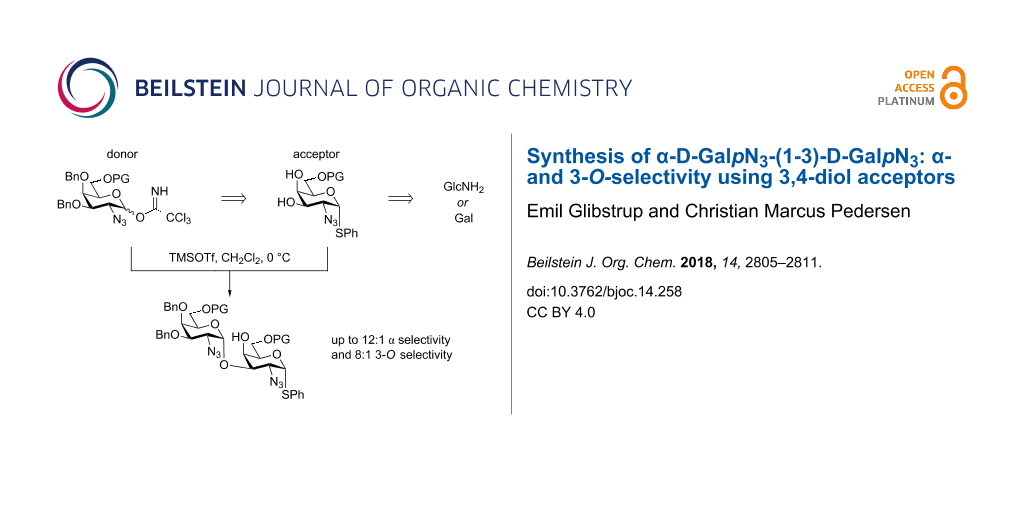
Abstract
The motif α-D-GalpNAc-(1-3)-D-GalpNAc is very common in Nature and hence its synthesis highly relevant. The synthesis of its azido precursor has been studied and optimized in terms of steps, yields and selectivity. It has been found that glycosylation of the 3,4-diol acceptor is an advantage over the use of a 4-O-protected acceptor and that both regio- and anomeric selectivity is enhanced by bulky 6-O-protective groups. The acceptors and donors are made from common building blocks, limiting protective manipulations, and in this context, unavoidable side reactions.

Graphical Abstract
Introduction
The disaccharide α-D-GalpNAc-(1-3)-β-D-GalpNAc is a widespread motif in glycobiology and present in several distinct entities. One of the most studied glycoconjugates, containing this sequence, is the Forssman antigen, in which this disaccharide is the terminal unit. The many biological roles attributed to this particular glycoconjugate and its appearance in some human tumors [1,2] has triggered the interest of carbohydrate chemists since the early days of complex oligosaccharides synthesis. The structure of the pentasaccharide part of the Forssman antigen was resolved in the seventies [3,4] and soon thereafter Paulsen and Bünsch finished the chemical synthesis of the pentasaccharide chain [5,6]. The pioneering work by Paulsen was later followed up by a total synthesis by the Ogawa group [7] and an oligosaccharide synthesis by the Magnusson group [8]. With the increasing understanding of glycobiology, the Forssman antigen has remained an interesting target for vaccine development [9] and further biological evaluations [10]. The disaccharide motif is also commonly found in viruses and bacteria. In bacteria, as an example, it has been found in pathogenic bacteria such as in Salmonella [11], Shigella [12], several Burkholderia [13], Escherichia coli [14], Vibrio chlorae [15], Edwardsiella ictaluri [16], Enterrococcus [17], Proteus mirabilis [18], and Streptococcus [19-22]. With the wide spread appearance of α-D-GalpNAc-(1-3)-β-D-GalpNAc in many pathogens, this motif has been synthesized many times for various purposes, e.g., as part of the lipopolysaccharide found in Shigella dysenteriae [23], as derivatives of the mucin O-glycan core structures for glycosidase studies [24], for the synthesis of T-antigen analogues [25], for the synthesis of E. coli O-antigens [26-29], for the development of Burkholderia vaccines [30-32], for the synthesis of PS A1 conjugate vaccine [33], and the synthesis of E. faecium wall teichoic acid fragments for vaccine development [34,35]. We have been interested in α-D-GalpNAc-(1-3)-β-D-GalpNAc as a part of our ongoing syntheses of S. pneumoniae lipoteichoic acid (LTA) derivatives [20,36,37]. During optimization of the synthesis, we realized that the synthesis of the α-D-GalpNAc-(1-3)-β-D-GalpNAc part worked more efficiently, when using the 3,4-diol acceptor instead of a fully protected 3-OH. As some protective group manipulations can be avoided and at the same time the yield and α-selectivity improved, we consider this finding important for oligosaccharide synthesis. Here we describe this regioselective glycosylation approach in detail.
Results and Discussion
In the initial strategy for synthesizing the disaccharide, the fully protected acceptor (1 or 2) was intended to be a key building block. However, an unexpected problem during its preparation occurred, i.e., benzylation of the axial 4-OH only gave the desired 4-O-Bn product as a minor product, whereas the benzoyl migration from the 3-O to the 4-O, followed by benzylation of the 3-O, was the major, if not the exclusive, product. Despite several different benzylation procedures, i.e., NaH and BnBr, TriBOT [38] and TfOH or BnBr and Ag2O, the 4-O-benzylation remained inaccessible, with a 3-O-benzoyl group present. Using the 6-O-Bn-protected variant (2 in Scheme 1), a 1:2 mixture of the desired vs migrated product could be obtained. When using the more bulky 6-OTBDPS as protective group (1 in Scheme 1) and benzylation with freshly prepared Ag2O and BnBr in the solvent mixture CH2Cl2/cyclohexane 1:4 as described by Wang et al. [39], a 91% isolated yield of the migrated product, i.e., 3-O-Bn, 4-O-Bz, was obtained (Scheme 1).
![[1860-5397-14-258-i1]](/bjoc/content/inline/1860-5397-14-258-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Undesired migration followed by benzylation of the 3-O-Bz GalN3 using several different benzylation procedures (LG: leaving group).
Scheme 1: Undesired migration followed by benzylation of the 3-O-Bz GalN3 using several different benzylation...
Following the apparent difference in nucleophilicity and accessibility of the axial 4-OH versus the equatorial 3-OH [40], we wondered, if this could be used to simplify the synthesis of the α-D-GalpN3-(1-3)-D-GalpN3 unit. When consulting the literature, surprisingly few reports were describing diol glycosylations targeting the galacto-configured sugar [41-43] and none for the synthesis of the α-D-GalpNAc-(1-3)-D-GalpNAc motif or its precursors.
We therefore set out to study this regioselective glycosylation in more detail and synthesized the necessary model donors and acceptors. Here, the simplicity of the approach showed its potential, as both, donor and acceptor, could be obtained from a common late stage building block, that in turn could be synthesized anomerically pure as described previously [44]. Alternatively, these components could be obtained via an azido phenylselenylation approach starting from D-galactose [45,46].
Starting from the common 2-azidoglucose precursor 7, the 4,6-O-benzylidene group was removed with TsOH to give the 4,6-diol in 95% yield, followed by selective 6-O-protection using TBDPS-Cl and imidazole (IM) in 90% yield. The 4,6-O-benzylidene motif could also be regioselectively opened using triethylsilane and BF3∙OEt2 to achieve the 6-O-Bn-protected variant in 78% yield (Scheme 2).
![[1860-5397-14-258-i2]](/bjoc/content/inline/1860-5397-14-258-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Simple synthesis of two acceptors and two donors from the same common and readily available building block.
Scheme 2: Simple synthesis of two acceptors and two donors from the same common and readily available buildin...
In order to invert the 4-OH, to achieve the galacto configuration, two approaches were attractive: one using our recently published method [44] or alternatively the Latrell–Dax inversion using nitrite [47,48]. When having the 6-O-TBDPS protection the Latrell–Dax inversion provided slightly higher yields of 1, whereas with the 6-O-Bn 8, the two methods provided comparable yields. Migration of the benzoyl group under the 4-O inversion conditions was irrelevant as it is removed in the following step. The proposed acceptors were obtained via Zemplén deprotection in 94% and 90% yield for the 6-O-TBDPS 9 and 6-O-Bn 10, respectively (Scheme 2). The acceptors could in turn be transformed into the corresponding donors in three simple steps by first benzylating the 3,4-diol (11 and 12), hydrolyzing the thiophenol using NBS (13 and 14), thereby making anomeric purity inconsequential, and finally formation of the donors, 15 and 16, as trichloroacetimidates [49]. With both, the donors and acceptors in hand, we set out to test our hypothesis of regioselective 3-O-α-glycosylation on the 3,4-diol 9, which is based on the superior nucleophilicity and accessibility of the 3-OH compared to the axial 4-OH. Initially, a small screening of the glycosylation conditions expected to affect the selectivity was performed.
As the donor 15 stems from the acceptor 9, it was decided to use a slight excess (1.2 equiv) of the “less precious” and recoverable acceptor 9, which, however, would result in lower yields when compared to glycosylations using excess of donor. In the initial experiment, using TMSOTf as the catalyst in CH2Cl2 at 0 °C, a pleasingly 8:1 3/4-O ratio and a 10:1 α/β-selectivity with a 57% isolated yield of the desired disaccharide 17, was obtained. Comparing the “normal procedure” with the “inverse procedure” [50], i.e., slow addition of donor 15 to acceptor 9 and catalyst, no discernable difference in the 3/4-O-regioselectivity or the α/β-ratio was observed, albeit a slightly lower isolated yield was obtained using the inverse procedure (Table 1, entry 1 vs entry 2). Changing the temperature, i.e., room temperature, 0 °C and −50 °C, had no effects on the 3/4-O-ratio, but resulted in a lower α/β-selectivity (5:1 at −50 °C; Table 1, entries 3 and 4) [51]. The reaction was also shown to be scalable giving comparable ratios and excellent isolated yields at both 1 mmol and 2.6 mmol scales (Table 1, entries 5 and 6). Increasing the excess of acceptor 9 to 1.7 equiv, showed no effect towards the ratios observed (Table 1, entry 6).
Table 1: Optimizing the α,3-O-selective glycosylation conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-258-i7.svg?max-width=637&scale=1.0)
|
|||||
| entry | scale [mmol] | temp. | 3/4-Oa | α/βa | isolated yields |
| 1 | 0.1 | 0 °C | 8:1 | 10:1 | 17 (57%) |
| 2c | 0.1 | 0 °C | 7:1 | 11:1 | 17 (47%) 18 (9%) |
| 3 | 0.1 | −50 °C | 7:1 | 5:1 | 17 (51%) 18 (10%) |
| 4 | 0.1 | rt | 7:1 | 10:1 | 17 (66%) 18 (11%) |
| 5 | 1.0 | 0 °C | 7:1 | 12:1 | 17 (67%) 18 (9%) |
| 6b | 2.6 | 0 °C | 8:1 | 11:1 | 17 (64%) 18 (9%) |
aDetermined by 1H NMR on the crude; b1.7 equiv acceptor; cinverse procedure.
With the conditions optimized, the selectivity was challenged using less bulky 6-O-protecting groups, such as a benzyl group, at either the donor, the acceptor or at both. With the 6-O-Bn donor 16 a slightly diminished, but still respectable, 49% yield of the desired product 19 was obtained (Scheme 3). However, when using the 6-O-TBDPS donor 15 (1.2 equiv) a 60% isolated yield of the desired disaccharide 20 was obtained. When using both the 6-O-Bn donor 10 and acceptor 16 we saw a lower selectivity than observed with the TBDPS–TBDPS variant 17, with an isolated yield of 48% of 21, but as an inseparable 1.7:1 mixture of the 3/4-O-glycosylated products. To put these results in to perspective, a few closely related glycosylations are shown in Scheme 4. Firstly, our own glycosylations using the 4-O-Bz variants, gave a lower yield, when using the TBDPS–TBDPS variant, of 36% [44]. The less sterically hindered Bn–TBDPS variant gave yields of 47%, which are comparable to the 3,4-diol glycosylation. Glycosylation with a closely related system having a 4-O-Bn, 6-O-TBDPS pattern gave 58% yield using 1.2 equiv of the donor, proving very comparable to our 60% yield for the same 4-O-Bn, 6-O-TBDPS system (Scheme 3) [34]. If utilizing the 3,4-diol glycosylation herein reported, the acceptor 10 can be synthesized in 4 less steps than otherwise required.
![[1860-5397-14-258-i3]](/bjoc/content/inline/1860-5397-14-258-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Challenging the α,3-O-selectivity with the different 6-O-protecting group variants.
Scheme 3: Challenging the α,3-O-selectivity with the different 6-O-protecting group variants.
![[1860-5397-14-258-i4]](/bjoc/content/inline/1860-5397-14-258-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Representative glycosylations with closely related systems [34,44].
Scheme 4: Representative glycosylations with closely related systems [34,44].
These results show, how the desired protecting group pattern can direct which glycosylation strategy to choose: In the less sterically hindered cases, a 4-O-protecting group, such as the benzoyl or benzyl, can be preferable. However, when a large 6-O-protective group, such as TBDPS, is used, it proves advantageous to do the glycosylation directly on the 3,4-diol.
Post-glycosylation modifications, i.e., benzylation of the free 4-OH on the TBDPS–TBDPS variant 17 using standard BnBr followed by NaH resulted in a 1:1 mixture of the desired product 22 and a side-product 23, in which the adjacent TBDPS has been cleaved off and the 6-OH subsequently benzylated (Scheme 5). This side reaction could, however, easily be avoided by using a procedure in which BnBr and TBAI was added and mixed before the addition of NaH. This modification gave a 91% yield of the product 22. Employing these conditions to the other three variants 24–26 allowed for much easier separation of the 3/4-O mixtures obtained from the glycosylations, especially for the Bn–Bn variant 26.
![[1860-5397-14-258-i5]](/bjoc/content/inline/1860-5397-14-258-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Capping the free 4-OH, allowing for easier separation of mixtures obtained during glycosylation.
Scheme 5: Capping the free 4-OH, allowing for easier separation of mixtures obtained during glycosylation.
Ultimately, we employed the strategy to synthesize a pseudotrisaccharide, found as the terminal part of the LTA from S. pneumoniae. The deprotection of disaccharide 22 to give 27 followed by formation of the trichloroacetimidate 28 proceeded smoothly, as shown in Scheme 6. The following glycosylation using MeCN as solvent relied on the nitrile effect to afford the desired β-anomer 29 in 74% yield.
![[1860-5397-14-258-i6]](/bjoc/content/inline/1860-5397-14-258-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Pseudotrisaccharide synthesis for LTA elucidation.
Scheme 6: Pseudotrisaccharide synthesis for LTA elucidation.
Conclusion
In conclusion we have shown, how the difference in nucleophilicity along with steric effects can be utilized to employ an unprecedented regioselective glycosylation on a 3,4-diol, resulting in up to 67% isolated yield. Following general guidelines can be given:
- With bulky 6-O-protective groups, a 4-O-protective group should be avoided, i.e., use the diol acceptor.
- Bulky 6-O-protective groups on the donor enhance the α-selectivity.
- Bulky 6-O-protective groups on either the donor or the acceptor enhance the 3-O-regioselectivity.
This simplification in synthesis allows for easier and faster access to these biologically abundant and relevant α-D-GalpNAc-(1-3)-D-GalpNAc motifs.
Supporting Information
| Supporting Information File 1: Full description of experimental procedures and characterization of new compounds. | ||
| Format: PDF | Size: 27.9 MB | Download |
References
-
Kawanami, J. J. Biochem. 1972, 72, 783–785. doi:10.1093/oxfordjournals.jbchem.a129960
Return to citation in text: [1] -
Ono, K.; Hattori, H.; Uemura, K.; Nakayama, J.; Ota, H.; Katsuyama, T. J. Histochem. Cytochem. 1994, 42, 659–665. doi:10.1177/42.5.7512587
Return to citation in text: [1] -
Siddiqui, B.; Hakomori, S.-I. J. Biol. Chem. 1971, 246, 5766–5769.
Return to citation in text: [1] -
Dabrowski, J.; Hanfland, P.; Egge, H. Biochemistry 1980, 19, 5652–5658. doi:10.1021/bi00565a030
Return to citation in text: [1] -
Paulsen, H.; Bünsch, A. Angew. Chem., Int. Ed. Engl. 1980, 19, 902–903. doi:10.1002/anie.198009021
Return to citation in text: [1] -
Paulsen, H.; Bünsch, A. Carbohydr. Res. 1982, 100, 143–167. doi:10.1016/S0008-6215(00)81032-0
Return to citation in text: [1] -
Nunomura, S.; Mori, M.; Ito, Y.; Ogawa, T. Tetrahedron Lett. 1989, 30, 6713–6716. doi:10.1016/S0040-4039(00)70658-7
Return to citation in text: [1] -
Nilsson, U.; Ray, A. K.; Magnusson, G. Carbohydr. Res. 1994, 252, 137–148. doi:10.1016/0008-6215(94)90011-6
Return to citation in text: [1] -
Feng, J.; Hevey, R.; Ling, C.-C. Carbohydr. Res. 2011, 346, 2650–2662. doi:10.1016/j.carres.2011.09.015
Return to citation in text: [1] -
Tanaka, H.; Takeuchi, R.; Jimbo, M.; Kuniya, N.; Takahashi, T. Chem. – Eur. J. 2013, 19, 3177–3187. doi:10.1002/chem.201203865
Return to citation in text: [1] -
Lüderitz, O. Angew. Chem., Int. Ed. Engl. 1970, 9, 649–663. doi:10.1002/anie.197006491
Return to citation in text: [1] -
Liu, B.; Knirel, Y. A.; Feng, L.; Perepelov, A. V.; Senchenkova, S. N.; Wang, Q.; Reeves, P. R.; Wang, L. FEMS Microbiol. Rev. 2008, 32, 627–653. doi:10.1111/j.1574-6976.2008.00114.x
Return to citation in text: [1] -
Ieranò, T.; Silipo, A.; Sturiale, L.; Garozzo, D.; Bryant, C.; Lanzetta, R.; Parrilli, M.; Aldridge, C.; Gould, F. K.; Corris, P. A.; Khan, C. M. A.; De Soyza, A.; Molinaro, A. Glycobiology 2009, 19, 1214–1223. doi:10.1093/glycob/cwp112
Return to citation in text: [1] -
Landersjo, C.; Widmalm, G. Biopolymers 2002, 64, 283–291. doi:10.1002/bip.10188
Return to citation in text: [1] -
Kocharova, N. A.; Knirel, Y. A.; Jansson, P.-E.; Weintraub, A. Carbohydr. Res. 2001, 332, 279–284. doi:10.1016/S0008-6215(01)00069-6
Return to citation in text: [1] -
Vinogradov, E.; Nossova, L.; Perry, M. B.; Kay, W. W. Carbohydr. Res. 2005, 340, 1509–1513. doi:10.1016/j.carres.2005.03.008
Return to citation in text: [1] -
Bychowska, A.; Theilacker, C.; Czerwicka, M.; Marszewska, K.; Huebner, J.; Holst, O.; Stepnowski, P.; Kaczyński, Z. Carbohydr. Res. 2011, 346, 2816–2819. doi:10.1016/j.carres.2011.09.026
Return to citation in text: [1] -
Perepelov, A. V.; Zabłotni, A.; Shashkov, A. S.; Knirel, Y. A.; Sidorczyk, Z. Carbohydr. Res. 2005, 340, 2305–2310. doi:10.1016/j.carres.2005.07.014
Return to citation in text: [1] -
Klein, R. A.; Hartmann, R.; Egge, H.; Behr, T.; Fischer, W. Carbohydr. Res. 1994, 256, 189–222. doi:10.1016/0008-6215(94)84209-4
Return to citation in text: [1] -
Gisch, N.; Kohler, T.; Ulmer, A. J.; Müthing, J.; Pribyl, T.; Fischer, K.; Lindner, B.; Hammerschmidt, S.; Zähringer, U. J. Biol. Chem. 2013, 288, 15654–15667. doi:10.1074/jbc.M112.446963
Return to citation in text: [1] [2] -
Gisch, N.; Schwudke, D.; Thomsen, S.; Heß, N.; Hakenbeck, R.; Denapaite, D. Sci. Rep. 2015, 5, No. 16718. doi:10.1038/srep16718
Return to citation in text: [1] -
Neiwert, O.; Holst, O.; Duda, K. A. Carbohydr. Res. 2014, 389, 192–195. doi:10.1016/j.carres.2013.12.018
Return to citation in text: [1] -
Paulsen, H.; Bünsch, H. Chem. Ber. 1981, 114, 3126–3145. doi:10.1002/cber.19811140918
Return to citation in text: [1] -
Hollinger, M.; Abraha, F.; Oscarson, S. Carbohydr. Res. 2011, 346, 1454–1466. doi:10.1016/j.carres.2011.03.036
Return to citation in text: [1] -
Sato, T.; Imamura, A.; Ando, H.; Ishida, H.; Kiso, M. Glycoconjugate J. 2009, 26, 83–98. doi:10.1007/s10719-008-9168-y
Return to citation in text: [1] -
Panchadhayee, R.; Misra, A. K. J. Carbohydr. Chem. 2010, 29, 39–50. doi:10.1080/07328300903477804
Return to citation in text: [1] -
Santra, A.; Ghosh, T.; Misra, A. K. Tetrahedron: Asymmetry 2012, 23, 1385–1392. doi:10.1016/j.tetasy.2012.08.006
Return to citation in text: [1] -
Ingle, A. B.; Chao, C.-S.; Hung, W.-C.; Mong, K.-K. T. Asian J. Org. Chem. 2014, 3, 870–876. doi:10.1002/ajoc.201402057
Return to citation in text: [1] -
Si, A.; Misra, A. K. Synthesis 2015, 47, 83–88. doi:10.1055/s-0034-1378911
Return to citation in text: [1] -
Bedini, E.; Cirillo, L.; Parrilli, M. Carbohydr. Res. 2012, 349, 24–32. doi:10.1016/j.carres.2011.12.007
Return to citation in text: [1] -
Ziaco, M.; De Castro, C.; Silipo, A.; Corsaro, M. M.; Molinaro, A.; Iadonisi, A.; Lanzetta, R.; Parrilli, M.; Bedini, E. Carbohydr. Res. 2015, 403, 182–191. doi:10.1016/j.carres.2014.04.018
Return to citation in text: [1] -
Bi, N.; Xiong, C.; Jin, G.; Guo, Z.; Gu, G. Carbohydr. Res. 2017, 451, 1–11. doi:10.1016/j.carres.2017.09.001
Return to citation in text: [1] -
Ghosh, S.; Nishat, S.; Andreana, P. R. J. Org. Chem. 2016, 81, 4475–4484. doi:10.1021/acs.joc.6b00195
Return to citation in text: [1] -
van der Es, D.; Groenia, N. A.; Laverde, D.; Overkleeft, H. S.; Huebner, J.; van der Marel, G. A.; Codée, J. D. C. Bioorg. Med. Chem. 2016, 24, 3893–3907. doi:10.1016/j.bmc.2016.03.019
Return to citation in text: [1] [2] [3] -
Zhou, Z.; Ding, W.; Li, C.; Wu, Z. J. Carbohydr. Chem. 2017, 36, 205–219. doi:10.1080/07328303.2017.1390576
Return to citation in text: [1] -
Pedersen, C. M.; Figueroa-Perez, I.; Lindner, B.; Ulmer, A. J.; Zähringer, U.; Schmidt, R. R. Angew. Chem., Int. Ed. 2010, 49, 2585–2590. doi:10.1002/anie.200906163
Return to citation in text: [1] -
Pedersen, C. M.; Figueroa-Perez, I.; Ulmer, A. J.; Zähringer, U.; Schmidt, R. R. Tetrahedron 2012, 68, 1052–1061. doi:10.1016/j.tet.2011.11.088
Return to citation in text: [1] -
Yamada, K.; Fujita, H.; Kunishima, M. Org. Lett. 2012, 14, 5026–5029. doi:10.1021/ol302222p
Return to citation in text: [1] -
Wang, L.; Hashidoko, Y.; Hashimoto, M. J. Org. Chem. 2016, 81, 4464–4474. doi:10.1021/acs.joc.6b00144
Return to citation in text: [1] -
Pedersen, C. M.; Olsen, J.; Brka, A. B.; Bols, M. Chem. – Eur. J. 2011, 17, 7080–7086. doi:10.1002/chem.201100020
Return to citation in text: [1] -
Geiger, J.; Reddy, B. G.; Winterfeld, G. A.; Weber, R.; Przybylski, M.; Schmidt, R. R. J. Org. Chem. 2007, 72, 4367–4377. doi:10.1021/jo061670b
Return to citation in text: [1] -
Cheshev, P. E.; Kononov, L. O.; Tsvetkov, Y. E.; Shashkov, A. S.; Nifantiev, N. E. Russ. J. Bioorg. Chem. 2002, 28, 419–429. doi:10.1023/A:1020428329743
Return to citation in text: [1] -
Hsu, Y.; Lu, X.-A.; Zulueta, M. M. L.; Tsai, C.-M.; Lin, K.-I.; Hung, S.-C.; Wong, C.-H. J. Am. Chem. Soc. 2012, 134, 4549–4552. doi:10.1021/ja300284x
Return to citation in text: [1] -
Glibstrup, E.; Pedersen, C. M. Org. Lett. 2016, 18, 4424–4427. doi:10.1021/acs.orglett.6b02241
Return to citation in text: [1] [2] [3] [4] -
Czernecki, S.; Ayadi, E.; Randriamandimby, D. J. Chem. Soc., Chem. Commun. 1994, 35–36. doi:10.1039/c39940000035
Return to citation in text: [1] -
Mironov, Y. V.; Sherman, A. A.; Nifantiev, N. E. Tetrahedron Lett. 2004, 45, 9107–9110. doi:10.1016/j.tetlet.2004.10.022
Return to citation in text: [1] -
Hederos, M.; Konradsson, P. J. Carbohydr. Chem. 2005, 24, 297–320. doi:10.1081/CAR-200061584
Return to citation in text: [1] -
Emmadi, M.; Kulkarni, S. S. J. Org. Chem. 2011, 76, 4703–4709. doi:10.1021/jo200342v
Return to citation in text: [1] -
Schmidt, R. R.; Michel, J. Angew. Chem., Int. Ed. Engl. 1980, 19, 731–732. doi:10.1002/anie.198007311
Return to citation in text: [1] -
Schmidt, R. R.; Toepfer, A. Tetrahedron Lett. 1991, 32, 3353–3356. doi:10.1016/S0040-4039(00)92704-7
Return to citation in text: [1] -
Kalikanda, J.; Li, Z. J. Org. Chem. 2011, 76, 5207–5218. doi:10.1021/jo1025157
Return to citation in text: [1]
| 41. | Geiger, J.; Reddy, B. G.; Winterfeld, G. A.; Weber, R.; Przybylski, M.; Schmidt, R. R. J. Org. Chem. 2007, 72, 4367–4377. doi:10.1021/jo061670b |
| 42. | Cheshev, P. E.; Kononov, L. O.; Tsvetkov, Y. E.; Shashkov, A. S.; Nifantiev, N. E. Russ. J. Bioorg. Chem. 2002, 28, 419–429. doi:10.1023/A:1020428329743 |
| 43. | Hsu, Y.; Lu, X.-A.; Zulueta, M. M. L.; Tsai, C.-M.; Lin, K.-I.; Hung, S.-C.; Wong, C.-H. J. Am. Chem. Soc. 2012, 134, 4549–4552. doi:10.1021/ja300284x |
| 44. | Glibstrup, E.; Pedersen, C. M. Org. Lett. 2016, 18, 4424–4427. doi:10.1021/acs.orglett.6b02241 |
| 45. | Czernecki, S.; Ayadi, E.; Randriamandimby, D. J. Chem. Soc., Chem. Commun. 1994, 35–36. doi:10.1039/c39940000035 |
| 46. | Mironov, Y. V.; Sherman, A. A.; Nifantiev, N. E. Tetrahedron Lett. 2004, 45, 9107–9110. doi:10.1016/j.tetlet.2004.10.022 |
| 1. | Kawanami, J. J. Biochem. 1972, 72, 783–785. doi:10.1093/oxfordjournals.jbchem.a129960 |
| 2. | Ono, K.; Hattori, H.; Uemura, K.; Nakayama, J.; Ota, H.; Katsuyama, T. J. Histochem. Cytochem. 1994, 42, 659–665. doi:10.1177/42.5.7512587 |
| 8. | Nilsson, U.; Ray, A. K.; Magnusson, G. Carbohydr. Res. 1994, 252, 137–148. doi:10.1016/0008-6215(94)90011-6 |
| 18. | Perepelov, A. V.; Zabłotni, A.; Shashkov, A. S.; Knirel, Y. A.; Sidorczyk, Z. Carbohydr. Res. 2005, 340, 2305–2310. doi:10.1016/j.carres.2005.07.014 |
| 34. | van der Es, D.; Groenia, N. A.; Laverde, D.; Overkleeft, H. S.; Huebner, J.; van der Marel, G. A.; Codée, J. D. C. Bioorg. Med. Chem. 2016, 24, 3893–3907. doi:10.1016/j.bmc.2016.03.019 |
| 7. | Nunomura, S.; Mori, M.; Ito, Y.; Ogawa, T. Tetrahedron Lett. 1989, 30, 6713–6716. doi:10.1016/S0040-4039(00)70658-7 |
| 19. | Klein, R. A.; Hartmann, R.; Egge, H.; Behr, T.; Fischer, W. Carbohydr. Res. 1994, 256, 189–222. doi:10.1016/0008-6215(94)84209-4 |
| 20. | Gisch, N.; Kohler, T.; Ulmer, A. J.; Müthing, J.; Pribyl, T.; Fischer, K.; Lindner, B.; Hammerschmidt, S.; Zähringer, U. J. Biol. Chem. 2013, 288, 15654–15667. doi:10.1074/jbc.M112.446963 |
| 21. | Gisch, N.; Schwudke, D.; Thomsen, S.; Heß, N.; Hakenbeck, R.; Denapaite, D. Sci. Rep. 2015, 5, No. 16718. doi:10.1038/srep16718 |
| 22. | Neiwert, O.; Holst, O.; Duda, K. A. Carbohydr. Res. 2014, 389, 192–195. doi:10.1016/j.carres.2013.12.018 |
| 34. | van der Es, D.; Groenia, N. A.; Laverde, D.; Overkleeft, H. S.; Huebner, J.; van der Marel, G. A.; Codée, J. D. C. Bioorg. Med. Chem. 2016, 24, 3893–3907. doi:10.1016/j.bmc.2016.03.019 |
| 44. | Glibstrup, E.; Pedersen, C. M. Org. Lett. 2016, 18, 4424–4427. doi:10.1021/acs.orglett.6b02241 |
| 5. | Paulsen, H.; Bünsch, A. Angew. Chem., Int. Ed. Engl. 1980, 19, 902–903. doi:10.1002/anie.198009021 |
| 6. | Paulsen, H.; Bünsch, A. Carbohydr. Res. 1982, 100, 143–167. doi:10.1016/S0008-6215(00)81032-0 |
| 16. | Vinogradov, E.; Nossova, L.; Perry, M. B.; Kay, W. W. Carbohydr. Res. 2005, 340, 1509–1513. doi:10.1016/j.carres.2005.03.008 |
| 51. | Kalikanda, J.; Li, Z. J. Org. Chem. 2011, 76, 5207–5218. doi:10.1021/jo1025157 |
| 3. | Siddiqui, B.; Hakomori, S.-I. J. Biol. Chem. 1971, 246, 5766–5769. |
| 4. | Dabrowski, J.; Hanfland, P.; Egge, H. Biochemistry 1980, 19, 5652–5658. doi:10.1021/bi00565a030 |
| 17. | Bychowska, A.; Theilacker, C.; Czerwicka, M.; Marszewska, K.; Huebner, J.; Holst, O.; Stepnowski, P.; Kaczyński, Z. Carbohydr. Res. 2011, 346, 2816–2819. doi:10.1016/j.carres.2011.09.026 |
| 44. | Glibstrup, E.; Pedersen, C. M. Org. Lett. 2016, 18, 4424–4427. doi:10.1021/acs.orglett.6b02241 |
| 12. | Liu, B.; Knirel, Y. A.; Feng, L.; Perepelov, A. V.; Senchenkova, S. N.; Wang, Q.; Reeves, P. R.; Wang, L. FEMS Microbiol. Rev. 2008, 32, 627–653. doi:10.1111/j.1574-6976.2008.00114.x |
| 14. | Landersjo, C.; Widmalm, G. Biopolymers 2002, 64, 283–291. doi:10.1002/bip.10188 |
| 49. | Schmidt, R. R.; Michel, J. Angew. Chem., Int. Ed. Engl. 1980, 19, 731–732. doi:10.1002/anie.198007311 |
| 11. | Lüderitz, O. Angew. Chem., Int. Ed. Engl. 1970, 9, 649–663. doi:10.1002/anie.197006491 |
| 15. | Kocharova, N. A.; Knirel, Y. A.; Jansson, P.-E.; Weintraub, A. Carbohydr. Res. 2001, 332, 279–284. doi:10.1016/S0008-6215(01)00069-6 |
| 50. | Schmidt, R. R.; Toepfer, A. Tetrahedron Lett. 1991, 32, 3353–3356. doi:10.1016/S0040-4039(00)92704-7 |
| 10. | Tanaka, H.; Takeuchi, R.; Jimbo, M.; Kuniya, N.; Takahashi, T. Chem. – Eur. J. 2013, 19, 3177–3187. doi:10.1002/chem.201203865 |
| 44. | Glibstrup, E.; Pedersen, C. M. Org. Lett. 2016, 18, 4424–4427. doi:10.1021/acs.orglett.6b02241 |
| 9. | Feng, J.; Hevey, R.; Ling, C.-C. Carbohydr. Res. 2011, 346, 2650–2662. doi:10.1016/j.carres.2011.09.015 |
| 13. | Ieranò, T.; Silipo, A.; Sturiale, L.; Garozzo, D.; Bryant, C.; Lanzetta, R.; Parrilli, M.; Aldridge, C.; Gould, F. K.; Corris, P. A.; Khan, C. M. A.; De Soyza, A.; Molinaro, A. Glycobiology 2009, 19, 1214–1223. doi:10.1093/glycob/cwp112 |
| 47. | Hederos, M.; Konradsson, P. J. Carbohydr. Chem. 2005, 24, 297–320. doi:10.1081/CAR-200061584 |
| 48. | Emmadi, M.; Kulkarni, S. S. J. Org. Chem. 2011, 76, 4703–4709. doi:10.1021/jo200342v |
| 25. | Sato, T.; Imamura, A.; Ando, H.; Ishida, H.; Kiso, M. Glycoconjugate J. 2009, 26, 83–98. doi:10.1007/s10719-008-9168-y |
| 23. | Paulsen, H.; Bünsch, H. Chem. Ber. 1981, 114, 3126–3145. doi:10.1002/cber.19811140918 |
| 24. | Hollinger, M.; Abraha, F.; Oscarson, S. Carbohydr. Res. 2011, 346, 1454–1466. doi:10.1016/j.carres.2011.03.036 |
| 39. | Wang, L.; Hashidoko, Y.; Hashimoto, M. J. Org. Chem. 2016, 81, 4464–4474. doi:10.1021/acs.joc.6b00144 |
| 40. | Pedersen, C. M.; Olsen, J.; Brka, A. B.; Bols, M. Chem. – Eur. J. 2011, 17, 7080–7086. doi:10.1002/chem.201100020 |
| 20. | Gisch, N.; Kohler, T.; Ulmer, A. J.; Müthing, J.; Pribyl, T.; Fischer, K.; Lindner, B.; Hammerschmidt, S.; Zähringer, U. J. Biol. Chem. 2013, 288, 15654–15667. doi:10.1074/jbc.M112.446963 |
| 36. | Pedersen, C. M.; Figueroa-Perez, I.; Lindner, B.; Ulmer, A. J.; Zähringer, U.; Schmidt, R. R. Angew. Chem., Int. Ed. 2010, 49, 2585–2590. doi:10.1002/anie.200906163 |
| 37. | Pedersen, C. M.; Figueroa-Perez, I.; Ulmer, A. J.; Zähringer, U.; Schmidt, R. R. Tetrahedron 2012, 68, 1052–1061. doi:10.1016/j.tet.2011.11.088 |
| 38. | Yamada, K.; Fujita, H.; Kunishima, M. Org. Lett. 2012, 14, 5026–5029. doi:10.1021/ol302222p |
| 33. | Ghosh, S.; Nishat, S.; Andreana, P. R. J. Org. Chem. 2016, 81, 4475–4484. doi:10.1021/acs.joc.6b00195 |
| 34. | van der Es, D.; Groenia, N. A.; Laverde, D.; Overkleeft, H. S.; Huebner, J.; van der Marel, G. A.; Codée, J. D. C. Bioorg. Med. Chem. 2016, 24, 3893–3907. doi:10.1016/j.bmc.2016.03.019 |
| 35. | Zhou, Z.; Ding, W.; Li, C.; Wu, Z. J. Carbohydr. Chem. 2017, 36, 205–219. doi:10.1080/07328303.2017.1390576 |
| 26. | Panchadhayee, R.; Misra, A. K. J. Carbohydr. Chem. 2010, 29, 39–50. doi:10.1080/07328300903477804 |
| 27. | Santra, A.; Ghosh, T.; Misra, A. K. Tetrahedron: Asymmetry 2012, 23, 1385–1392. doi:10.1016/j.tetasy.2012.08.006 |
| 28. | Ingle, A. B.; Chao, C.-S.; Hung, W.-C.; Mong, K.-K. T. Asian J. Org. Chem. 2014, 3, 870–876. doi:10.1002/ajoc.201402057 |
| 29. | Si, A.; Misra, A. K. Synthesis 2015, 47, 83–88. doi:10.1055/s-0034-1378911 |
| 30. | Bedini, E.; Cirillo, L.; Parrilli, M. Carbohydr. Res. 2012, 349, 24–32. doi:10.1016/j.carres.2011.12.007 |
| 31. | Ziaco, M.; De Castro, C.; Silipo, A.; Corsaro, M. M.; Molinaro, A.; Iadonisi, A.; Lanzetta, R.; Parrilli, M.; Bedini, E. Carbohydr. Res. 2015, 403, 182–191. doi:10.1016/j.carres.2014.04.018 |
| 32. | Bi, N.; Xiong, C.; Jin, G.; Guo, Z.; Gu, G. Carbohydr. Res. 2017, 451, 1–11. doi:10.1016/j.carres.2017.09.001 |
© 2018 Glibstrup and Pedersen; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)