.6, 30.9, 28.9, 28.6, 26.0 (× 3), 18.4, −5.4, −5.6; IR: 2952, 1728, 1471 cm−1; HRMS–ESI m/z: [M + Na]+ calcd for C29H44O6SiNa 539.2805; found, 539.2802.
Synthesis of tetracycle 12b. A solution of the norbornene derivative 7b (70 mg, 0.18 mmol) in degassed toluene (6 mL) was heated with Grubbs’ catalyst G-I (15 mg, 0.018 mmol) under ethylene atmosphere at 65 °C for 12 h. After completion (TLC) of the metathesis reaction, dimethyl acetylenedicarboxylate (0.04 mL, 0.27 mmol) was added to the reaction mixture. The reaction mixture was then heated for 12 h till the Diels–Alder reaction of the diene 9b generated in situ was complete. The solvent was removed under vacuo and the product was purified by column chromatography (15% EA/PE) to afford the tetracycle 12b (66 mg, 66%) as a colorless oil; 1H NMR (300 MHz) δ 5.99–5.87 (m, 1H), 5.63–5.57 (m, 1H), 5.35–5.34 (m, 1H), 4.99–4.94 (m, 2H), 3.74 (s, 3H), 3.73 (s, 3H), 3.58–3.48 (m, 2H), 3.25–3.16 (m, 2H), 3.10–3.07 (m, 1H), 2.85–2.75 (m, 1H), 2.14–2.07 (m, 2H), 2.04 (s, 3H), 2.02–1.85 (m, 2H), 1.69–1.59 (m, 4H), 1.53–1.25 (m, 3H), 0.94 (s, 9H), 0.02 (s, 3H), −0.03 (s, 3H); 13C NMR (75 MHz) δ 170.1, 169.3, 168.7, 139.5, 136.9, 134.0, 132.5, 115.9, 115.0, 73.4, 65.1, 60.6, 57.6, 56.2, 53.1, 52.4, 52.1, 40.9, 37.6, 36.2, 34.9, 34.7, 28.2, 26.2 (× 3), 22.1, 21.8, 17.9, −5.8, −6.1; IR: 2950, 1737, 1434, 1249 cm−1; HRMS–ESI m/z: [M + Na]+ calcd for C31H46O7SiNa 581.2911; found, 581.2914.
Synthesis of the tetracycle 20. The dienyne 18 (100 mg, 0.25 mmol) in degassed anhydrous toluene (7 mL) was treated with Grubbs’ catalyst G-II (22 mg, 0.025 mmol) at 65 °C for 5 h. On completion of the reaction (TLC), dimethyl acetylenedicarboxylate (0.06 mL, 0.37 mmol) was added to the resulting reaction mixture. The mixture was heated at 65 °C for 8 h. Removal of the solvent under vacuo followed by column chromatography (15% EA/PE) afforded the Diels–Alder adduct 20 (102 mg, 76%) as a colorless oil; 1H NMR (300 MHz) δ 6.06–6.02 (m, 1H), 6.00–5.94 (m, 1H), 5.24–5.21 (m, 1H), 5.03–4.95 (m, 2H), 3.80 (s, 3H), 3.75 (s, 3H), 3.53 (s, 2H), 3.19–3.11 (m, 1H), 3.09–3.02 (m, 1H), 3.00–2.86 (m, 1H), 2.43–2.34 (m, 1H), 2.14 (s, 3H), 2.08–1.98 (m, 2H), 1.96–1.78 (m, 2H), 1.55–1.50 (m, 2H), 1.46–1.41 (m, 1H), 1.34–1.25 (m, 1H), 1.12–0.99 (m, 1H), 0.91 (s, 9H), 0.04 (s, 3H), 0.02 (s, 3H); 13C NMR (75 MHz) δ 170.0, 169.0, 167.5, 140.1, 139.3, 139.2, 129.0, 115.3, 109.8, 74.4, 65.6, 62.3, 58.1, 56.7, 54.7, 52.4, 52.3, 41.9, 40.1, 35.9, 35.6, 27.6, 26.2 (× 3), 22.9, 21.1, 18.2, −5.8, −5.9; IR: 2950, 1731, 1434, 1257 cm−1; HRMS–ESI m/z: [M + Na]+ calcd for C30H44O7SiNa 567.2754; found, 567.2756.
Acknowledgements
SG is grateful to Indian National Science Academy, New Delhi for financial support through INSA Senior Scientist program. We are grateful to DST, Government of India for financial support for National Single Crystal Diffractometer facility at the Department of Inorganic Chemistry of this institute. We sincerely thank Mr. Mukesh Choudhary of Institute of Chemical Technology, Mumbai for carrying out some preliminary experiments.
References
-
Arjona, O.; Csáky, A. G.; Plumet, J. Eur. J. Org. Chem. 2003, 611–622. doi:10.1002/ejoc.200390100
Return to citation in text: [1] -
Holub, N.; Blechert, S. Chem. – Asian J. 2007, 2, 1064–1082. doi:10.1002/asia.200700072
Return to citation in text: [1] -
Bose, S.; Ghosh, S. Proc. Indian Natl. Sci. Acad. 2014, 80, 37–54. doi:10.16943/ptinsa/2014/v80i1/55085
Return to citation in text: [1] -
Kotha, S.; Meshram, M.; Khedkar, P.; Banerjee, S.; Deodhar, D. Beilstein J. Org. Chem. 2015, 11, 1833–1864. doi:10.3762/bjoc.11.199
Return to citation in text: [1] -
Stille, J. R.; Santarsiero, B. D.; Grubbs, R. H. J. Org. Chem. 1990, 55, 843–862. doi:10.1021/jo00290a013
Return to citation in text: [1] -
Zuercher, W. J.; Hashimoto, M.; Grubbs, R. H. J. Am. Chem. Soc. 1996, 118, 6634–6640. doi:10.1021/ja9606743
Return to citation in text: [1] -
Stragies, R.; Blechert, S. Synlett 1998, 169–170. doi:10.1055/s-1998-1592
Return to citation in text: [1] -
Arjona, O.; Csákÿ, A. G.; Murcia, M. C.; Plumet, J. Tetrahedron Lett. 2000, 41, 9777–9779. doi:10.1016/S0040-4039(00)01721-4
Return to citation in text: [1] -
Weatherhead, G. S.; Ford, J. G.; Alexanian, E. J.; Schrock, R. R.; Hoveyda, A. H. J. Am. Chem. Soc. 2000, 122, 1828–1829. doi:10.1021/ja993681a
Return to citation in text: [1] -
Arjona, O.; Csákÿ, A. G.; Medel, R.; Plumet, J. J. Org. Chem. 2002, 67, 1380–1383. doi:10.1021/jo016000e
Return to citation in text: [1] -
Sakurai, H.; Daiko, T.; Hirao, T. Science 2003, 301, 1878. doi:10.1126/science.1088290
Return to citation in text: [1] -
Arjona, O.; Csákÿ, A. G.; León, V.; Medel, R.; Plumet, J. Tetrahedron Lett. 2004, 45, 565–567. doi:10.1016/j.tetlet.2003.10.197
Return to citation in text: [1] -
Holtsclaw, J.; Koreeda, M. Org. Lett. 2004, 6, 3719–3722. doi:10.1021/ol048650l
Return to citation in text: [1] -
Funel, J.-A.; Prunet, J. Synlett 2005, 235–238. doi:10.1055/s-2004-837200
Return to citation in text: [1] -
Chandler, C. L.; Phillips, A. J. Org. Lett. 2005, 7, 3493–3495. doi:10.1021/ol051199t
Return to citation in text: [1] -
Maechling, S.; Norman, S. E.; Mckendrick, J. E.; Basra, S.; Köppner, K.; Blechert, S. Tetrahedron Lett. 2006, 47, 189–192. doi:10.1016/j.tetlet.2005.10.155
Return to citation in text: [1] -
Hart, A. C.; Phillips, A. J. J. Am. Chem. Soc. 2006, 128, 1094–1095. doi:10.1021/ja057899a
Return to citation in text: [1] -
Phillips, A. J.; Hart, A. C.; Henderson, J. A. Tetrahedron Lett. 2006, 47, 3743–3745. doi:10.1016/j.tetlet.2006.03.124
Return to citation in text: [1] -
Calvet, G.; Blanchard, N.; Kouklovsky, C. Org. Lett. 2007, 9, 1485–1488. doi:10.1021/ol0702066
Return to citation in text: [1] -
Henderson, J. A.; Phillips, A. J. Angew. Chem., Int. Ed. 2008, 47, 8499–8501. doi:10.1002/anie.200803593
Return to citation in text: [1] -
Nguyen, N. N. M.; Leclère, M.; Stogaitis, N.; Fallis, A. G. Org. Lett. 2010, 12, 1684–1687. doi:10.1021/ol100150f
Return to citation in text: [1] -
Lam, J. K.; Pham, H. V.; Houk, K. N.; Vanderwal, C. D. J. Am. Chem. Soc. 2013, 135, 17585–17594. doi:10.1021/ja409618p
Return to citation in text: [1] -
Malik, C. K.; Ghosh, S. Org. Lett. 2007, 9, 2537–2540. doi:10.1021/ol070906a
Return to citation in text: [1] -
Maity, S.; Ghosh, S. Tetrahedron Lett. 2008, 49, 1133–1136. doi:10.1016/j.tetlet.2007.12.064
Return to citation in text: [1] -
Mondal, S.; Malik, C. K.; Ghosh, S. Tetrahedron Lett. 2008, 49, 5649–5651. doi:10.1016/j.tetlet.2008.07.083
Return to citation in text: [1] -
Malik, C. K.; Yadav, R. N.; Drew, M. G. B.; Ghosh, S. J. Org. Chem. 2009, 74, 1957–1963. doi:10.1021/jo802077t
Return to citation in text: [1] -
Malik, C. K.; Hossain, M. F.; Ghosh, S. Tetrahedron Lett. 2009, 50, 3063–3066. doi:10.1016/j.tetlet.2009.04.033
Return to citation in text: [1] -
Mondal, S.; Yadav, R. N.; Ghosh, S. Tetrahedron Lett. 2009, 50, 5277–5279. doi:10.1016/j.tetlet.2009.07.012
Return to citation in text: [1] -
Maity, S.; Ghosh, S. Tetrahedron 2009, 65, 9202–9210. doi:10.1016/j.tet.2009.09.029
Return to citation in text: [1] -
Matcha, K.; Maity, S.; Malik, C. K.; Ghosh, S. Tetrahedron Lett. 2010, 51, 2754–2757. doi:10.1016/j.tetlet.2010.03.074
Return to citation in text: [1] -
Yadav, R. N.; Mondal, S.; Ghosh, S. Tetrahedron Lett. 2011, 52, 1942–1945. doi:10.1016/j.tetlet.2011.02.054
Return to citation in text: [1] -
Bose, S.; Ghosh, M.; Ghosh, S. J. Org. Chem. 2012, 77, 6345–6350. doi:10.1021/jo300945b
Return to citation in text: [1] -
Datta, R.; Bose, S.; Viththlbhai, P. B.; Ghosh, S. Tetrahedron Lett. 2014, 55, 3538–3540. doi:10.1016/j.tetlet.2014.04.091
Return to citation in text: [1] [2] -
Villar, H.; Frings, M.; Bolm, C. Chem. Soc. Rev. 2007, 36, 55–66. doi:10.1039/B508899M
Return to citation in text: [1] -
Diver, S. T.; Giessert, A. J. Chem. Rev. 2004, 104, 1317–1382. doi:10.1021/cr020009e
Return to citation in text: [1] -
Mori, M. Adv. Synth. Catal. 2007, 349, 121–135. doi:10.1002/adsc.200600484
Return to citation in text: [1] -
Mori, M. Materials 2010, 3, 2087–2140. doi:10.3390/ma3032087
Return to citation in text: [1] -
Banti, D.; North, M. Adv. Synth. Catal. 2002, 344, 694–704.
Return to citation in text: [1] [2] -
Banti, D.; Groaz, E.; North, M. Tetrahedron 2004, 60, 8043–8052. doi:10.1016/j.tet.2004.06.114
Return to citation in text: [1] [2] -
Groaz, E.; Banti, D.; North, M. Eur. J. Org. Chem. 2007, 3727–3745. doi:10.1002/ejoc.200700291
Return to citation in text: [1] [2] -
Kotha, S.; Ravikumar, O. Tetrahedron Lett. 2014, 55, 5781–5784. doi:10.1016/j.tetlet.2014.08.108
Return to citation in text: [1] [2] -
Kotha, S.; Ravikumar, O. Eur. J. Org. Chem. 2014, 5582–5590. doi:10.1002/ejoc.201402273
Return to citation in text: [1] [2] -
Spandl, R. J.; Rudyk, H.; Spring, D. R. Chem. Commun. 2008, 3001–3003. doi:10.1039/B807278G
Return to citation in text: [1] [2] [3] -
Breitler, S.; Han, Y.; Corey, E. J. Org. Lett. 2017, 19, 6686–6687. doi:10.1021/acs.orglett.7b03412
Return to citation in text: [1] -
Wright, J.; Drtina, G. J.; Roberts, R. A.; Paquette, L. A. J. Am. Chem. Soc. 1988, 110, 5806–5817. doi:10.1021/ja00225a036
Return to citation in text: [1] -
Wender, P. A.; Singh, S. K. Tetrahedron Lett. 1990, 31, 2517–2520. doi:10.1016/0040-4039(90)80114-2
Return to citation in text: [1] -
Hudlicky, T.; Fleming, A.; Radesca, L. J. Am. Chem. Soc. 1989, 111, 6691–6707. doi:10.1021/ja00199a032
Return to citation in text: [1] -
CCDC 1847091 contains supplementary crystallographic data for the compound 13.These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Datta, R.; Ghosh, S. J. Org. Chem. 2017, 82, 7675–7682. doi:10.1021/acs.joc.7b01179
Return to citation in text: [1]
| 1. | Arjona, O.; Csáky, A. G.; Plumet, J. Eur. J. Org. Chem. 2003, 611–622. doi:10.1002/ejoc.200390100 |
| 2. | Holub, N.; Blechert, S. Chem. – Asian J. 2007, 2, 1064–1082. doi:10.1002/asia.200700072 |
| 3. | Bose, S.; Ghosh, S. Proc. Indian Natl. Sci. Acad. 2014, 80, 37–54. doi:10.16943/ptinsa/2014/v80i1/55085 |
| 4. | Kotha, S.; Meshram, M.; Khedkar, P.; Banerjee, S.; Deodhar, D. Beilstein J. Org. Chem. 2015, 11, 1833–1864. doi:10.3762/bjoc.11.199 |
| 38. | Banti, D.; North, M. Adv. Synth. Catal. 2002, 344, 694–704. |
| 39. | Banti, D.; Groaz, E.; North, M. Tetrahedron 2004, 60, 8043–8052. doi:10.1016/j.tet.2004.06.114 |
| 40. | Groaz, E.; Banti, D.; North, M. Eur. J. Org. Chem. 2007, 3727–3745. doi:10.1002/ejoc.200700291 |
| 34. | Villar, H.; Frings, M.; Bolm, C. Chem. Soc. Rev. 2007, 36, 55–66. doi:10.1039/B508899M |
| 35. | Diver, S. T.; Giessert, A. J. Chem. Rev. 2004, 104, 1317–1382. doi:10.1021/cr020009e |
| 36. | Mori, M. Adv. Synth. Catal. 2007, 349, 121–135. doi:10.1002/adsc.200600484 |
| 37. | Mori, M. Materials 2010, 3, 2087–2140. doi:10.3390/ma3032087 |
| 23. | Malik, C. K.; Ghosh, S. Org. Lett. 2007, 9, 2537–2540. doi:10.1021/ol070906a |
| 24. | Maity, S.; Ghosh, S. Tetrahedron Lett. 2008, 49, 1133–1136. doi:10.1016/j.tetlet.2007.12.064 |
| 25. | Mondal, S.; Malik, C. K.; Ghosh, S. Tetrahedron Lett. 2008, 49, 5649–5651. doi:10.1016/j.tetlet.2008.07.083 |
| 26. | Malik, C. K.; Yadav, R. N.; Drew, M. G. B.; Ghosh, S. J. Org. Chem. 2009, 74, 1957–1963. doi:10.1021/jo802077t |
| 27. | Malik, C. K.; Hossain, M. F.; Ghosh, S. Tetrahedron Lett. 2009, 50, 3063–3066. doi:10.1016/j.tetlet.2009.04.033 |
| 28. | Mondal, S.; Yadav, R. N.; Ghosh, S. Tetrahedron Lett. 2009, 50, 5277–5279. doi:10.1016/j.tetlet.2009.07.012 |
| 29. | Maity, S.; Ghosh, S. Tetrahedron 2009, 65, 9202–9210. doi:10.1016/j.tet.2009.09.029 |
| 30. | Matcha, K.; Maity, S.; Malik, C. K.; Ghosh, S. Tetrahedron Lett. 2010, 51, 2754–2757. doi:10.1016/j.tetlet.2010.03.074 |
| 31. | Yadav, R. N.; Mondal, S.; Ghosh, S. Tetrahedron Lett. 2011, 52, 1942–1945. doi:10.1016/j.tetlet.2011.02.054 |
| 32. | Bose, S.; Ghosh, M.; Ghosh, S. J. Org. Chem. 2012, 77, 6345–6350. doi:10.1021/jo300945b |
| 33. | Datta, R.; Bose, S.; Viththlbhai, P. B.; Ghosh, S. Tetrahedron Lett. 2014, 55, 3538–3540. doi:10.1016/j.tetlet.2014.04.091 |
| 49. | Datta, R.; Ghosh, S. J. Org. Chem. 2017, 82, 7675–7682. doi:10.1021/acs.joc.7b01179 |
| 5. | Stille, J. R.; Santarsiero, B. D.; Grubbs, R. H. J. Org. Chem. 1990, 55, 843–862. doi:10.1021/jo00290a013 |
| 6. | Zuercher, W. J.; Hashimoto, M.; Grubbs, R. H. J. Am. Chem. Soc. 1996, 118, 6634–6640. doi:10.1021/ja9606743 |
| 7. | Stragies, R.; Blechert, S. Synlett 1998, 169–170. doi:10.1055/s-1998-1592 |
| 8. | Arjona, O.; Csákÿ, A. G.; Murcia, M. C.; Plumet, J. Tetrahedron Lett. 2000, 41, 9777–9779. doi:10.1016/S0040-4039(00)01721-4 |
| 9. | Weatherhead, G. S.; Ford, J. G.; Alexanian, E. J.; Schrock, R. R.; Hoveyda, A. H. J. Am. Chem. Soc. 2000, 122, 1828–1829. doi:10.1021/ja993681a |
| 10. | Arjona, O.; Csákÿ, A. G.; Medel, R.; Plumet, J. J. Org. Chem. 2002, 67, 1380–1383. doi:10.1021/jo016000e |
| 11. | Sakurai, H.; Daiko, T.; Hirao, T. Science 2003, 301, 1878. doi:10.1126/science.1088290 |
| 12. | Arjona, O.; Csákÿ, A. G.; León, V.; Medel, R.; Plumet, J. Tetrahedron Lett. 2004, 45, 565–567. doi:10.1016/j.tetlet.2003.10.197 |
| 13. | Holtsclaw, J.; Koreeda, M. Org. Lett. 2004, 6, 3719–3722. doi:10.1021/ol048650l |
| 14. | Funel, J.-A.; Prunet, J. Synlett 2005, 235–238. doi:10.1055/s-2004-837200 |
| 15. | Chandler, C. L.; Phillips, A. J. Org. Lett. 2005, 7, 3493–3495. doi:10.1021/ol051199t |
| 16. | Maechling, S.; Norman, S. E.; Mckendrick, J. E.; Basra, S.; Köppner, K.; Blechert, S. Tetrahedron Lett. 2006, 47, 189–192. doi:10.1016/j.tetlet.2005.10.155 |
| 17. | Hart, A. C.; Phillips, A. J. J. Am. Chem. Soc. 2006, 128, 1094–1095. doi:10.1021/ja057899a |
| 18. | Phillips, A. J.; Hart, A. C.; Henderson, J. A. Tetrahedron Lett. 2006, 47, 3743–3745. doi:10.1016/j.tetlet.2006.03.124 |
| 19. | Calvet, G.; Blanchard, N.; Kouklovsky, C. Org. Lett. 2007, 9, 1485–1488. doi:10.1021/ol0702066 |
| 20. | Henderson, J. A.; Phillips, A. J. Angew. Chem., Int. Ed. 2008, 47, 8499–8501. doi:10.1002/anie.200803593 |
| 21. | Nguyen, N. N. M.; Leclère, M.; Stogaitis, N.; Fallis, A. G. Org. Lett. 2010, 12, 1684–1687. doi:10.1021/ol100150f |
| 22. | Lam, J. K.; Pham, H. V.; Houk, K. N.; Vanderwal, C. D. J. Am. Chem. Soc. 2013, 135, 17585–17594. doi:10.1021/ja409618p |
| 33. | Datta, R.; Bose, S.; Viththlbhai, P. B.; Ghosh, S. Tetrahedron Lett. 2014, 55, 3538–3540. doi:10.1016/j.tetlet.2014.04.091 |
| 43. | Spandl, R. J.; Rudyk, H.; Spring, D. R. Chem. Commun. 2008, 3001–3003. doi:10.1039/B807278G |
| 44. | Breitler, S.; Han, Y.; Corey, E. J. Org. Lett. 2017, 19, 6686–6687. doi:10.1021/acs.orglett.7b03412 |
| 45. | Wright, J.; Drtina, G. J.; Roberts, R. A.; Paquette, L. A. J. Am. Chem. Soc. 1988, 110, 5806–5817. doi:10.1021/ja00225a036 |
| 46. | Wender, P. A.; Singh, S. K. Tetrahedron Lett. 1990, 31, 2517–2520. doi:10.1016/0040-4039(90)80114-2 |
| 47. | Hudlicky, T.; Fleming, A.; Radesca, L. J. Am. Chem. Soc. 1989, 111, 6691–6707. doi:10.1021/ja00199a032 |
| 38. | Banti, D.; North, M. Adv. Synth. Catal. 2002, 344, 694–704. |
| 39. | Banti, D.; Groaz, E.; North, M. Tetrahedron 2004, 60, 8043–8052. doi:10.1016/j.tet.2004.06.114 |
| 40. | Groaz, E.; Banti, D.; North, M. Eur. J. Org. Chem. 2007, 3727–3745. doi:10.1002/ejoc.200700291 |
| 41. | Kotha, S.; Ravikumar, O. Tetrahedron Lett. 2014, 55, 5781–5784. doi:10.1016/j.tetlet.2014.08.108 |
| 42. | Kotha, S.; Ravikumar, O. Eur. J. Org. Chem. 2014, 5582–5590. doi:10.1002/ejoc.201402273 |
| 43. | Spandl, R. J.; Rudyk, H.; Spring, D. R. Chem. Commun. 2008, 3001–3003. doi:10.1039/B807278G |
| 43. | Spandl, R. J.; Rudyk, H.; Spring, D. R. Chem. Commun. 2008, 3001–3003. doi:10.1039/B807278G |
| 41. | Kotha, S.; Ravikumar, O. Tetrahedron Lett. 2014, 55, 5781–5784. doi:10.1016/j.tetlet.2014.08.108 |
| 42. | Kotha, S.; Ravikumar, O. Eur. J. Org. Chem. 2014, 5582–5590. doi:10.1002/ejoc.201402273 |
| 48. | CCDC 1847091 contains supplementary crystallographic data for the compound 13.These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
© 2018 Datta and Ghosh; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)