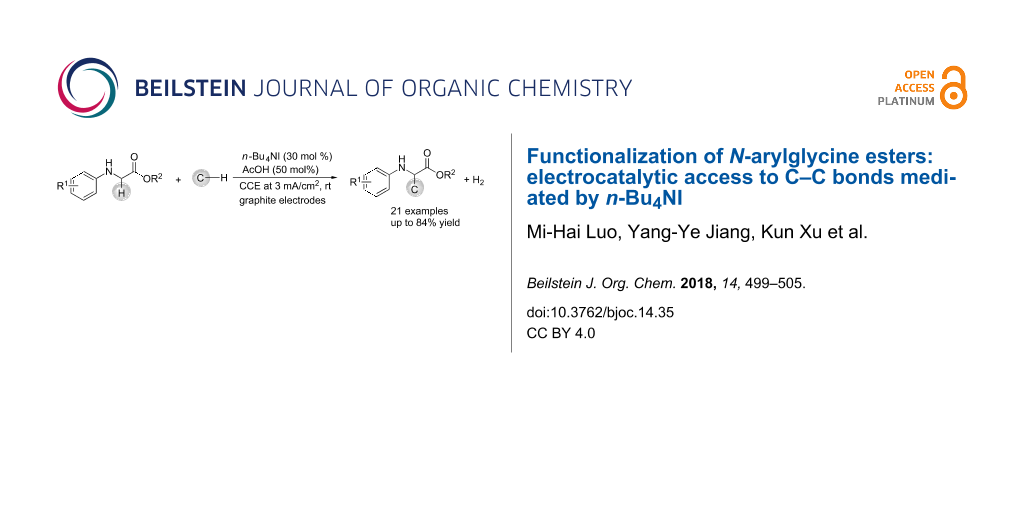
Abstract
An efficient electrocatalytic functionalization of N-arylglycine esters is reported. The protocol proceeds in an undivided cell under constant current conditions employing the simple, cheap and readily available n-Bu4NI as the mediator. In addition, it is demonstrated that the mediated process is superior to the direct electrochemical functionalization.

Graphical Abstract
Introduction
The oxidative cross dehydrogenative coupling (CDC) of two C–H bonds has emerged as an versatile and powerful strategy for forming new C–C bonds in organic chemistry due to its step and atom economic characteristic as well as avoiding the prefunctionalization of substrates [1-5]. Most of the CDC reactions occur between the benzylic C–H bonds and α-C–H bonds adjacent to a heteroatom (N or O) [6-10]. However, the oxidative C–C formation of secondary amines, especially amino acids has been more important for studying properties and functions of natural and non-natural amino acids [11]. Consequently, efficient and selective construction of C–C bonds of amino acids has always been paid much attention in industrial and academic setting and many advances have been made [12-17]. Li et al. first reported the functionalization of glycine derivatives with malonates using a stoichiometric quantity of Cu(OAc)2 as catalyst and oxidant [12]. Later on, arylation, vinylation and alkynylation of glycine derivatives were also accomplished by the same group (Scheme 1) [13]. Using the Cu(OAc)2/pyrrolidine dual catalysts system, Huang developed the oxidative cross coupling of glycine derivatives with acetone in the presence of TBHP or DDQ as terminal oxidants [14]. The protocol was also extended to reactions with 2-alkylquinoline [15] and phenols [16] using O2 and di-tert-butyl peroxide (DTBP) as oxidant, respectively (Scheme 1). A CuCl-catalyzed oxidative cross coupling of glycine derivatives with indoles has been developed by Hou et al., wherein simple copper salts were used as catalysts and oxygen as the co-oxidant (Scheme 1) [17].
![[1860-5397-14-35-i1]](/bjoc/content/inline/1860-5397-14-35-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Cross dehydrogenative coupling of N-arylglycine esters with C–H nucleophiles.
Scheme 1: Cross dehydrogenative coupling of N-arylglycine esters with C–H nucleophiles.
Alternatively, photocatalytic versions of CDC reactions of glycine derivatives with C-nucleophiles were also developed [18,19]. For example, combining the visible light catalyst Ru(bpy)3Cl2, and the transition metal catalyst Cu(OTf)2, Wu and co-workers achieved aerobic oxidative coupling of secondary amines with β-keto esters to form C(sp3)–C(sp3) bonds (Scheme 1) [18].
Although much advance has been made for the functionalization of glycine derivatives, most of these strategies mentioned above require stoichiometric or excess amounts of chemical oxidants and transition metal (photo)catalysts. The utilization of stoichiometric or excess amounts of chemical oxidants results in producing waste, which is not atomic economically and environmentally benign. In addition, over-oxidation of products likely occurs in the presence of excess amounts of oxidant. On the other hand, the toxicity of residual traces of transition metal (photo)catalyst in products is also highly concerned. Consequently, metal-free and environmentally friendly oxidative C–C bond formation is highly desired.
Electrochemistry has proved to be an environmentally benign method to achieve the formation of a new chemical bond and a functional group conversion by using electrons as redox reagent rather than terminal chemicals [20-27]. In this context, we have applied simple halide ions as redox catalysts to achieve electrochemical C–H bond functionalization, leading to the formation of new C–C, C–N, C–O and C–S bonds [28-31]. Herein, we report the electrochemical α-C–H functionalization of N-arylglycine esters with C–H nucleophiles using n-Bu4NI as redox catalyst (Scheme 1). The chemistry was performed in an undivided cell under constant current electrolysis. It was observed that n-Bu4NI promotes the reaction dramatically and higher yields of α-functionalized products were afforded compared with the direct electrolysis.
Results and Discussion
Initially, N-arylglycine ester 1a and C–H nucleophile 1,3,5-trimethoxybenzene (2a) were chosen as model compounds to optimize the electrolytic conditions. As shown in Table 1, when constant current electrolysis (CCE) of 1a and 2a was performed in an undivided cell equipped with 0.1 M LiClO4 in CH3CN in the presence of AcOH using two graphite plates as anode and cathode, the desired product 3aa was isolated in 27% yield (Table 1, entry 1). Replacing CH3CN by other solvents, such as methanol, ethanol or CH2Cl2 failed to improve the reaction efficiency and only trace amounts of 3aa were detected (Table 1, entries 2–4). Further solvent screening disclosed that a 1:2 ratio of CH3CN and CH2Cl2 was better, giving 3aa in 66% yield (Table 1, entries 5 and 6). Exploring the influence of current density on the CDC reaction indicated that 3 mA/cm2 was suitable; lower or higher current density led to a slightly lower yields of 3aa (entry 5, vs entries 7 and 8). It was observed that the additive plays an important role. Among several additives examined, AcOH was proved to be the best, although TFA also gave comparable yields of 3aa (Table 1, entries 5 and 11–13), whereas, it gave only 38% yield of 3aa when the reaction was carried out in the absence of AcOH (Table 1, entry 9). Investigation of the anode proved that graphite was superior to Pt and DSA (Dimensionally Stable Anode, Table 1, entries 14 and 15). Finally, to further improve the reaction efficiency, several halide-containing mediators as redox catalyst were evaluated. To our delight, when n-Bu4NI was utilized as a redox catalyst, the yield of 3aa increased to 81% (Table 1, entries 16–19).
Table 1: Optimization of reaction conditionsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-35-i7.svg?max-width=637&scale=1.18182)
|
||||||
|---|---|---|---|---|---|---|
| entry | solvent | mediator (equiv) | J (mA/cm2) | additive (equiv) | cathode/anode | yield (%)b |
| 1 | CH3CN | –c | 3 | AcOH (0.5) | C/C | 27 |
| 2 | MeOH | –c | 3 | AcOH (0.5) | C/C | trace |
| 3 | EtOH | –c | 3 | AcOH (0.5) | C/C | trace |
| 4d | CH2Cl2 | –c | 3 | AcOH(0.5) | C/C | trace |
| 5 | CH3CN/CH2Cl2 (1:2) | –c | 3 | AcOH (0.5) | C/C | 66 |
| 6 | CH3CN/CH2Cl2 (2:1) | –c | 3 | AcOH (0.5) | C/C | 56 |
| 7 | CH3CN/CH2Cl2 (1:2) | –c | 1 | AcOH (0.5) | C/C | 47 |
| 8 | CH3CN/CH2Cl2(1:2) | –c | 5 | AcOH (0.5) | C/C | 50 |
| 9 | CH3CN/CH2Cl2 (1:2) | –c | 3 | AcOH (0.0) | C/C | 38 |
| 10 | CH3CN/CH2Cl2 (1:2) | –c | 3 | AcOH (1.0) | C/C | 56 |
| 11 | CH3CN/CH2Cl2 (1:2) | –c | 3 | TFA (0.5) | C/C | 60 |
| 12 | CH3CN/CH2Cl2 (1:2) | –c | 3 | H2SO4 (0.5) | C/C | 13 |
| 13 | CH3CN/CH2Cl2 (1:2) | –c | 3 | Na2CO3 (0.5) | C/C | 40 |
| 14 | CH3CN/CH2Cl2 (1:2) | –c | 3 | AcOH (0.5) | C/Pt | 48 |
| 15 | CH3CN/CH2Cl2 (1:2) | –c | 3 | AcOH (0.5) | C/DSA | 41 |
| 16 | CH3CN/CH2Cl2 (1:2) | n-Bu4NI (0.3) | 3 | AcOH (0.5) | C/C | 81 |
| 17 | CH3CN/CH2Cl2 (1:2) | n-Bu4NBr (0.3) | 3 | AcOH (0.5) | C/C | 48 |
| 18 | CH3CN/CH2Cl2 (1:2) | NH4I (0.3) | 3 | AcOH (0.5) | C/C | 54 |
| 19 | CH3CN/CH2Cl2 (1:2) | NH4Br (0.3) | 3 | AcOH (0.5) | C/C | 20 |
aConditions: 1a (1.0 mmol), 2a (1.2 mmol) in 15 mL solution, LiClO4 (0.1 M), room temperature, electrode C represents graphite plate. bIsolated yield. cWithout mediator. dn-Bu4NBF4 was used as supporting electrolyte.
On the basis of the screening of reaction conditions, we could conclude that the electrocatalytic oxidative coupling should be performed in a mixed solution of CH3CN and CH2Cl2 (v/v = 1:2), in the presence of n-Bu4NI (30 mol %) as the mediator, AcOH (50 mol %) as the additive and using graphite plate as electrodes under constant current at 3 mA/cm2. With the optimized conditions in hand, we then investigated the scope of N-arylglycine esters in the reaction with 2a. As a comparison, direct electrochemical coupling of N-arylglycine esters with 2a was also performed. As shown in Scheme 2, substituents including either electron-donating groups (such as methyl and methoxy) or electron-withdrawing groups (Cl, Br and CF3) in the 4-position of the aryl group were tolerated and gave moderate to good yields of 3ba–3ea (61–69% yields), whereas, only 47–58% yields of the desired 3ba–3ea were isolated under the direct electrolytic conditions. However, 3fa was afforded in 39% and 12% yields, respectively, when the electrochemical reaction was performed in the presence or absence of n-Bu4NI as the redox catalyst. The reason for the low yield of 3fa is not clear yet. Steric factors seem to play an important role in the electrochemical CDC reaction of N-arylglycine esters with 2a. When the methyl group was situated at 2- or 3-position of the aniline, instead of in the 4-position, the corresponding products 3ga and 3ha were afforded in 36% and 27% yields, respectively. However, 3ga and 3ha were isolated in 14% and 11% yields, respectively, under the direct electrolytic conditions. In the cases of N-arylglycine methyl ester 1i and N-arylglycine benzyl ester 1j, the electrocatalytic functionalization afforded excellent yields of 3ia (76%) and 3ja (84%), whereas, less efficiency (40–43% yields) was observed without the assistance of n-Bu4NI.
![[1860-5397-14-35-i2]](/bjoc/content/inline/1860-5397-14-35-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Electrochemical CDC reaction of 2a and various N-arylglycine esters. Reaction conditions for the indirect electrolysis: 1 (0.5 mmol), 2a (0.6 mmol), n-Bu4NI (30 mol %), 0.1 M LiClO4/CH3CN (5 mL) and CH2Cl2 (10 mL), AcOH (50 mol %), current density (3 mA/cm2), graphite anode and cathode, at room temperature; reaction conditions for direct electrolysis: 1 (0.5 mmol), 2a (0.6 mmol), 0.1 M LiClO4/CH3CN (5 mL) and CH2Cl2 (10 mL), AcOH (50 mol %), current density (3 mA/cm2), graphite anode and cathode, at room temperature; yields in parenthesis obtained from direct electrolysis.
Scheme 2: Electrochemical CDC reaction of 2a and various N-arylglycine esters. Reaction conditions for the in...
Next, the reactivity of different C–H nucleophiles was also investigated. As shown in Scheme 3, β-keto ester and malonates worked well to afford the desired products 3ab–3ad in moderate yields. In addition, naphthanols were also compatible in this transformation, giving the corresponding products 3ae–3ag in good yields. Notably, when styrene and 1-ethynylbenzene were subjected to reaction with 1a under the standard indirect electrolytic conditions, quinoline-2-carboxylate 3ah was isolated in 64% and 58% yield, respectively. Substituted styrenes and 1-ethynylbenzene were also tolerated well, giving corresponding products 3ai–3aj in 45–50% yields. The formations of 3ah–3aj likely derives from an azo-Diels–Alder reaction of styrene or ethynylbenzene with an imine intermediate, in situ generated from anodic oxidation of 1a, followed by additional oxidation [32-36].
![[1860-5397-14-35-i3]](/bjoc/content/inline/1860-5397-14-35-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of 2 using n-Bu4NI as mediator. Reaction conditions:1a (0.5 mmol), 2 (0.6 mmol), n-Bu4NI (30 mol %), 0.1 M LiClO4/CH3CN (5 mL) and CH2Cl2 (10 mL), AcOH (50 mol %), current density (3 mA/cm2), graphite anode and cathode. Isolated yields are given.
Scheme 3: Scope of 2 using n-Bu4NI as mediator. Reaction conditions:1a (0.5 mmol), 2 (0.6 mmol), n-Bu4NI (30 ...
To prove the practicability of the protocol, a scaled-up reaction was also carried out. As illustrated in Scheme 4, when 6 mmol of ethyl p-tolylglycinate (1a) was allowed to react with 1,3,5-trimethoxybenzene (2a) under the standard conditions, adduct 3aa was isolated in a 75% yield, without obvious losing of yield.
![[1860-5397-14-35-i4]](/bjoc/content/inline/1860-5397-14-35-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
To better understand the reaction mechanism, control experiments were performed. As shown in Scheme 5, the anodic oxidation of 1a in the absence of a C–H nucleophile under the standard conditions gives imine intermediate product 5, which was detected by TLC and GC–MS. Moreover, when separated synthesized 5 was subjected to react with 2a, the corresponding product 3aa was isolated in 89% yield. These control experiments indicate that 5 is a possible reaction intermediate.
![[1860-5397-14-35-i5]](/bjoc/content/inline/1860-5397-14-35-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Based on these control experiments described above, as well as related references [4], a plausible mechanism for the electrocatalytic cross dehydrogenative coupling of N-arylglycine esters 1 with C–H nucleophiles 2 is outlined in Scheme 6. The anodic oxidation of iodide generates the active species I2 or I+. Followed by a homogeneous reaction with N-arylglycine esters, N-iodo intermediate 4 was generated. Eliminating a molecule of HI affords imine intermediate 5. In the presence of acetic acid, the protonated 5-H+ undergoes nucleophilic addition with C–H nucleophiles 2 to give the desired products 3.
![[1860-5397-14-35-i6]](/bjoc/content/inline/1860-5397-14-35-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: A plausible mechanism for the electrocatalytic cross dehydrogenative coupling of N-arylglycine esters with C-nucleophiles.
Scheme 6: A plausible mechanism for the electrocatalytic cross dehydrogenative coupling of N-arylglycine este...
Conclusion
In summary, an efficient electrocatalytic cross dehydrogenative coupling of arylglycine esters with C–H nucleophiles has been developed. This protocol employs simple n-Bu4NI as the redox catalyst, avoiding utilization of transition metals and excess amounts of external oxidant, thereby providing an environmentally benign method to the CDC reaction. In addition, it was observed that the electrocatalytic process is superior to the direct electrolysis. Further application of this electrochemical protocol for the formation of new C–C bonds is still on the way in our group.
Experimental
Instruments and reagents
All melting points were measured with an electrothermal melting point apparatus and are uncorrected. NMR spectra were recorded using a 400 MHz or 300 MHz spectrometer (400 MHz 1H frequency, 100 MHz 13C frequency; 300 MHz 1H frequency, 75 MHz 13C frequency). Chemical shifts are given as δ values (internal standard: TMS). Coupling constants are reported in Hz. All starting materials and solvents were obtained from commercial sources and used without further purification. Products were purified by chromatography on silica gel (petroleum ether/EtOAc).
Typical procedure for the electrocatalytic cross dehydrogenative coupling of N-arylglycine ester and C–H nucleophiles
An undivided cell was equipped with a carbon anode (2 × 2 cm2) and a carbon cathode (2 × 2 cm2) and connected to a DC regulated power supply. To the cell was added N-arylglycine ester (0.5 mmol), C–H nucleophiles (0.6 mmol), n-Bu4NI (0.15 mmol) and 5 mL of 0.1 M LiClO4/CH3CN and 10 mL CH2Cl2. The mixture was electrolyzed using constant current conditions (≈3 mA/cm2) at room temperature under magnetic stirring. When TLC analysis indicated that the electrolysis was complete (witnessed by the disappearance of the N-arylglycine ester), the solvent was removed under reduced pressure. The residue was poured into a saturated aqueous solution of Na2S2O3 and the product was then extracted with DCM (3 × 20 mL), dried over Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel using a mixture of petroleum ether/EtOAc (v/v = 3:1) as eluent to afford the desired pure product.
Typical procedure for the direct electrochemical cross dehydrogenative coupling of N-arylglycine ester and C–H nucleophiles
The procedure was identical to that of electrocatalytic synthesis, but without the addition of n-Bu4NI as the mediator.
Supporting Information
| Supporting Information File 1: General procedure and analytical data. | ||
| Format: PDF | Size: 2.6 MB | Download |
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 21472011, 21272021) and the National Key Technology R&D Program (2017YFB0307502, 2011BAD23B01). ZCC also thank financial support from the Open Project Program of Beijing Key Laboratory of Flavor Chemistry, Beijing Technology and Business University (BTBU).
References
-
Girard, S. A.; Knauber, T.; Li, C.-J. Angew. Chem., Int. Ed. 2014, 53, 74. doi:10.1002/anie.201304268
Return to citation in text: [1] -
Scheuermann, C. J. Chem. – Asian J. 2010, 5, 436. doi:10.1002/asia.200900487
Return to citation in text: [1] -
Ritleng, V.; Sirlin, C.; Pfeffer, M. Chem. Rev. 2002, 102, 1731. doi:10.1021/cr0104330
Return to citation in text: [1] -
Xu, H.-J.; Lu, Y.; Farmer, M. E.; Wang, H.-W.; Zhao, D.; Kang, Y.-S.; Sun, W.-Y.; Yu, J.-Q. J. Am. Chem. Soc. 2017, 139, 2200. doi:10.1021/jacs.6b13269
Return to citation in text: [1] [2] -
Jiang, H.; He, J.; Liu, T.; Yu, J.-Q. J. Am. Chem. Soc. 2016, 138, 2055. doi:10.1021/jacs.5b13462
Return to citation in text: [1] -
Chatani, N.; Asaumi, T.; Yorimitsu, S.; Ikeda, T.; Kakiuchi, F.; Murai, S. J. Am. Chem. Soc. 2001, 123, 10935. doi:10.1021/ja011540e
Return to citation in text: [1] -
Li, Z.; Li, C.-J. J. Am. Chem. Soc. 2005, 127, 6968. doi:10.1021/ja0516054
Return to citation in text: [1] -
Condie, A. G.; González-Gómez, J. C.; Stephenson, C. R. J. J. Am. Chem. Soc. 2010, 132, 1464. doi:10.1021/ja909145y
Return to citation in text: [1] -
Freeman, D. B.; Furst, L.; Condie, A. G.; Stephenson, C. R. J. Org. Lett. 2012, 14, 94. doi:10.1021/ol202883v
Return to citation in text: [1] -
Hari, D. P.; König, B. Org. Lett. 2011, 13, 3852. doi:10.1021/ol201376v
Return to citation in text: [1] -
Twyman, R. M. Principles of proteomics; BIOS Scientific Publishers: New York, 2004.
Return to citation in text: [1] -
Zhao, L.; Li, C.-J. Angew. Chem., Int. Ed. 2008, 47, 7075. doi:10.1002/anie.200801367
Return to citation in text: [1] [2] -
Li, C.-J. Acc. Chem. Res. 2009, 42, 335. doi:10.1021/ar800164n
Return to citation in text: [1] [2] -
Xie, J.; Huang, Z.-Z. Angew. Chem., Int. Ed. 2010, 49, 10181. doi:10.1002/anie.201004940
Return to citation in text: [1] [2] -
Zhu, Z.-Q.; Bai, P.; Huang, Z.-Z. Org. Lett. 2014, 16, 4881. doi:10.1021/ol502402s
Return to citation in text: [1] [2] -
Salman, M.; Zhu, Z.-Q.; Huang, Z.-Z. Org. Lett. 2016, 18, 1526. doi:10.1021/acs.orglett.6b00162
Return to citation in text: [1] [2] -
Huo, C.; Wang, C.; Wu, M.; Jia, X.; Xie, H.; Yuan, Y. Adv. Synth. Catal. 2014, 356, 411. doi:10.1002/adsc.201300535
Return to citation in text: [1] [2] -
Gao, X.-W.; Meng, Q.-Y.; Xiang, M.; Chen, B.; Feng, K.; Tung, C.-H.; Wu, L.-Z. Adv. Synth. Catal. 2013, 355, 2158. doi:10.1002/adsc.201300311
Return to citation in text: [1] [2] -
Yang, X.; Li, L.; Li, Y.; Zhang, Y. J. Org. Chem. 2016, 81, 12433. doi:10.1021/acs.joc.6b02683
Return to citation in text: [1] -
Sperry, J. B.; Wright, D. L. Chem. Soc. Rev. 2006, 35, 605. doi:10.1039/b512308a
Return to citation in text: [1] -
Francke, R.; Little, R. D. Chem. Soc. Rev. 2014, 43, 2492. doi:10.1039/c3cs60464k
Return to citation in text: [1] -
Horn, E. J.; Rosen, B. R.; Baran, P. S. ACS Cent. Sci. 2016, 2, 302. doi:10.1021/acscentsci.6b00091
Return to citation in text: [1] -
Yang, Q.-L.; Li, Y.-Q.; Ma, C.; Fang, P.; Zhang, X.-J.; Mei, T.-S. J. Am. Chem. Soc. 2017, 139, 3293. doi:10.1021/jacs.7b01232
Return to citation in text: [1] -
Schäfer, H. J. Angew. Chem., Int. Ed. 2017, 56, 15502. doi:10.1002/anie.201707804
Return to citation in text: [1] -
Zhao, H.-B.; Hou, Z.-W.; Liu, Z.-J.; Zhou, Z.-F.; Song, J.; Xu, H.-C. Angew. Chem., Int. Ed. 2017, 56, 587. doi:10.1002/anie.201610715
Return to citation in text: [1] -
Wang, P.; Tang, S.; Huang, P.; Lei, A. Angew. Chem., Int. Ed. 2017, 56, 3009. doi:10.1002/anie.201700012
Return to citation in text: [1] -
Jiang, Y.; Xu, K.; Zeng, C. Chem. Rev. 2017, in press. doi:10.1021/acs.chemrev.7b00271
Return to citation in text: [1] -
Li, W.-C.; Zeng, C.-C.; Hu, L.-M.; Tian, H.-Y.; Little, R. D. Adv. Synth. Catal. 2013, 355, 2884. doi:10.1002/adsc.201300502
Return to citation in text: [1] -
Chen, J.; Yan, W.-Q.; Lam, C. M.; Zeng, C.-C.; Hu, L.-M.; Little, R. D. Org. Lett. 2015, 17, 986. doi:10.1021/acs.orglett.5b00083
Return to citation in text: [1] -
Liang, S.; Zeng, C.-C.; Luo, X.-G.; Ren, F.-z.; Tian, H.-Y.; Sun, B.-G.; Little, R. D. Green Chem. 2016, 18, 2222. doi:10.1039/C5GC02626A
Return to citation in text: [1] -
Kang, L.-S.; Luo, M.-H.; Lam, C. M.; Hu, L.-M.; Little, R. D.; Zeng, C.-C. Green Chem. 2016, 18, 3767. doi:10.1039/C6GC00666C
Return to citation in text: [1] -
Ni, M.; Zhang, Y.; Gong, T.; Feng, B. Adv. Synth. Catal. 2017, 359, 824. doi:10.1002/adsc.201601066
Return to citation in text: [1] -
Gandeepan, P.; Rajamalli, P.; Cheng, C.-H. Asian J. Org. Chem. 2014, 3, 303. doi:10.1002/ajoc.201300262
Return to citation in text: [1] -
Xie, Z.; Jia, J.; Liu, X.; Liu, L. Adv. Synth. Catal. 2016, 358, 919. doi:10.1002/adsc.201501015
Return to citation in text: [1] -
Liu, J.; Wang, Y.; Yu, L.; Huo, C.; Wang, X.; Jia, X. Adv. Synth. Catal. 2014, 356, 3214. doi:10.1002/adsc.201400005
Return to citation in text: [1] -
Rohlmann, R.; Stopka, T.; Richter, H.; Garcı́a Mancheño, O. J. Org. Chem. 2013, 78, 6050. doi:10.1021/jo4007199
Return to citation in text: [1]
| 1. | Girard, S. A.; Knauber, T.; Li, C.-J. Angew. Chem., Int. Ed. 2014, 53, 74. doi:10.1002/anie.201304268 |
| 2. | Scheuermann, C. J. Chem. – Asian J. 2010, 5, 436. doi:10.1002/asia.200900487 |
| 3. | Ritleng, V.; Sirlin, C.; Pfeffer, M. Chem. Rev. 2002, 102, 1731. doi:10.1021/cr0104330 |
| 4. | Xu, H.-J.; Lu, Y.; Farmer, M. E.; Wang, H.-W.; Zhao, D.; Kang, Y.-S.; Sun, W.-Y.; Yu, J.-Q. J. Am. Chem. Soc. 2017, 139, 2200. doi:10.1021/jacs.6b13269 |
| 5. | Jiang, H.; He, J.; Liu, T.; Yu, J.-Q. J. Am. Chem. Soc. 2016, 138, 2055. doi:10.1021/jacs.5b13462 |
| 12. | Zhao, L.; Li, C.-J. Angew. Chem., Int. Ed. 2008, 47, 7075. doi:10.1002/anie.200801367 |
| 32. | Ni, M.; Zhang, Y.; Gong, T.; Feng, B. Adv. Synth. Catal. 2017, 359, 824. doi:10.1002/adsc.201601066 |
| 33. | Gandeepan, P.; Rajamalli, P.; Cheng, C.-H. Asian J. Org. Chem. 2014, 3, 303. doi:10.1002/ajoc.201300262 |
| 34. | Xie, Z.; Jia, J.; Liu, X.; Liu, L. Adv. Synth. Catal. 2016, 358, 919. doi:10.1002/adsc.201501015 |
| 35. | Liu, J.; Wang, Y.; Yu, L.; Huo, C.; Wang, X.; Jia, X. Adv. Synth. Catal. 2014, 356, 3214. doi:10.1002/adsc.201400005 |
| 36. | Rohlmann, R.; Stopka, T.; Richter, H.; Garcı́a Mancheño, O. J. Org. Chem. 2013, 78, 6050. doi:10.1021/jo4007199 |
| 12. | Zhao, L.; Li, C.-J. Angew. Chem., Int. Ed. 2008, 47, 7075. doi:10.1002/anie.200801367 |
| 13. | Li, C.-J. Acc. Chem. Res. 2009, 42, 335. doi:10.1021/ar800164n |
| 14. | Xie, J.; Huang, Z.-Z. Angew. Chem., Int. Ed. 2010, 49, 10181. doi:10.1002/anie.201004940 |
| 15. | Zhu, Z.-Q.; Bai, P.; Huang, Z.-Z. Org. Lett. 2014, 16, 4881. doi:10.1021/ol502402s |
| 16. | Salman, M.; Zhu, Z.-Q.; Huang, Z.-Z. Org. Lett. 2016, 18, 1526. doi:10.1021/acs.orglett.6b00162 |
| 17. | Huo, C.; Wang, C.; Wu, M.; Jia, X.; Xie, H.; Yuan, Y. Adv. Synth. Catal. 2014, 356, 411. doi:10.1002/adsc.201300535 |
| 4. | Xu, H.-J.; Lu, Y.; Farmer, M. E.; Wang, H.-W.; Zhao, D.; Kang, Y.-S.; Sun, W.-Y.; Yu, J.-Q. J. Am. Chem. Soc. 2017, 139, 2200. doi:10.1021/jacs.6b13269 |
| 11. | Twyman, R. M. Principles of proteomics; BIOS Scientific Publishers: New York, 2004. |
| 20. | Sperry, J. B.; Wright, D. L. Chem. Soc. Rev. 2006, 35, 605. doi:10.1039/b512308a |
| 21. | Francke, R.; Little, R. D. Chem. Soc. Rev. 2014, 43, 2492. doi:10.1039/c3cs60464k |
| 22. | Horn, E. J.; Rosen, B. R.; Baran, P. S. ACS Cent. Sci. 2016, 2, 302. doi:10.1021/acscentsci.6b00091 |
| 23. | Yang, Q.-L.; Li, Y.-Q.; Ma, C.; Fang, P.; Zhang, X.-J.; Mei, T.-S. J. Am. Chem. Soc. 2017, 139, 3293. doi:10.1021/jacs.7b01232 |
| 24. | Schäfer, H. J. Angew. Chem., Int. Ed. 2017, 56, 15502. doi:10.1002/anie.201707804 |
| 25. | Zhao, H.-B.; Hou, Z.-W.; Liu, Z.-J.; Zhou, Z.-F.; Song, J.; Xu, H.-C. Angew. Chem., Int. Ed. 2017, 56, 587. doi:10.1002/anie.201610715 |
| 26. | Wang, P.; Tang, S.; Huang, P.; Lei, A. Angew. Chem., Int. Ed. 2017, 56, 3009. doi:10.1002/anie.201700012 |
| 27. | Jiang, Y.; Xu, K.; Zeng, C. Chem. Rev. 2017, in press. doi:10.1021/acs.chemrev.7b00271 |
| 6. | Chatani, N.; Asaumi, T.; Yorimitsu, S.; Ikeda, T.; Kakiuchi, F.; Murai, S. J. Am. Chem. Soc. 2001, 123, 10935. doi:10.1021/ja011540e |
| 7. | Li, Z.; Li, C.-J. J. Am. Chem. Soc. 2005, 127, 6968. doi:10.1021/ja0516054 |
| 8. | Condie, A. G.; González-Gómez, J. C.; Stephenson, C. R. J. J. Am. Chem. Soc. 2010, 132, 1464. doi:10.1021/ja909145y |
| 9. | Freeman, D. B.; Furst, L.; Condie, A. G.; Stephenson, C. R. J. Org. Lett. 2012, 14, 94. doi:10.1021/ol202883v |
| 10. | Hari, D. P.; König, B. Org. Lett. 2011, 13, 3852. doi:10.1021/ol201376v |
| 28. | Li, W.-C.; Zeng, C.-C.; Hu, L.-M.; Tian, H.-Y.; Little, R. D. Adv. Synth. Catal. 2013, 355, 2884. doi:10.1002/adsc.201300502 |
| 29. | Chen, J.; Yan, W.-Q.; Lam, C. M.; Zeng, C.-C.; Hu, L.-M.; Little, R. D. Org. Lett. 2015, 17, 986. doi:10.1021/acs.orglett.5b00083 |
| 30. | Liang, S.; Zeng, C.-C.; Luo, X.-G.; Ren, F.-z.; Tian, H.-Y.; Sun, B.-G.; Little, R. D. Green Chem. 2016, 18, 2222. doi:10.1039/C5GC02626A |
| 31. | Kang, L.-S.; Luo, M.-H.; Lam, C. M.; Hu, L.-M.; Little, R. D.; Zeng, C.-C. Green Chem. 2016, 18, 3767. doi:10.1039/C6GC00666C |
| 16. | Salman, M.; Zhu, Z.-Q.; Huang, Z.-Z. Org. Lett. 2016, 18, 1526. doi:10.1021/acs.orglett.6b00162 |
| 18. | Gao, X.-W.; Meng, Q.-Y.; Xiang, M.; Chen, B.; Feng, K.; Tung, C.-H.; Wu, L.-Z. Adv. Synth. Catal. 2013, 355, 2158. doi:10.1002/adsc.201300311 |
| 19. | Yang, X.; Li, L.; Li, Y.; Zhang, Y. J. Org. Chem. 2016, 81, 12433. doi:10.1021/acs.joc.6b02683 |
| 15. | Zhu, Z.-Q.; Bai, P.; Huang, Z.-Z. Org. Lett. 2014, 16, 4881. doi:10.1021/ol502402s |
| 18. | Gao, X.-W.; Meng, Q.-Y.; Xiang, M.; Chen, B.; Feng, K.; Tung, C.-H.; Wu, L.-Z. Adv. Synth. Catal. 2013, 355, 2158. doi:10.1002/adsc.201300311 |
| 14. | Xie, J.; Huang, Z.-Z. Angew. Chem., Int. Ed. 2010, 49, 10181. doi:10.1002/anie.201004940 |
| 17. | Huo, C.; Wang, C.; Wu, M.; Jia, X.; Xie, H.; Yuan, Y. Adv. Synth. Catal. 2014, 356, 411. doi:10.1002/adsc.201300535 |
© 2018 Luo et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)