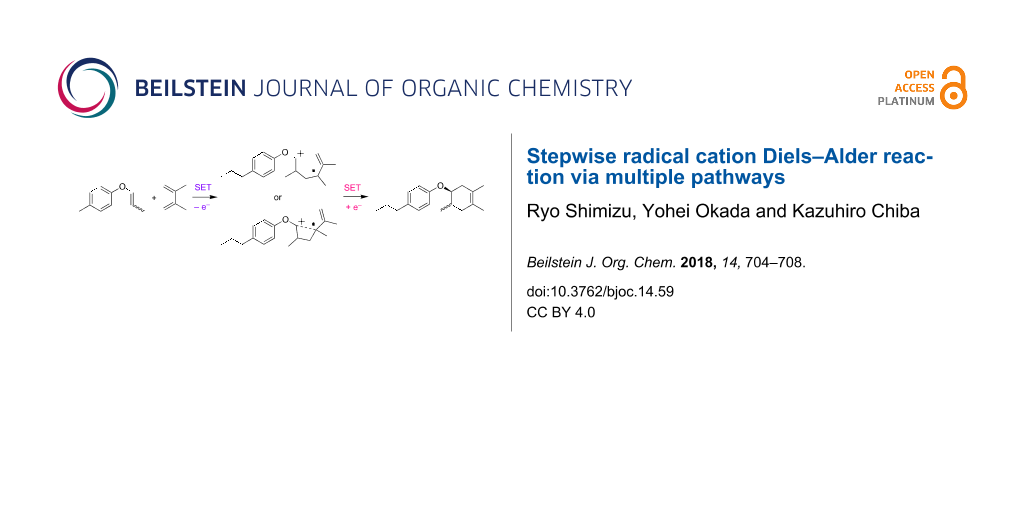
Abstract
Herein we disclose the radical cation Diels–Alder reaction of aryl vinyl ethers by electrocatalysis, which is triggered by an oxidative SET process. The reaction clearly proceeds in a stepwise fashion, which is a rare mechanism in this class. We also found that two distinctive pathways, including “direct” and “indirect”, are possible to construct the Diels–Alder adduct.

Graphical Abstract
Introduction
Umpolung, also known as polarity inversion, is a powerful approach in synthetic organic chemistry to trigger reactions that are otherwise difficult or impossible. In an umpolung reaction, the normal reactivity of the molecules being studied is reversed, e.g., electrophilicity is generated from a nucleophile. The single electron transfer (SET) process has been recognized as the most straightforward way to induce umpolung, which has recently been carried out by means of photo- [1-6] and electrochemical [7-12] approaches. A typical example involves an oxidative SET of an electron-rich and thus nucleophilic C–C double bond. The oxidative SET produces a radical cation species, which offers electrophilic reactivity for subsequent transformations. Enol ether radical cations are among the simplest members of this class and thus have been widely used in synthetic organic chemistry [13-15].
The Diels–Alder reaction is a classic reaction, and one of the most powerful methods to construct six-membered ring systems. A “normal” Diels–Alder reaction must consider an electronic matching of the substrates, with an electron-rich diene and electron-deficient dienophile as the general combination. Although having both an electron-rich diene and dienophile is a less- or non-effective combination, oxidative SET has proven to be able to overcome this mismatch and such a strategy is well-known as a radical cation Diels–Alder reaction. The use of trans-anethole as a model electron-rich dienophile is the representative example in this class. It was first reported by Bauld in 1986 [16] and was elegantly revisited by Yoon in 2011 [17] in the field of photoredox catalysis (Scheme 1). The reaction was further studied by Ferreira and Shores [18], followed by a unique mechanistic investigation by Rappé [19]. Although most recent examples of the radical cation Diels–Alder reaction have employed styrenes, the scope is not limited to such electron-rich dienophiles. Bauld and Yoon demonstrated that aryl vinyl ethers, enol ether equivalents, and aryl vinyl sulfides are also promising dienophiles for the reactions (Scheme 2) [17,20-25].
![[1860-5397-14-59-i1]](/bjoc/content/inline/1860-5397-14-59-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Radical cation Diels–Alder reaction of trans-anethole [17].
Scheme 1: Radical cation Diels–Alder reaction of trans-anethole [17].
![[1860-5397-14-59-i2]](/bjoc/content/inline/1860-5397-14-59-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Radical cation Diels–Alder reactions of aryl vinyl ether and sulfides [17,25].
Scheme 2: Radical cation Diels–Alder reactions of aryl vinyl ether and sulfides [17,25].
We have been developing oxidative SET-triggered cycloadditions of enol ethers by electrocatalysis [26-32] in lithium perchlorate/nitromethane electrolyte solution [33]. The reactions involve a radical cation chain process and are complete using a catalytic amount of electricity. During the course of our study, we discovered that radical cation Diels–Alder reactions are also possible by electrocatalysis, however, the scope was limited to styrenes [34]. Described herein is a stepwise radical cation Diels–Alder reaction of enol ethers by electrocatalysis, which proceeds via multiple unique pathways.
Results and Discussion
The present work began with the synthesis of the aryl vinyl ether 1 from p-propylphenol in 2 steps (Scheme S1 and Figure S1 in Supporting Information File 1). Both E- and Z-forms were readily purified by silica gel column chromatography. When the anodic oxidation of the Z-form 1Z was carried out in the presence of 2,3-dimethyl-1,3-butadiene (2), the corresponding Diels–Alder adduct 3 was obtained in excellent yield (Scheme 3). The reaction was completed using a catalytic amount of electricity and therefore should involve a radical cation chain process. Since Diels–Alder adduct 3 was an approximately cis/trans = 1:2 mixture, the reaction must proceed in a stepwise fashion. This stereochemistry was confirmed when the E-form 1E also gave the same synthetic outcome. Namely, the Diels–Alder adduct 3 was obtained as an approximately cis/trans = 1:2 mixture, indicating that the reaction was indeed stepwise.
![[1860-5397-14-59-i3]](/bjoc/content/inline/1860-5397-14-59-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Radical cation Diels–Alder reaction of aryl vinyl ether (1). Conditions: 1.0 M LiClO4/CH3NO2, carbon felt electrodes, 1.2 V vs Ag/AgCl, 0.7 F/mol.
Scheme 3: Radical cation Diels–Alder reaction of aryl vinyl ether (1). Conditions: 1.0 M LiClO4/CH3NO2, carbo...
When the reaction was carefully monitored by gas chromatography–mass spectrometry (GC–MS), we found that an adduct was generated in the early stage that was not the Diels–Alder product 3. Therefore, we intentionally stopped the reaction after 0.1 F/mol of electricity had passed and attempted to identify the adduct. We found that it was a vinylcyclobutane 4, whose formation was only observed in the early stage of the reaction and was completely consumed after 0.7 F/mol of electricity was passed (Scheme 4 and Figure 1). Vinylcyclobutanes are known to rearrange to cyclohexenes via thermal and/or photochemical processes and therefore, we carried out the anodic oxidation of vinylcyclobutane 4 in the absence of 2,3-dimethyl-1,3-butadiene (2, Scheme 5 and Supporting Information File 1, Figure S2). Interestingly, the rearrangement took place effectively to give Diels–Alder adduct 3 as an approximately cis/trans = 1:4 mixture, a significant difference from the synthetic outcome mentioned above (Scheme 3). Notably, no rearrangement was observed without electricity and the vinyl substituent was found to be essential for the transformation since no cyclohexane 6 was obtained from the cyclobutane 5 (Scheme 6). We also found that the Diels–Alder adduct 3 with an approximately cis/trans = 1:1 mixture was obtained in the early stage of the reaction (Supporting Information File 1, Figure S3).
![[1860-5397-14-59-i4]](/bjoc/content/inline/1860-5397-14-59-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Oxidative SET-triggered reaction of aryl vinyl ether 1c. Conditions: 1.0 M LiClO4/CH3NO2, carbon felt electrodes, 1.2 V vs Ag/AgCl, 0.1 F/mol.
Scheme 4: Oxidative SET-triggered reaction of aryl vinyl ether 1c. Conditions: 1.0 M LiClO4/CH3NO2, carbon fe...
![[1860-5397-14-59-1]](/bjoc/content/figures/1860-5397-14-59-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: GC–MS Monitoring of the oxidative SET-triggered reaction of aryl vinyl ether 1c.
Figure 1: GC–MS Monitoring of the oxidative SET-triggered reaction of aryl vinyl ether 1c.
![[1860-5397-14-59-i5]](/bjoc/content/inline/1860-5397-14-59-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Oxidative SET-triggered rearrangement of vinyl cyclobutane 4. Conditions: 1.0 M LiClO4/CH3NO2, carbon felt electrodes, 1.2 V vs Ag/AgCl, 0.5 F/mol.
Scheme 5: Oxidative SET-triggered rearrangement of vinyl cyclobutane 4. Conditions: 1.0 M LiClO4/CH3NO2, carb...
![[1860-5397-14-59-i6]](/bjoc/content/inline/1860-5397-14-59-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Unsuccessful rearrangement of cyclobutanes. Conditions: 1.0 M LiClO4/CH3NO2, carbon felt electrodes, a) no electricity, b)1.2 V vs Ag/AgCl, 0.1 F/mol.
Scheme 6: Unsuccessful rearrangement of cyclobutanes. Conditions: 1.0 M LiClO4/CH3NO2, carbon felt electrodes...
Taken together, we can now propose a mechanism for the radical cation Diels–Alder reaction of the aryl vinyl ether 1 (Scheme 7). On the basis of the oxidation potentials, oxidative SET would selectively take place from the aryl vinyl ether 1 even in the presence of 2,3-dimethyl-1,3-butadiene (2) to generate the radical cation 1·+, in which the stereochemistry derived from the starting material is lost. Indeed, the anodic oxidation of either the Z- (1Z) or the E-form (1E) in the absence of 2,3-dimethyl-1,3-butadiene (2) led to isomerization. In other words, inversion of the configuration of the radical cation 1·+ proceeds readily, leading to a loss in stereoselectivity. The radical cation 1·+ is then trapped by 2,3-dimethyl-1,3-butadiene (2) to construct the acyclic radical cation intermediate A·+, which is potentially converted into the aromatic radical cation with a six-membered ring (3·+) or a four-membered ring (4·+) via rapid intramolecular SET processes. It can be rationalized that the six-membered ring closure of the acyclic radical cation intermediate (A·+) would be a stepwise process, leading to the Diels–Alder adduct 3 as an approximately cis/trans = 1:1 mixture. However, since the oxidative SET-triggered rearrangement of the vinylcyclobutane 4 gives the Diels–Alder adduct 3 as an approximately cis/trans = 1:4 mixture, this should proceed via a different intermediate from the acyclic radical cation intermediate (A·+). Here, we propose the cyclic radical cation intermediate B·+ for the rearrangement, imparting stereoselectivity for the six-membered ring closure. The anodic oxidation of the Diels–Alder adduct 3 does not give either the retro-reaction product 1 or the vinylcyclobutane 4 and therefore, it seems that the overall transformation involves irreversible steps. Thus, the radical cation Diels–Alder reaction of aryl vinyl ether 1 effectively proceeds either via “direct” or “indirect” pathways, affording the corresponding adduct 3 in excellent yield.
![[1860-5397-14-59-i7]](/bjoc/content/inline/1860-5397-14-59-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Proposed mechanism for the radical cation Diels–Alder reaction of aryl vinyl ether 1.
Scheme 7: Proposed mechanism for the radical cation Diels–Alder reaction of aryl vinyl ether 1.
Conclusion
In conclusion, we have demonstrated that the radical cation Diels–Alder reaction initiated by electrocatalysis in lithium perchlorate/nitromethane electrolyte solution is not limited to styrenes but was also effective for the aryl vinyl ether. While the mechanism is still controversial, most reported radical cation Diels–Alder reactions are highly stereoselective, even including stereospecific concerted examples. On the other hand, our current results clearly indicate that a stepwise mechanism is also effective for this class. Furthermore, the radical cation Diels–Alder reaction of the aryl vinyl ether is found to proceed via multiple unique pathways, including direct and indirect manners. Our findings described herein are beneficial to design novel SET-triggered cycloadditions, which are under development in our laboratory.
Supporting Information
| Supporting Information File 1: Additional scheme and figures, general remarks, synthesis and characterization data, including copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 1.4 MB | Download |
References
-
Twilton, J.; Le, C.; Zhang, P.; Shaw, M. H.; Evans, R. W.; MacMillan, D. W. C. Nat. Rev. Chem. 2017, 1, No. 0052. doi:10.1038/s41570-017-0052
Return to citation in text: [1] -
Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r
Return to citation in text: [1] -
Skubi, K. L.; Blum, T. R.; Yoon, T. P. Chem. Rev. 2016, 116, 10035–10074. doi:10.1021/acs.chemrev.6b00018
Return to citation in text: [1] -
Schultz, D. M.; Yoon, T. P. Science 2014, 343, 1239176. doi:10.1126/science.1239176
Return to citation in text: [1] -
Kärkäs, M. D.; Porco, J. A., Jr.; Stephenson, C. R. J. Chem. Rev. 2016, 116, 9683–9747. doi:10.1021/acs.chemrev.5b00760
Return to citation in text: [1] -
Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/B913880N
Return to citation in text: [1] -
Yoshida, J.-i.; Shimizu, A.; Hayashi, R. Chem. Rev. 2018. doi:10.1021/acs.chemrev.7b00475
Return to citation in text: [1] -
Yoshida, J.-i.; Kataoka, K.; Horcajada, R.; Nagaki, A. Chem. Rev. 2008, 108, 2265–2299. doi:10.1021/cr0680843
Return to citation in text: [1] -
Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397
Return to citation in text: [1] -
Horn, E. J.; Rosen, B. R.; Baran, P. S. ACS Cent. Sci. 2016, 2, 302–308. doi:10.1021/acscentsci.6b00091
Return to citation in text: [1] -
Francke, R.; Little, R. D. Chem. Soc. Rev. 2014, 43, 2492–2521. doi:10.1039/c3cs60464k
Return to citation in text: [1] -
Sperry, J. B.; Wright, D. L. Chem. Soc. Rev. 2006, 35, 605–621. doi:10.1039/b512308a
Return to citation in text: [1] -
Feng, R.; Smith, J. A.; Moeller, K. D. Acc. Chem. Res. 2017, 50, 2346–2352. doi:10.1021/acs.accounts.7b00287
Return to citation in text: [1] -
Moeller, K. D. Synlett 2009, 1208–1218. doi:10.1055/s-0028-1088126
Return to citation in text: [1] -
Tang, F.; Moeller, K. D. Tetrahedron 2009, 65, 10863–10875. doi:10.1016/j.tet.2009.09.028
Return to citation in text: [1] -
Reynolds, D. W.; Bauld, N. L. Tetrahedron 1986, 42, 6189–6194. doi:10.1016/S0040-4020(01)88079-0
Return to citation in text: [1] -
Lin, S.; Ischay, M. A.; Fry, C. G.; Yoon, T. P. J. Am. Chem. Soc. 2011, 133, 19350–19353. doi:10.1021/ja2093579
Return to citation in text: [1] [2] [3] [4] -
Stevenson, S. M.; Shores, M. P.; Ferreira, E. M. Angew. Chem., Int. Ed. 2015, 54, 6506–6510. doi:10.1002/anie.201501220
Return to citation in text: [1] -
Higgins, R. F.; Fatur, S. M.; Shepard, S. G.; Stevenson, S. M.; Boston, D. J.; Ferreira, E. M.; Damrauer, N. H.; Rappé, A. K.; Shores, M. P. J. Am. Chem. Soc. 2016, 138, 5451–5464. doi:10.1021/jacs.6b02723
Return to citation in text: [1] -
Reynolds, D. W.; Harirchian, B.; Chiou, H. S.; Marsh, B. K.; Bauld, N. L. J. Phys. Org. Chem. 1989, 2, 57–88. doi:10.1002/poc.610020108
Return to citation in text: [1] -
Pabon, R. A.; Bellville, D. J.; Bauld, N. L. J. Am. Chem. Soc. 1984, 106, 2730–2731. doi:10.1021/ja00321a059
Return to citation in text: [1] -
Harirchian, B.; Bauld, N. L. J. Am. Chem. Soc. 1989, 111, 1826–1828. doi:10.1021/ja00187a041
Return to citation in text: [1] -
Aplin, J. T.; Bauld, N. L. J. Chem. Soc., Perkin Trans. 2 1997, 853–855. doi:10.1039/a605317c
Return to citation in text: [1] -
Harirchian, B.; Bauld, N. L. Tetrahedron Lett. 1987, 28, 927–930. doi:10.1016/S0040-4039(00)95876-3
Return to citation in text: [1] -
Lin, S.; Lies, S. D.; Gravatt, C. S.; Yoon, T. P. Org. Lett. 2017, 19, 368–371. doi:10.1021/acs.orglett.6b03545
Return to citation in text: [1] [2] -
Okada, Y.; Chiba, K. Chem. Rev. 2018. doi:10.1021/acs.chemrev.7b00400
Return to citation in text: [1] -
Yamaguchi, Y.; Okada, Y.; Chiba, K. J. Org. Chem. 2013, 78, 2626–2638. doi:10.1021/jo3028246
Return to citation in text: [1] -
Okada, Y.; Nishimoto, A.; Akaba, R.; Chiba, K. J. Org. Chem. 2011, 76, 3470–3476. doi:10.1021/jo200490q
Return to citation in text: [1] -
Okada, Y.; Chiba, K. Electrochim. Acta 2011, 56, 1037–1042. doi:10.1016/j.electacta.2010.10.042
Return to citation in text: [1] -
Okada, Y.; Akaba, R.; Chiba, K. Org. Lett. 2009, 11, 1033–1035. doi:10.1021/ol802984n
Return to citation in text: [1] -
Arata, M.; Miura, T.; Chiba, K. Org. Lett. 2007, 9, 4347–4350. doi:10.1021/ol7019845
Return to citation in text: [1] -
Chiba, K.; Miura, T.; Kim, S.; Kitano, Y.; Tada, M. J. Am. Chem. Soc. 2001, 123, 11314–11315. doi:10.1021/ja016885b
Return to citation in text: [1] -
Imada, Y.; Yamaguchi, Y.; Shida, N.; Okada, Y.; Chiba, K. Chem. Commun. 2017, 53, 3960–3963. doi:10.1039/C7CC00664K
Return to citation in text: [1] -
Okada, Y.; Yamaguchi, Y.; Ozaki, A.; Chiba, K. Chem. Sci. 2016, 7, 6387–6393. doi:10.1039/C6SC02117D
Return to citation in text: [1]
| 1. | Twilton, J.; Le, C.; Zhang, P.; Shaw, M. H.; Evans, R. W.; MacMillan, D. W. C. Nat. Rev. Chem. 2017, 1, No. 0052. doi:10.1038/s41570-017-0052 |
| 2. | Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r |
| 3. | Skubi, K. L.; Blum, T. R.; Yoon, T. P. Chem. Rev. 2016, 116, 10035–10074. doi:10.1021/acs.chemrev.6b00018 |
| 4. | Schultz, D. M.; Yoon, T. P. Science 2014, 343, 1239176. doi:10.1126/science.1239176 |
| 5. | Kärkäs, M. D.; Porco, J. A., Jr.; Stephenson, C. R. J. Chem. Rev. 2016, 116, 9683–9747. doi:10.1021/acs.chemrev.5b00760 |
| 6. | Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/B913880N |
| 17. | Lin, S.; Ischay, M. A.; Fry, C. G.; Yoon, T. P. J. Am. Chem. Soc. 2011, 133, 19350–19353. doi:10.1021/ja2093579 |
| 16. | Reynolds, D. W.; Bauld, N. L. Tetrahedron 1986, 42, 6189–6194. doi:10.1016/S0040-4020(01)88079-0 |
| 13. | Feng, R.; Smith, J. A.; Moeller, K. D. Acc. Chem. Res. 2017, 50, 2346–2352. doi:10.1021/acs.accounts.7b00287 |
| 14. | Moeller, K. D. Synlett 2009, 1208–1218. doi:10.1055/s-0028-1088126 |
| 15. | Tang, F.; Moeller, K. D. Tetrahedron 2009, 65, 10863–10875. doi:10.1016/j.tet.2009.09.028 |
| 34. | Okada, Y.; Yamaguchi, Y.; Ozaki, A.; Chiba, K. Chem. Sci. 2016, 7, 6387–6393. doi:10.1039/C6SC02117D |
| 7. | Yoshida, J.-i.; Shimizu, A.; Hayashi, R. Chem. Rev. 2018. doi:10.1021/acs.chemrev.7b00475 |
| 8. | Yoshida, J.-i.; Kataoka, K.; Horcajada, R.; Nagaki, A. Chem. Rev. 2008, 108, 2265–2299. doi:10.1021/cr0680843 |
| 9. | Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397 |
| 10. | Horn, E. J.; Rosen, B. R.; Baran, P. S. ACS Cent. Sci. 2016, 2, 302–308. doi:10.1021/acscentsci.6b00091 |
| 11. | Francke, R.; Little, R. D. Chem. Soc. Rev. 2014, 43, 2492–2521. doi:10.1039/c3cs60464k |
| 12. | Sperry, J. B.; Wright, D. L. Chem. Soc. Rev. 2006, 35, 605–621. doi:10.1039/b512308a |
| 17. | Lin, S.; Ischay, M. A.; Fry, C. G.; Yoon, T. P. J. Am. Chem. Soc. 2011, 133, 19350–19353. doi:10.1021/ja2093579 |
| 26. | Okada, Y.; Chiba, K. Chem. Rev. 2018. doi:10.1021/acs.chemrev.7b00400 |
| 27. | Yamaguchi, Y.; Okada, Y.; Chiba, K. J. Org. Chem. 2013, 78, 2626–2638. doi:10.1021/jo3028246 |
| 28. | Okada, Y.; Nishimoto, A.; Akaba, R.; Chiba, K. J. Org. Chem. 2011, 76, 3470–3476. doi:10.1021/jo200490q |
| 29. | Okada, Y.; Chiba, K. Electrochim. Acta 2011, 56, 1037–1042. doi:10.1016/j.electacta.2010.10.042 |
| 30. | Okada, Y.; Akaba, R.; Chiba, K. Org. Lett. 2009, 11, 1033–1035. doi:10.1021/ol802984n |
| 31. | Arata, M.; Miura, T.; Chiba, K. Org. Lett. 2007, 9, 4347–4350. doi:10.1021/ol7019845 |
| 32. | Chiba, K.; Miura, T.; Kim, S.; Kitano, Y.; Tada, M. J. Am. Chem. Soc. 2001, 123, 11314–11315. doi:10.1021/ja016885b |
| 17. | Lin, S.; Ischay, M. A.; Fry, C. G.; Yoon, T. P. J. Am. Chem. Soc. 2011, 133, 19350–19353. doi:10.1021/ja2093579 |
| 20. | Reynolds, D. W.; Harirchian, B.; Chiou, H. S.; Marsh, B. K.; Bauld, N. L. J. Phys. Org. Chem. 1989, 2, 57–88. doi:10.1002/poc.610020108 |
| 21. | Pabon, R. A.; Bellville, D. J.; Bauld, N. L. J. Am. Chem. Soc. 1984, 106, 2730–2731. doi:10.1021/ja00321a059 |
| 22. | Harirchian, B.; Bauld, N. L. J. Am. Chem. Soc. 1989, 111, 1826–1828. doi:10.1021/ja00187a041 |
| 23. | Aplin, J. T.; Bauld, N. L. J. Chem. Soc., Perkin Trans. 2 1997, 853–855. doi:10.1039/a605317c |
| 24. | Harirchian, B.; Bauld, N. L. Tetrahedron Lett. 1987, 28, 927–930. doi:10.1016/S0040-4039(00)95876-3 |
| 25. | Lin, S.; Lies, S. D.; Gravatt, C. S.; Yoon, T. P. Org. Lett. 2017, 19, 368–371. doi:10.1021/acs.orglett.6b03545 |
| 33. | Imada, Y.; Yamaguchi, Y.; Shida, N.; Okada, Y.; Chiba, K. Chem. Commun. 2017, 53, 3960–3963. doi:10.1039/C7CC00664K |
| 19. | Higgins, R. F.; Fatur, S. M.; Shepard, S. G.; Stevenson, S. M.; Boston, D. J.; Ferreira, E. M.; Damrauer, N. H.; Rappé, A. K.; Shores, M. P. J. Am. Chem. Soc. 2016, 138, 5451–5464. doi:10.1021/jacs.6b02723 |
| 18. | Stevenson, S. M.; Shores, M. P.; Ferreira, E. M. Angew. Chem., Int. Ed. 2015, 54, 6506–6510. doi:10.1002/anie.201501220 |
| 17. | Lin, S.; Ischay, M. A.; Fry, C. G.; Yoon, T. P. J. Am. Chem. Soc. 2011, 133, 19350–19353. doi:10.1021/ja2093579 |
| 25. | Lin, S.; Lies, S. D.; Gravatt, C. S.; Yoon, T. P. Org. Lett. 2017, 19, 368–371. doi:10.1021/acs.orglett.6b03545 |
© 2018 Shimizu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)