1Unité de Chimie et Biocatalyse, Département de Biologie Structurale et Chimie, Institut Pasteur, 28 rue du Dr. Roux, 75724 Paris Cedex 15, France
2Unité Mixte de Recherche 3523, Centre National de la Recherche Scientifique, 28 rue du Dr. Roux, 75724 Paris Cedex 15, France
3Université Paris Descartes, Sorbonne Paris Cité, 12 rue de l'École de Médecine, 75006 Paris, France
Corresponding author email
Associate Editor: B. Stoltz Beilstein J. Org. Chem.2018,14, 2846–2852.https://doi.org/10.3762/bjoc.14.263 Received 09 Aug 2018,
Accepted 26 Oct 2018,
Published 15 Nov 2018
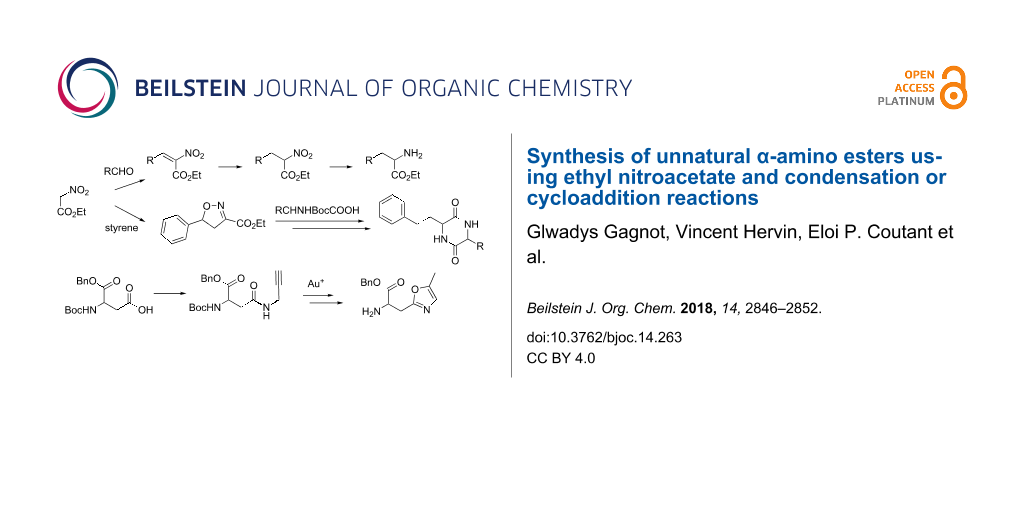
We report here on the use of ethyl nitroacetate as a glycine template to produce α-amino esters. This started with a study of its condensation with various arylacetals to give ethyl 3-aryl-2-nitroacrylates followed by a reduction (NaBH4 and then zinc/HCl) into α-amino esters. The scope of this method was explored as well as an alternative with arylacylals instead. We also focused on various [2 + 3] cycloadditions, one leading to a spiroacetal, which led to the undesired ethyl 5-(benzamidomethyl)isoxazole-3-carboxylate. The addition of ethyl nitroacetate on a 5-methylene-4,5-dihydrooxazole using cerium(IV) ammonium nitrate was also explored and the synthesis of other oxazole-bearing α-amino esters was achieved using gold(I) chemistry.
In the course of our work on an original synthesis of imidazo[1,2-a]pyrazin-3(7H)-one luciferins [1], a large variety of racemic α-amino esters was required to prepare a corresponding array of analogues. As we reviewed recently, nitroacetates are amongst the principal glycine templates used to prepare α-amino esters 1[2]. The retrosynthetic pathway depicted in Scheme 1 requires a reduction of ethyl nitroacrylates 2, which are made from condensation reactions between aldehydes 3 or acetals 5 and ethyl nitroacetate (4). However, the high yield condensations with aldehydes 3 which were reported for some examples [3,4] require more than stoichiometric amounts of titanium tetrachloride thus leading to considerable amounts of metal-containing chemical waste. Moreover, far lower yields were reported in many other instances when using this reagent [5,6]. In an attempt to improve the generality of this synthetic pathway and diminish its requirement for metals, we studied some alternatives, such as the condensations between ethyl nitroacetate (4) and acetals 5 or other approaches further described in the following.
Scheme 1:
α-Amino esters from ethyl nitroacetate (4).
Scheme 1:
α-Amino esters from ethyl nitroacetate (4).
As depicted in Table 1, we first studied the scope of the condensation between ethyl nitroacetate (4) and aryldimethylacetals 5a–s which are easily obtained in situ from the corresponding arylaldehydes 3a–s. As seen by 1H NMR monitoring, the treatment of arylaldehydes 3a–s with trimethyl orthoformate and an acid-bearing resin in dry methanol led to full conversion into the corresponding acetals 5a–s. Then, as reported [7,8], heating the crude acetals 5a–s and ethyl nitroacetate (4) in the presence of acetic anhydride afforded a mixture of compounds containing variable amounts of the expected acrylates 2a–s. No attempts were made to purify these as they were immediately subjected to a reduction of its double bond using sodium borohydride [9,10] in refluxing isopropanol in order to properly isolate the corresponding α-nitro esters 6a–s. Isopropanol was used instead of ethanol in order to decrease the incidence of a recurrent side product arising from a decarboxylation or a retro condensation of the partially reduced ester function (this side product was characterized in 1H NMR by two triplets at 4.6 ppm and 3.4 ppm, but eluded our purification efforts). Moreover, one minute in refluxing isopropanol was found to be sufficient to complete the reduction especially for the fairly insoluble acrylate 2e, and also avoided most of the undesired transesterification byproducts that could form upon long reaction times. As seen in Table 1, modest yields of compounds 6a–i were isolated anyway, especially for meta-substituted phenyl-bearing products 6c, 6f and 6h. Even if the reduction was less than perfect, the low yields originate from the initial condensation between acetals 5 and 4. Indeed, when considering the phenyl derivative of 6a, a sobering 35% isolated yield was obtained, in stark contrast to the reported 95% yield published in 1980 [7]. In our hands, 1H NMR analysis of the crude condensation product pointed out the occurrence of the intermediate acrylate 2a along with a large amount of methyl benzoate, a well-known side product in this reaction [7,8]. From the aryl acetals 5g–l featuring electron-withdrawing groups, low to non-detectable amounts of the condensations products 2g–l were observed by 1H NMR analysis, and this was reflected in the isolated yield of the corresponding α-nitroesters 6g–i as well as the lack of compounds 6j–l. Similarly, no condensation was observed when starting with the pyridyl-bearing acetal 5m. These disappointing results are plausibly due to two factors: (i) As mentioned above, the condensation of ethyl nitroacetate (4) with aryl acetals 5 takes place along with an O-alkylation reaction which leads, via a rearrangement, to the corresponding aryl ester byproduct [7,8]. The proportion between the C-alkylation (leading to the expected acrylate) and this O-alkylation is most certainly governed by the electronic effects of the substituent on the aryl group. Indeed, the best overall yields are observed when starting from the electronically similar 2-methoxy acetal 5b or the 4-methoxy analog 5d, as well as the 4-benzyloxy acetal 5e. (ii) Secondly, the reduction of acrylates 2a–s to compounds 6a–s was achieved with sodium borohydride and the resulting basic conditions could be detrimental to the stability of some of these α-nitro acrylates. In the past, such reductions have been achieved under fairly uncommon conditions (NaBH4 in a mixture of isopropanol and chloroform over a large proportion of silica gel) [11,12]. However, when tried, no real overall improvements were observed with these conditions. Other series of trials were made to improve the overall yields of the furan-bearing α-nitro esters 6n–r. We first tried to avoid the preparation of the acetals 5n or 5o and used the previously reported direct condensation between furfural (3n) and ethyl nitroacetate (4) [13]. Unfortunately, we could not reproduce the 95% yield reported for compound 2n, and under a thoroughly inert atmosphere (as advised) we obtained 48–53% isolated yields at best. In any case, this approach did shorten the synthetic pathway by one step and upon the reduction of the resulting acrylates 2n or 2o using sodium borohydride, the α-nitro esters 6n and 6o were isolated in the rather modest yields indicated in Table 1. We then focused on the model reduction of acrylate 2n into 6n. Since all our attempts to achieve a palladium-catalyzed hydrogenation failed, we tried other borohydride salts. The use of tetramethylammonium borohydride did not increase the overall yield of 6n, however, a rather substantial improvement was observed when using sodium cyanoborohydride. Indeed, from pure acrylate 2n, a 70% yield of 6n was obtained, and in one pot starting from aldehyde 3n, a 48% overall yield of 6n was achieved. Moreover, without using an inert atmosphere for the initial condensation between furfural (3n) and ethyl nitroacetate (4) the overall yield of 6n dropped to 37% even when using sodium cyanoborohydride. The optimized conditions were then applied to aldehyde 3r and afforded a significantly improved 46% yield of 6r, in comparison with our single trial via 5r, which ended up with less than 20% of an impure sample of 6r. Finally, from the isolated α-nitro esters 6a–s, their reduction into the corresponding α-amino esters 1a–s, using zinc and hydrochloric acid in ethanol usually proceeded in good yield, although care had to be taken during work-up as zinc complexes required the addition of an excess of ammonia to fully break in the course of the extraction.
Table 1:
Synthesis and reductions of α-nitro acrylates into α-amino esters.
Ar
% 6a
% 6b
% 1
a
C6H5
35c
–
b
2-MeOC6H4
53
90
c
3-MeOC6H4
21
92
d
4-MeOC6H4
55
95
e
4-BnOC6H4
51
67
f
3-MeC6H4
16
80
g
2-FC6H4
6
94
h
3-FC6H4
0.9
–
i
4-FC6H4
13
94
j
2-CF3C6H4
0d
–
k
3-CF3C6H4
<5d
–
l
4-CF3C6H4
<5d
–
m
2-pyridyl
0d
–
n
furan-2-yl
27
38/48e/70f
94
o
furan-3-yl
–
34
88
p
5-methyl-furan-2-yl
60
–
75
q
5-ethyl-furan-2-yl
39
–
56
r
4,5-dimethyl-furan-2-yl
<20
46e
79
s
thiophen-2-yl
33
–
58
aIsolated yield from 3a–s, via acetals 5a–s. bIsolated yield via the direct condensation between 3a–s and 4. c39% yield from (diethoxymethyl)benzene. dAs seen by 1H NMR analysis. eUsing NaBH3CN; see text. fAlso using NaBH3CN but from pure 2n; see text.
In an attempt to overcome the lack of condensation between ethyl nitroacetate (4) and electron-poor substrates 5j–l, we focused on the model preparation of the trifluoromethyl-bearing α-nitro ester 6j from acylals 7 depicted in Scheme 2. As well reviewed [14], acylals can be prepared from aldehydes and anhydrides using a variety of acids as catalysts. In our case, 1H NMR monitoring of the reaction between 2-(trifluoromethyl)benzaldehyde (3j) and acetic anhydride using indium(III) chloride [15] as a Lewis acid catalyst [16] without any solvent at room temperature pointed out a complete conversion into acylal 7 overnight. A similar reaction using pivaloyl anhydride and either indium(III) chloride or tetrafluoroboric acid as a catalyst had to be heated at 60 °C for a few hours to secure a similar conversion into pivalal 8. From intermediates 7 and 8, and as previously reported in the case of malonates [17-19], we then hoped for an improvement of their condensation with ethyl nitroacetate (4). The 1H NMR monitoring of the reaction between compounds 7 or 8 and ethyl nitroacetate (4) in the presence of a catalytic amount of indium(III) chloride pointed out the occurrence of tangibly more of the expected acrylate 2j, although along with many byproducts. Indeed, in a typical experiment, upon reduction of the crude reaction product obtained from 8 and ethyl nitroacetate (4), a discouraging 13% yield of the corresponding α-nitro ester 6j was isolated. The use of α-nitro esters to obtain disubstituted α-amino esters such as compound 10 via an alkylation step has been described [20]. In order to reach such α-amino esters, we tried their preparation via a C-methylation of α-nitroester 6a in DMF using sodium hydride and methyl iodide. Upon purification, this gave 46% of the nitro compound 9 with 94% purity (as assessed by 1H NMR). Despite this modest yield, the ensuing reduction using zinc and hydrochloric acid in ethanol overnight gave a sufficient amount of the (pure) target phenyl-bearing α-amino ester 10 which had been previously obtained by catalytic hydrogenation using palladium [20]. The same transformation sequences were used starting with compound 6n and provided the furan-bearing α-amino ester 12 in 32% overall yield via the nitro compound 11. We also investigated the reported [4] 1,4-addition of a methyl on compound 2n to prepare the β-methylated derivative 13. In our hands, a rather modest 43% yield of the expected adduct 13 was achieved from purified acrylate 2n. Again, the ensuing reduction of 13 using zinc and hydrochloric acid gave the target α-amino ester 14 in an 85% yield.
Scheme 2:
Preparations of α-amino esters 10, 12 and 14.
Scheme 2:
Preparations of α-amino esters 10, 12 and 14.
To avoid the recourse to more rare and/or expensive heterocyclic aldehydes, we also tried synthetic approaches based on [2 + 3] cycloadditions. As depicted in Scheme 3, the carbon dioxide-producing reaction [21] between two equivalents of ethyl nitroacetate (4) and styrene (15), gave the isoxazoline 16 in a 72% yield as a latent α-amino ester [22-24]. From this compound, a reductive cleavage of the isoxazoline ring was initiated using palladium over charcoal and a large excess of ammonium formate in refluxing ethanol. The analysis of the resulting mixture by LC/MS and 1H NMR pointed out the occurrence of the expected [25] oxime 17 but along with an unexpected sizable proportion of the α-amino ester 18. Accordingly, the (filtrated) ethanolic solution was then treated with zinc and hydrochloric acid in ethanol to complete the reduction and the amino ester 18 was isolated in a 72% yield. In order to illustrate the synthetic potential of the isoxazoline 16 as an already protected amino acid moiety, we prepared the piperazine-2,5-diones 23a,b in four steps. This was achieved by the hydrolysis of the ester function of 16, followed by its coupling with glycine or phenylalanine ethyl esters (respectively 20a and 20b) using 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate (TBTU) as a coupling agent to give the corresponding amides 21a,b. The isoxazole ring of these compounds was then cleaved using palladium and ammonium formate to give the corresponding oximes which were immediately reduced into amines 22a,b, using zinc and hydrochloric acid, and a thermal cyclization of these crude products gave the piperazine-2,5-diones 23a,b in, respectively, 31 and 40% overall yield from compound 16.
Scheme 3:
Syntheses of α-amino ester 18 and piperazinediones 23a,b.
Scheme 3:
Syntheses of α-amino ester 18 and piperazinediones 23a,b.
A second [2 + 3] cycloaddition-based approach is described in Scheme 4. It started with the preparation of the methylene-bearing dipolarophile 25 from propargylamide 24 using gold(I) chemistry, which turned out to be tolerant to a wide variety of dry solvents (dichloromethane, tetrahydrofuran, toluene, dimethylformamide, or acetonitrile) [26,27]. As for a related report [28] describing [2 + 3] cycloadditions between chlorooxime 26 and other methylene-bearing compounds, its reaction with compound 25 gave the spiroacetal 27. However, in the present case this cycloadduct was only detectable by 1H NMR analysis of the crude reaction mixture. Indeed, a slow ring-opening reaction took place upon standing in solution to mainly give the isoxazole isomer 28 along with much less of the target oxazole-bearing α-hydroximino ester 29. Extensive trials to alter the selectivity of the ring opening using heat, adsorption over silica, acids (BF3·OEt2, AcOH) or bases (NEt3, LDA, EtONa) all failed to change the ratio of compounds 28 and 29, which were isolated in 5 and 28% yield, respectively. Despite the potential synthetic interest [29,30] of isoxazole 28, we did not pursue this further, but focused on another approach involving an oxidative addition of ethyl nitroacetate (4) on the methylene-bearing dipolarophiles 25 mediated by cerium(IV) ammonium nitrate (CAN). This was inspired by reports describing CAN-mediated carbon–carbon bond formation reactions between ethyl nitroacetate (4) and tri-O-acetylglycals [31,32] or other sugar-derived alkenes [33]. We first tried the conditions described in the literature (0 °C, mixture of methanol and dimethylformamide as a solvent), without much success in our case. Quite a few trials followed, changing the solvent to a dichloromethane/dimethylformamide mixture or dimethylformamide alone, or modifying the reaction conditions (at room temperature, 0 °C or −20 °C), but none resulted in a flagrant improvement. Indeed, as precisely described in the experimental part, the (impure) target nitroester 30 was isolated once in a disappointing 22% yield and quite a few byproducts were noticed. A control experiment omitting the ethyl nitroacetate (4) allowed us to identify amongst these: the oxazole derivative 31 resulting from an isomerization of 25 as well as alcohol 32 resulting from an oxidation of compound 25. Accordingly, this greatly dampened our hope to improve this transformation (trials with manganese(III) acetate were not successful either). We then resorted to a different approach to prepare the oxazole-bearing α-amino ester 36 from 2,4,5-trimethyloxazole (33). Deprotonation of this compound was achieved using lithium diisopropylamide (LDA) and this was followed by the addition of diethyl oxalate to give a mixture of compounds including the ketoester 34. Then, treatment of this mixture with hydroxylamine allowed the isolation of the target α-hydroximino ester 35 in a 7% overall yield. Two dimensional NMR experiments confirmed the depicted structure for compound 35 and trace amounts of the other isomers were detected in other chromatographic fractions but could not be fully purified. In any case, from the α-hydroximino ester 35, a reduction using zinc and hydrochloric acid gave the target oxazole-bearing α-amino ester 36.
Scheme 4:
Syntheses of α-hydroximino ester 29 and α-amino ester 36.
Scheme 4:
Syntheses of α-hydroximino ester 29 and α-amino ester 36.
Finally, as depicted in Scheme 5, the oxazole-bearing α-amino ester 43 was prepared from the aspartic acid derivative 37 through the propargylamide 38 followed by a gold(I)-catalyzed cyclization to form the oxazoline derivative 40. Concerning the amidation step, propargylamide 38 was obtained in 78% yield provided that an excess of triethylamine was avoided (otherwise, as determined by a control experiment, substantial amounts of the relatively stable succinyl derivative 39[34] resulting from a triethylamine-triggered cyclization of 38 were isolated) [35-37]. The treatment of compound 38 with a catalytic amount of gold(I) in warm toluene provided us with the oxazoline 40 in an 80% yield. However, this compound turned out to be unstable, either on standing, probably because of an autoxidation, as reported in other instances [27], or in CDCl3, probably because of acid traces. To achieve its isomerization, a literature search pointed out the use of an excess of DBU and heat [38,39]. However, boiling compound 40 in toluene in the presence of an excess of DBU led, after chromatography, to only 24% of the benzyl ester 41. Since, amongst few side reactions, we suspected a benzylester cleavage, we undertook this reaction under argon in ethanol at 110 °C using a microwave reactor along with only one equivalent of DBU and these changes provided us with the ethyl ester 42 in a 51% yield. Finally, a far more simple procedure was found by just adding a catalytic amount of hydrogen chloride in 1,4-dioxane to the toluene solution containing compound 40. This afforded, after overnight stirring, the isomerized compound 41 in a 69% yield. Finally, the deprotection of the amine function was achieved with the use of an excess of hydrogen chloride in 1,4-dioxane to give the target α-amino ester 43.
In the course of our attempts to extend the use of ethyl nitroacetate (4) to prepare α-amino ethyl esters via condensation reactions with aldehydes 3 or dimethylacetals 5, some severe limitations were encountered. Indeed, the ubiquitous occurrence of aryl methyl esters, arising from an unwanted O-alkylation of ethyl nitroacetate (4), plagued all our efforts to improve the latter synthetic pathway [2]. This side reaction pretty much limited the approach to electron-rich substrates and even our attempts to use the acylals 7 or 8, easily made from 2-trifluoromethylbenzaldehyde (3j), were very moderately successful. Such phenomenon probably accounts for the modest yields reported in many instances even when using titanium tetrachloride to achieve this condensation [5,6]. Concerning the reduction of the nitroacrylates 2 into the α-nitro esters 6, tangible but still modest yield improvements were observed when using sodium cyanoborohydride instead of sodium borohydride in some cases. This actually illustrates the sensitivity of this reduction which, along with the condensation, are quite limiting. As described above, the recourse to cycloaddition-based approaches allowed us to explore some original chemistry aiming at the preparation of oxazole-bearing α-amino esters which was of interest per se. Indeed, the previously unreported acid-catalyzed conditions to achieve the isomerization of the methylene-bearing oxazoline 40 into oxazole 41 should be useful in many other instances. In any case, as described in a following report [40], to overcome some of the limitations described here, we then focused on an exhaustive investigation of malonate-based strategies and reached an even more diverse set of α-amino esters.
Supporting Information
Supporting Information File 1:
Experimental and copies of spectra.
This work was supported by the Agence Nationale de la Recherche (ANR), grant ANR-11-CRNT-0004, in the context of the investment program ‘GLOBAL CARE’, an association of the Instituts Carnot ‘Pasteur-Maladies Infectieuses’, ‘Curie-Cancer’, ‘Voir et Entendre’, ‘Institut du Cerveau et de la moelle Épinière’ and the ‘Consortium pour l’Accélération de l’Innovation et de son Transfert dans le domaine du Lymphome’ (CALYM). This project also benefited from the Valoexpress funding call of the Institut Pasteur. Prof. Christian Bréchot, Dr. Muriel Delepierre and Dr. Daniel Larzul, from the Institut Pasteur are acknowledged for their interest and support.
References
Coutant, E. P.; Janin, Y. L. Chem. – Eur. J.2015,21, 17158–17171. doi:10.1002/chem.201501531
Return to citation in text:
[1]
Hervin, V. O.; Coutant, E. P.; Gagnot, G.; Janin, Y. L. Synthesis2017,49, 4093–4110. doi:10.1055/s-0036-1589506
Return to citation in text:
[1]
[2]
Fornicola, R. S.; Oblinger, E.; Montgomery, J. J. Org. Chem.1998,63, 3528–3529. doi:10.1021/jo980477h
Return to citation in text:
[1]
[2]
Nakamura, S.; Uchiyama, M.; Ohwada, T. J. Am. Chem. Soc.2003,125, 5282–5283. doi:10.1021/ja0343151
Return to citation in text:
[1]
[2]
Nakamura, S.; Sugimoto, H.; Ohwada, T. J. Am. Chem. Soc.2007,129, 1724–1732. doi:10.1021/ja067682w
Return to citation in text:
[1]
[2]
Kochetkov, K. A.; Babievskii, K. K.; Belikov, V. M.; Garbalinskaya, N. S.; Bakhmutov, V. I. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.)1980,29, 458–461. doi:10.1007/bf00949634
Return to citation in text:
[1]
[2]
[3]
[4]
Kochetkov, K. A.; Babievskii, K. K.; Nalivaiko, E. V.; Garbalinskaya, N. S.; Belikov, V. M. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.)1981,30, 466–469. doi:10.1007/bf00949597
Return to citation in text:
[1]
[2]
[3]
Meyers, A. I.; Sircar, J. C. J. Org. Chem.1967,32, 4134–4136. doi:10.1021/jo01287a116
Return to citation in text:
[1]
Ogiwara, Y.; Takahashi, K.; Kitazawa, T.; Sakai, N. J. Org. Chem.2015,80, 3101–3110. doi:10.1021/acs.joc.5b00011
Return to citation in text:
[1]
Li, Z.; Duan, Z.; Wang, H.; Tian, R.; Zhu, Q.; Wu, Y. Synlett2008, 2535–2539. doi:10.1055/s-2008-1078216
Return to citation in text:
[1]
Harel, T.; Rozen, S. J. Org. Chem.2007,72, 6500–6503. doi:10.1021/jo0709450
Return to citation in text:
[1]
[2]
Leslie-Smith, M. G.; Paton, R. M.; Webb, N. Tetrahedron Lett.1994,49, 9251–9254. doi:10.1016/0040-4039(94)88480-3
Return to citation in text:
[1]
Cecchi, L.; De Sarlo, F.; Machetti, F. Tetrahedron Lett.2005,46, 7877–7879. doi:10.1016/j.tetlet.2005.09.110
Return to citation in text:
[1]
Cecchi, L.; De Sarlo, F.; Machetti, F. Eur. J. Org. Chem.2006, 4852–4860. doi:10.1002/ejoc.200600475
Return to citation in text:
[1]
Griffioen, G.; Van Doorren, T.; Rojas de la Parra, V.; Marchand, A.; Allasia, S.; Kilonda, A.; Chaltin, P. Indole amide derivatives and related compounds for use in the treatment of neurodegenerative diseases. PCT Patent Application WO/2010/142801 A1, Dec 16, 2010.
Return to citation in text:
[1]
Weyrauch, J. P.; Hashmi, A. S. K.; Schuster, A.; Hengst, T.; Schetter, S.; Littmann, A.; Rudolph, M.; Hamzic, M.; Visus, J.; Rominger, F.; Frey, W.; Bats, J. W. Chem. – Eur. J.2010,16, 956–963. doi:10.1002/chem.200902472
Return to citation in text:
[1]
Hashmi, A. S. K.; Blanco Jaimes, M. C.; Schuster, A. M.; Rominger, F. J. Org. Chem.2012,77, 6394–6408. doi:10.1021/jo301288w
Return to citation in text:
[1]
[2]
Bantreil, X.; Vaxelaire, C.; Godet, T.; Parker, E.; Sauer, C.; Belmont, P. Org. Biomol. Chem.2011,9, 4831–4841. doi:10.1039/c1ob05354j
Return to citation in text:
[1]
Mioskowski, C.; De Lamo Marin, S.; Maruani, M.; Gill, M. Analogs of 4-hydroxyisoleucine and uses thereof. U.S. Patent Application 2006/0199853 A1, Sept 7, 2006.
Return to citation in text:
[1]
Mioskowski, C.; Wagner, A.; De Lamo Marin, S.; Catala, C.; Becht, J. M. Method for preparing 4-hydroxyisoleucine diastereoisomers and enantiomers and derivatives thereof. PCT Patent Application WO2004/052836 A, June 24, 2004.
Return to citation in text:
[1]
Sommermann, T.; Kim, B. G.; Peters, K.; Peters, E.-M.; Linker, T. Chem. Commun.2004, 2624–2625. doi:10.1039/b410120k
Return to citation in text:
[1]
Elamparuthi, E.; Kim, B. G.; Yin, J.; Maurer, M.; Linker, T. Tetrahedron2008,64, 11925–11937. doi:10.1016/j.tet.2008.08.109
Return to citation in text:
[1]
Kim, B. G.; Schilde, U.; Linker, T. Synthesis2005, 1507–1513. doi:10.1055/s-2005-865325
Return to citation in text:
[1]
Barra, M.; Roy, O.; Traikia, M.; Taillefumier, C. Org. Biomol. Chem.2010,8, 2941–2955. doi:10.1039/b923275c
Return to citation in text:
[1]
Matsoukas, J.; Cordopatis, P.; Theodoropoulos, D. J. Org. Chem.1977,42, 2105–2108. doi:10.1021/jo00432a017
Return to citation in text:
[1]
Schön, I.; Szirtes, T.; Rill, A.; Balogh, G.; Vadász, Z.; Seprődi, J.; Teplán, I.; Chino, N.; Yoshizawa Kumogaye, K.; Sakakibara, S. J. Chem. Soc., Perkin Trans. 11991, 3213–3223. doi:10.1039/p19910003213
Return to citation in text:
[1]
Flaih, N.; Gadjou, C.; Lafont, O.; Galons, H. Synlett2000, 896–898. doi:10.1055/s-2000-6701
Return to citation in text:
[1]
Tiecco, M.; Testaferri, L.; Tingoli, M.; Marini, F. J. Org. Chem.1993,58, 1349–1354. doi:10.1021/jo00058a011
Return to citation in text:
[1]
Bull, R. J.; Ray, N. C. 2-(9H-Xanthen-9-yl)-oxazol derivatives as m3 muscarinic receptor antagonists for the treatment of asthma and chronic obstructive lung disease. PCT Patent Application WO 2009/098455 A1, Aug 13, 2009.
Return to citation in text:
[1]
Coutant, E. P.; Hervin, V.; Gagnot, G.; Ford, C.; Baatallah, R.; Janin, Y. L. Beilstein J. Org. Chem.2018,14, 2853–2859. doi:10.3762/bjoc.14.264
Return to citation in text:
[1]
References 35-37
35.
Matsoukas, J.; Cordopatis, P.; Theodoropoulos, D. J. Org. Chem.1977,42, 2105–2108. doi:10.1021/jo00432a017
Tiecco, M.; Testaferri, L.; Tingoli, M.; Marini, F. J. Org. Chem.1993,58, 1349–1354. doi:10.1021/jo00058a011
39.
Bull, R. J.; Ray, N. C. 2-(9H-Xanthen-9-yl)-oxazol derivatives as m3 muscarinic receptor antagonists for the treatment of asthma and chronic obstructive lung disease. PCT Patent Application WO 2009/098455 A1, Aug 13, 2009.
Kochetkov, K. A.; Babievskii, K. K.; Belikov, V. M.; Garbalinskaya, N. S.; Bakhmutov, V. I. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.)1980,29, 458–461. doi:10.1007/bf00949634
8.
Kochetkov, K. A.; Babievskii, K. K.; Nalivaiko, E. V.; Garbalinskaya, N. S.; Belikov, V. M. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.)1981,30, 466–469. doi:10.1007/bf00949597
Kochetkov, K. A.; Babievskii, K. K.; Belikov, V. M.; Garbalinskaya, N. S.; Bakhmutov, V. I. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.)1980,29, 458–461. doi:10.1007/bf00949634
8.
Kochetkov, K. A.; Babievskii, K. K.; Nalivaiko, E. V.; Garbalinskaya, N. S.; Belikov, V. M. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.)1981,30, 466–469. doi:10.1007/bf00949597
Kochetkov, K. A.; Babievskii, K. K.; Belikov, V. M.; Garbalinskaya, N. S.; Bakhmutov, V. I. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.)1980,29, 458–461. doi:10.1007/bf00949634
8.
Kochetkov, K. A.; Babievskii, K. K.; Nalivaiko, E. V.; Garbalinskaya, N. S.; Belikov, V. M. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.)1981,30, 466–469. doi:10.1007/bf00949597
Kochetkov, K. A.; Babievskii, K. K.; Belikov, V. M.; Garbalinskaya, N. S.; Bakhmutov, V. I. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.)1980,29, 458–461. doi:10.1007/bf00949634
Mioskowski, C.; De Lamo Marin, S.; Maruani, M.; Gill, M. Analogs of 4-hydroxyisoleucine and uses thereof. U.S. Patent Application 2006/0199853 A1, Sept 7, 2006.
30.
Mioskowski, C.; Wagner, A.; De Lamo Marin, S.; Catala, C.; Becht, J. M. Method for preparing 4-hydroxyisoleucine diastereoisomers and enantiomers and derivatives thereof. PCT Patent Application WO2004/052836 A, June 24, 2004.
Weyrauch, J. P.; Hashmi, A. S. K.; Schuster, A.; Hengst, T.; Schetter, S.; Littmann, A.; Rudolph, M.; Hamzic, M.; Visus, J.; Rominger, F.; Frey, W.; Bats, J. W. Chem. – Eur. J.2010,16, 956–963. doi:10.1002/chem.200902472
27.
Hashmi, A. S. K.; Blanco Jaimes, M. C.; Schuster, A. M.; Rominger, F. J. Org. Chem.2012,77, 6394–6408. doi:10.1021/jo301288w
Griffioen, G.; Van Doorren, T.; Rojas de la Parra, V.; Marchand, A.; Allasia, S.; Kilonda, A.; Chaltin, P. Indole amide derivatives and related compounds for use in the treatment of neurodegenerative diseases. PCT Patent Application WO/2010/142801 A1, Dec 16, 2010.



![[1860-5397-14-263-i1]](/bjoc/content/inline/1860-5397-14-263-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-263-i6.svg?max-width=637&scale=1.0)
![[1860-5397-14-263-i2]](/bjoc/content/inline/1860-5397-14-263-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-14-263-i3]](/bjoc/content/inline/1860-5397-14-263-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-14-263-i4]](/bjoc/content/inline/1860-5397-14-263-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-14-263-i5]](/bjoc/content/inline/1860-5397-14-263-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)