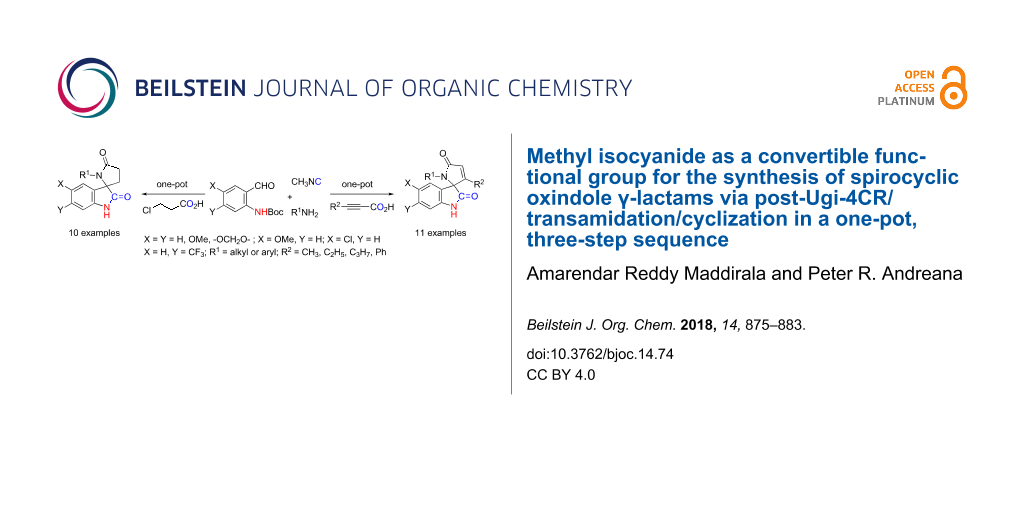
Abstract
The synthesis of spiro[indoline-3,2'-pyrrole]-2,5'(1'H)-diones and spiro[indoline-3,2'-pyrrolidine]-2,5'-diones, via a post-Ugi-domino transamidation/cyclization sequential process, has been achieved in three sequential steps utilizing a one-pot reaction protocol. The variation in carboxylic acid substrates allows for the generation of new chiral racemic quaternary carbon centers under basic conditions providing molecular diversity and a small library of spirocyclic oxindoles.

Graphical Abstract
Introduction
The Ugi-multicomponent coupling reaction [1,2], followed by post-modification transformations involving tandem reaction sequences [3] and the Ugi–deprotection–cyclization (UDC) strategies [4-8] have been exploited as powerful tools allowing access to biological and pharmaceutical high-value heterocyclic scaffolds [9-11]. These reactions are appealing in that they are atom economical, simple and generate ample molecular diversity with the ease of using readily available starting materials. Developing new, post-modified Ugi-four-component reaction (Ugi-4CR) transformations in domino cyclization [12-15] sequences are very important for achieving unprecedented chemical bonds and functionality towards the construction of synthetic scaffolds.
The synthesis of spirocyclic oxindoles has always been of key interest to organic chemists because of significant biological activity [16-20] and their presence in naturally occurring molecules [21-23]. Significant efforts have been made to design creative synthetic strategies for spirocyclic oxindole molecules, of which, isatin-based domino reactions [24-30] have proved to be very versatile [31] and readily achievable [32-37]. However, finding a simple and efficient synthetic method for these molecules that allows for structural diversity is also important but not necessarily trivial. For these and other reasons, we became interested in synthesizing spiro[indoline-3,2'-pyrrole]-2,5'(1'H)-dione and spiro[indoline-3,2'-pyrrolidine]-2,5'-dione scaffolds (a class of spirocyclic oxindole γ-lactams).
There have been other groups in the past, including our own research group, who have reported on post-modified Ugi-four-component synthetic strategies (Scheme 1) towards the synthesis of 2-oxindoles and spiro[indoline-3,2'-pyrrole]-2,5'(1'H)-diones and spiro[indoline-3,2'-pyrrolidine]-2,5'-diones. Zhu et al. [38] reported 3-substituted-2-indolinones via a microwave-assisted post-Ugi-4CR/Buchwald–Hartwig reaction and another similar approach was illustrated by Van der Eycken et al. [39] for spiro[indoline-3,2'-pyrrole]-2,5'(1'H)-diones. In previous efforts to study 3-substituted 2-indolinones through a three-step post-Ugi-4CR/Bechamp type-reduction followed by a transamidation sequence strategy [40], we came across interesting observations. We noted that when methyl isocyanide [41,42] was used for the Ugi-4CR and the subsequent post-intramolecular transamidation was performed under acidic conditions, particularly in the presence of TFA, the reaction led to 3-substituted 2-indolinones in a three-step process [40]. In this work, we discovered that methyl isocyanide [43] operates under a mechanism of convertible isocyanides (CICs) [44-49], and could be thought of as a synthetic equivalent to ‘CO’ for insertion into the 2-indolinone backbone (shown in Scheme 1). To further elaborate on this observation and for understanding the role of methyl isocyanide as a CIC, we designed an efficient synthetic strategy for spiro[indoline-3,2'-pyrrole]-2,5'(1'H)-diones and spiro[indoline-3,2'-pyrrolidine]-2,5'-diones via a one-pot, three-step reaction sequence. Advantages of this strategy include: a) minimal number of synthetic steps, b) avoidance of tedious work-up procedures including purification, and c) use of starting materials either readily available or facile to synthesize.
![[1860-5397-14-74-i1]](/bjoc/content/inline/1860-5397-14-74-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Previously reported post-Ugi-4CR methods for the synthesis of 2-oxindoles and spirocyclic 2-oxindoles.
Scheme 1: Previously reported post-Ugi-4CR methods for the synthesis of 2-oxindoles and spirocyclic 2-oxindol...
Here within, we document that the reaction sequence for spirocyclic oxindole γ-lactams (Scheme 2) follows a three-step sequential strategy involving: a) an Ugi-4CR, b) an acid-promoted intramolecular transamidation, and c) a base-mediated cyclization giving spiro[indoline-3,2'-pyrrole]-2,5'(1'H)-diones. We propose that the acid-mediated Boc deprotection in the Ugi intermediate 5 leads to aniline intermediate I which can simultaneously undergo a CIC cyclization through an intramolecular transamidation process giving compound 6 and, in the process, extrude methylamine as a gaseous byproduct. Furthermore, compound 6 is proposed to undergo an intramolecular cyclization, under basic conditions, yielding target compounds 7 (Scheme 2).
![[1860-5397-14-74-i2]](/bjoc/content/inline/1860-5397-14-74-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Post-Ugi-4CR/transamidation/cyclization sequence.
Scheme 2: Post-Ugi-4CR/transamidation/cyclization sequence.
Results and Discussion
Our initial synthetic attempts began with the Ugi-4CR involving stoichiometrically equal amounts of 2-(Boc-amino)benzaldehyde (1a), aniline (2a), tetrolic acid (3a), and methyl isocyanide (4) in methanol at room temperature to generate adduct 5a; confirmed using mass spectroscopic analysis (Scheme 3). Intermediate 5a, which was not purified, was prone to undergo an intramolecular transamidation when 50% TFA was used to remove the Boc group in DCM at room temperature for 5 h ultimately yielding 6a and methylamine as the gaseous byproduct. Following the neutralization of TFA, unpurified compound 6a was tested for the ability to cyclize under basic conditions. We began our cyclization studies with 6a by dissolving it in acetonitrile following by the addition of 2 equiv K2CO3 and then refluxing for 1 h. The reaction was monitored by TLC, which indicated the disappearance of the starting material and formation of a new spot. Post-work-up and purification, spectroscopic analysis of the product matched nicely with the 5-endo-dig cyclization product 7a and not the 4-exo-dig-cyclization compound 7a' (Scheme 3). We rationalized, as per Baldwin’s rules, that the 5-endo-dig cyclization of a 1,4-Michael addition was more favorable than the 4-exo-dig-cyclization [50,51]. The structure of compound 7a was definitively confirmed by X-ray analysis (Figure 1).
![[1860-5397-14-74-i3]](/bjoc/content/inline/1860-5397-14-74-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Base-promoted intramolecular 5-endo-dig cyclization.
Scheme 3: Base-promoted intramolecular 5-endo-dig cyclization.
![[1860-5397-14-74-1]](/bjoc/content/figures/1860-5397-14-74-1.png?scale=2.0&max-width=1024&background=FFFFFF)
We then elected to carry out yield optimization studies of the cyclization reaction using a number of solvents, reagents and reaction temperatures (Table 1) and also to explore the possibility of a possible 4-exo-dig outcome. The study validated that all the reaction conditions, with the exception of AgOTf/DCM (Table 1, entry 6), led to the formation of the desired 5-endo-dig cyclization product 7a and none of the 4-exo-dig cyclization product 7a' was observed. In Table 1, entries 2–5 and 7 show moderate product yields whereas entries 1 and 8 show superior yields of >80%.
Table 1: Optimization conditions for the intramolecular cyclization (Scheme 3, 6→7a and 7a').a
| entry | reagent | solvent | temperature | time | yield 7a (%) | yield 7a' (%) |
|---|---|---|---|---|---|---|
| 1 | K2CO3 | MeCN | reflux | 1 h | 82 | n.o. |
| 2 | K2CO3 | methanol | rt | 2 h | 72 | n.o. |
| 3 | K2CO3 | toluene | reflux | 2 h | 70 | n.o. |
| 4 | Cs2CO3 | toluene | reflux | 2 h | 72 | n.o. |
| 5 | Et3N | DCM | rt | 4 h | 65 | n.o. |
| 6 | AgOTf | DCM | rt | 24 h | n.r. | n.r. |
| 7 | KOt-Bu | THF | rt | 30 min | 75 | n.o. |
| 8 | KOt-Bu | MeCN | rt | 30 min | 80 | n.o. |
an.o.: not observed, n.r.: no reaction; highlighted entries denote best results.
With the optimized reaction conditions in hand (Table 1, entries 1 and 8), the substrate scope was explored using a one-pot Ugi-4CR/transamidation/cyclization sequence employing various combinations of readily available and synthetically accessible starting materials (Figure 2). Under all circumstances, the intramolecular cyclization proceeded smoothly using K2CO3/MeCN reflux conditions and products 7b–k were obtained in good to excellent yields (Scheme 4). Although the results shown only reflect the use of K2CO3/MeCN reflux conditions, similar, if not exact reaction outcomes were observed when conditions from Table 1, entry 8 were used.
![[1860-5397-14-74-2]](/bjoc/content/figures/1860-5397-14-74-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Readily and synthetically accessible starting materials.
Figure 2: Readily and synthetically accessible starting materials.
![[1860-5397-14-74-i4]](/bjoc/content/inline/1860-5397-14-74-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction scope with varying combinations of substrates.
Scheme 4: Reaction scope with varying combinations of substrates.
To further explore the utility of our methodology, we examined similar reaction conditions for the synthesis of spiro[indoline-3,2'-pyrrolidine]-2,5'-dione scaffolds (Scheme 5). For this reaction we used 2-(Boc-amino)benzaldehyde 1c, aniline (2a), 3-chloropropanoic acid (3e) and methyl isocyanide (4) in a one-pot reaction process to generate compound 6b from 5b. In all cases, the Michael acceptor [52] (intermediate II) was generated in situ from 6b, under basic conditions (Table 1, entry 1, K2CO3/MeCN/reflux), followed by the intramolecular cyclization proceeding through a 1,4-Michael addition to form an exclusive 5-endo-trig cyclization of 5-chloro-1'-phenylspiro[indoline-3,2'-pyrrolidine]-2,5'-dione (8a, Scheme 5). Compound 8a was unequivocally confirmed by both, mass spectral analysis and NMR.
![[1860-5397-14-74-i5]](/bjoc/content/inline/1860-5397-14-74-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of 5-chloro-1'-phenylspiro[indoline-3,2'-pyrrolidine]-2,5'-dione (8a).
Scheme 5: Synthesis of 5-chloro-1'-phenylspiro[indoline-3,2'-pyrrolidine]-2,5'-dione (8a).
Encouraged by the results, we prepared a library of spiro[indoline-3,2'-pyrrolidine]-2,5'-diones 8b–i from readily available starting materials in which overall yields were determined to be moderate to good (Figure 3). Furthermore, the applicability and utility of this process was demonstrated through the synthesis of a 5-HT6 receptor antagonist 8j (Scheme 6) [53].
![[1860-5397-14-74-3]](/bjoc/content/figures/1860-5397-14-74-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Small molecule library of spiro[indoline-3,2'-pyrrolidine]-2,5'-dione analogs.
Figure 3: Small molecule library of spiro[indoline-3,2'-pyrrolidine]-2,5'-dione analogs.
![[1860-5397-14-74-i6]](/bjoc/content/inline/1860-5397-14-74-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Method applicability for the one-pot synthesis of 5-HT6 receptor antagonist 8j [53].
Scheme 6: Method applicability for the one-pot synthesis of 5-HT6 receptor antagonist 8j [53].
Conclusion
In conclusion, we have investigated and developed an efficient process towards spirocyclic α,β-unsaturated γ-lactam oxindoles and spirocyclic γ-lactams using a one-pot three-step post-Ugi-4CR intramolecular transamidation/cyclization approach. We utilized this strategy for the synthesis of a small library of spiro[indoline-3,2'-pyrrole]-2,5'(1'H)-dione and spiro[indoline-3,2'-pyrrolidine]-2,5'-dione analogs illustrating important utility toward biologically relevant compounds. To this point, our strategy was further extended and applied towards the synthesis of a well-known 5-HT6 receptor antagonist 8j. We also successfully utilized methyl isocyanide, as a CIC, for spiro[indoline-3,2'-pyrrole]-2,5'(1'H)-diones and spiro[indoline-3,2'-pyrrolidine]-2,5'-diones. Immediate plans are underway to test the inhibitory properties of the newly synthesized compounds in in vitro assays.
Experimental
All reagents and solvents that were purchased were used without further purification unless otherwise stated. Reaction progress was monitored by thin-layer chromatography (TLC). Spots on TLC were visualized using UV light. Column chromatography was performed using normal-phase silica gel. Yields refer to chromatographically and spectroscopically pure compounds. 1H and 13C NMR were recorded using a Bruker Avance III 600 MHz. The residual DMSO-d6 1H quintet at δ 2.50 ppm and residual 13C septet at δ 39.51 ppm, CDCl3 1H singlet at δ 7.27 ppm and 13C triplet at δ 77.23 ppm were used as standards for 1H NMR and 13C NMR spectra, respectively. Signal patterns are indicated as s: singlet; d: doublet; t: triplet; q: quartet; m: multiplet; dd: doublet of doublets; and br: broad. Coupling constants are reported in hertz (Hz). High resolution mass spectra (HRMS) were obtained using a Bruker Maxis 4G mass spectrometer.
General procedure for 7a–k: Into a clear solution of 2-(Boc-amino)benzaldehyde 1 (1 mmol) in methanol (5 mL) was added amine 2 (1 mmol) and stirred for 5 minutes at room temperature. Carboxylic acid 3 (1 mmol) and methyl isocyanide (4, 1 mmol) were then added simultaneously. The mixture was stirred until no noticeable amounts of starting material were visible by TLC. Upon completion of the reaction, methanol was evaporated under reduced pressure and the crude Ugi products, without any purification, dissolved in a CH2Cl2 and trifluoroacetic acid mixture (1:1, 2 mL) and subsequently stirred at room temperature for 5 h. The reaction progress was monitored by TLC and upon completion of the reaction the solvent was evaporated under reduced pressure. Without any purification, the crude compound was dissolved in acetonitrile (2 mL) and K2CO3 (2 mmol) was added. The reaction was allowed to stir under refluxing conditions for 1 h and the reaction was monitored for completion using TLC. Upon noted completion of the reaction, the mixture was cooled to room temperature and the solvent was evaporated under reduced pressure. The crude compound(s) was subjected to flash column chromatography (EtOAc/hexanes) to yield pure compounds 7a–k.
General procedure for 8a–j: Into a clear solution of 2-(Boc-amino)benzaldehyde 1 (1 mmol) in methanol (5 mL) was added amine 2 (1 mmol) and stirred for 5 minutes at room temperature. Then, 3-chloropropanoic acid (3, 1 mmol) and methyl isocyanide (4, 1 mmol) were added to the reaction pot. The reaction was allowed to stir until no noticeable amounts of starting material were visible using TLC. Upon completion of the reaction, methanol was evaporated under reduced pressure and the crude Ugi products, without any purification, was dissolved in a CH2Cl2 and trifluoroacetic acid mixture (1:1, 2 mL). The reaction was then allowed to stir at room temperature for 5 h and monitored by TLC. Upon completion of the reaction, the solvent was evaporated under reduced pressure and the crude compound, without any purification, was dissolved in acetonitrile (2 mL) and K2CO3 (2 mmol) was added. The reaction was then allowed to stir under refluxing conditions for 1 h and reaction completion was monitored using TLC. The reaction mixture was then allowed to cool to room temperature and the solvent evaporated under reduced pressure. The crude products were subjected to flash column chromatography (EtOAc/hexanes) to yield pure compounds 8a–j.
Supporting Information
| Supporting Information File 1: 1H and 13C NMR spectroscopic data for compounds 7a–k and 8a–j, and X-ray crystal structure details for 7a. | ||
| Format: PDF | Size: 1.2 MB | Download |
References
-
Ugi, I.; Steinbrückner, C. Angew. Chem. 1960, 72, 267–268. doi:10.1002/ange.19600720709
Return to citation in text: [1] -
Dömling, A.; Ugi, I. Angew. Chem., Int. Ed. 2000, 39, 3168–3210. doi:10.1002/1521-3773(20000915)39:18<3168::AID-ANIE3168>3.0.CO;2-U
Return to citation in text: [1] -
Sharma, U. K.; Sharma, N.; Vachhani, D. D.; Van der Eycken, E. V. Chem. Soc. Rev. 2015, 44, 1836–1860. doi:10.1039/C4CS00253A
Return to citation in text: [1] -
Hulme, C.; Peng, J.; Louridas, B.; Menard, P.; Krolikowski, P.; Kumar, N. V. Tetrahedron Lett. 1998, 39, 8047–8050. doi:10.1016/S0040-4039(98)01770-5
Return to citation in text: [1] -
Huang, Y.; Khoury, K.; Chanas, T.; Dömling, A. Org. Lett. 2012, 14, 5916–5919. doi:10.1021/ol302837h
Return to citation in text: [1] -
Xu, Z.; Shaw, A. Y.; Dietrich, J.; Cappelli, A. P.; Nichol, G.; Hulme, C. Mol. Diversity 2012, 16, 73–79. doi:10.1007/s11030-011-9354-x
Return to citation in text: [1] -
Hulme, C.; Ma, L.; Romano, J.; Morrissette, M. Tetrahedron Lett. 1999, 40, 7925–7928. doi:10.1016/S0040-4039(99)01580-4
Return to citation in text: [1] -
Hulme, C.; Ma, L.; Cherrier, M.-P.; Romano, J. J.; Morton, G.; Duquenne, C.; Salvino, J.; Labaudiniere, R. Tetrahedron Lett. 2000, 41, 1883–1887. doi:10.1016/S0040-4039(00)00052-6
Return to citation in text: [1] -
Tempest, P.; Ma, V.; Thomas, S.; Hua, Z.; Kelly, M. G.; Hulme, C. Tetrahedron Lett. 2001, 42, 4959–4962. doi:10.1016/S0040-4039(01)00919-4
Return to citation in text: [1] -
Dömling, A.; Wang, W.; Wang, K. Chem. Rev. 2012, 112, 3083–3135. doi:10.1021/cr100233r
Return to citation in text: [1] -
Jiang, B.; Rajale, T.; Wever, W.; Tu, S.-J.; Li, G. Chem. – Asian J. 2010, 5, 2318–2335. doi:10.1002/asia.201000310
Return to citation in text: [1] -
Bourgault, J. P.; Maddirala, A. R.; Andreana, P. R. Org. Biomol. Chem. 2014, 12, 8125–8127. doi:10.1039/C4OB01148A
Return to citation in text: [1] -
Koopmanschap, G.; Ruijter, E.; Orru, R. V. A. Beilstein J. Org. Chem. 2014, 10, 544–598. doi:10.3762/bjoc.10.50
Return to citation in text: [1] -
Santra, S.; Andreana, P. R. Angew. Chem., Int. Ed. 2011, 50, 9418–9422. doi:10.1002/anie.201103567
Return to citation in text: [1] -
Santra, S.; Andreana, P. R. J. Org. Chem. 2011, 76, 2261–2264. doi:10.1021/jo102305q
Return to citation in text: [1] -
De Silva, R. A.; Santra, S.; Andreana, P. R. Org. Lett. 2008, 10, 4541–4544. doi:10.1021/ol801841m
Return to citation in text: [1] -
Kumar, A.; Gupta, G.; Bishnoi, A. K.; Saxena, R.; Saini, K. S.; Konwar, R.; Kumar, S.; Dwivedi, A. Bioorg. Med. Chem. 2015, 23, 839–848. doi:10.1016/j.bmc.2014.12.037
Return to citation in text: [1] -
Santos, M. M. M. Tetrahedron 2014, 70, 9735–9757. doi:10.1016/j.tet.2014.08.005
Return to citation in text: [1] -
Yu, B.; Yu, D.-Q.; Liu, H.-M. Eur. J. Med. Chem. 2015, 97, 673–698. doi:10.1016/j.ejmech.2014.06.056
Return to citation in text: [1] -
Yu, B.; Yu, Z.; Qi, P.-P.; Yu, D.-Q.; Liu, H.-M. Eur. J. Med. Chem. 2015, 95, 35–40. doi:10.1016/j.ejmech.2015.03.020
Return to citation in text: [1] -
Dong, H.; Song, S.; Li, J.; Xu, C.; Zhang, H.; Ouyang, L. Bioorg. Med. Chem. Lett. 2015, 25, 3585–3591. doi:10.1016/j.bmcl.2015.06.076
Return to citation in text: [1] -
Fu, P.; Kong, F.; Li, X.; Wang, Y.; Zhu, W. Org. Lett. 2014, 16, 3708–3711. doi:10.1021/ol501523d
Return to citation in text: [1] -
Jossang, A.; Jossang, P.; Hadi, H. A.; Sevenet, T.; Bodo, B. J. Org. Chem. 1991, 56, 6527–6530. doi:10.1021/jo00023a016
Return to citation in text: [1] -
Bacher, N.; Tiefenthaler, M.; Sturm, S.; Stuppner, H.; Ausserlechner, M. J.; Kofler, R.; Konwalinka, G. Br. J. Haematol. 2006, 132, 615–622. doi:10.1111/j.1365-2141.2005.05907.x
Return to citation in text: [1] -
Singh, G. S.; Desta, Z. Y. Chem. Rev. 2012, 112, 6104–6155. doi:10.1021/cr300135y
Return to citation in text: [1] -
Zhou, B.; Yang, Y.; Shi, J.; Luo, Z.; Li, Y. J. Org. Chem. 2013, 78, 2897–2907. doi:10.1021/jo302655u
Return to citation in text: [1] -
Lv, H.; Tiwari, B.; Mo, J.; Xing, C.; Chi, Y. R. Org. Lett. 2012, 14, 5412–5415. doi:10.1021/ol302475g
Return to citation in text: [1] -
Zhang, B.; Feng, P.; Sun, L.-H.; Cui, Y.; Ye, S.; Jiao, N. Chem. – Eur. J. 2012, 18, 9198–9203. doi:10.1002/chem.201201375
Return to citation in text: [1] -
Dalpozzo, R.; Bartoli, G.; Bencivenni, G. Chem. Soc. Rev. 2012, 41, 7247–7290. doi:10.1039/c2cs35100e
Return to citation in text: [1] -
Meloche, J. L.; Ashfeld, B. L. Angew. Chem., Int. Ed. 2017, 56, 6604–6608. doi:10.1002/anie.201701147
Return to citation in text: [1] -
Zhu, G.; Liu, S.; Wu, S.; Peng, L.; Qu, J.; Wang, B. J. Org. Chem. 2017, 82, 4317–4327. doi:10.1021/acs.joc.7b00316
Return to citation in text: [1] -
Wang, C.; Zhu, S.; Wang, G.; Li, Z.; Hui, X.-P. Eur. J. Org. Chem. 2016, 5653–5658. doi:10.1002/ejoc.201600958
Return to citation in text: [1] -
Liu, Y.; Wang, H.; Wan, J. Asian J. Org. Chem. 2013, 2, 374–386. doi:10.1002/ajoc.201200180
Return to citation in text: [1] -
Sun, Y.; Sun, J.; Yan, C.-G. Beilstein J. Org. Chem. 2013, 9, 8–14. doi:10.3762/bjoc.9.2
Return to citation in text: [1] -
Miyamoto, H.; Hirano, T.; Okawa, Y.; Nakazaki, A.; Kobayashi, S. Tetrahedron 2013, 69, 9481–9493. doi:10.1016/j.tet.2013.08.057
Return to citation in text: [1] -
Samineni, R.; Madapa, J.; Srihari, P.; Mehta, G. Org. Lett. 2017, 19, 3119–3122. doi:10.1021/acs.orglett.7b01233
Return to citation in text: [1] -
Yang, P.; Wang, X.; Chen, F.; Zhang, Z.-B.; Chen, C.; Peng, L.; Wang, L.-X. J. Org. Chem. 2017, 82, 3908–3916. doi:10.1021/acs.joc.6b03090
Return to citation in text: [1] -
Bonnaterre, F.; Bois-Choussy, M.; Zhu, J. Org. Lett. 2006, 8, 4351–4354. doi:10.1021/ol061755z
Return to citation in text: [1] -
Sharma, N.; Li, Z.; Sharma, U. K.; Van der Eycken, E. Org. Lett. 2014, 16, 3884–3887. doi:10.1021/ol5019079
Return to citation in text: [1] -
Maddirala, A. R.; Andreana, P. R. Eur. J. Org. Chem. 2016, 196–209. doi:10.1002/ejoc.201501273
Return to citation in text: [1] [2] -
Gautier, A. Justus Liebigs Ann. Chem. 1868, 146, 119–124. doi:10.1002/jlac.18681460107
Return to citation in text: [1] -
Eckert, H.; Nestl, A.; Ugi, I. Methyl Isocyanide. Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd, 2001. doi:10.1002/047084289X.rm198
Return to citation in text: [1] -
Schuster, R. E.; Scott, J. E.; Casanova, J., Jr. Org. Synth. 1966, 46, 75. doi:10.15227/orgsyn.046.0075
Return to citation in text: [1] -
Santra, S.; Andreana, T.; Bourgault, J.-P.; Andreana, P. R. Convertible Isocyanides: Application in Small Molecule Synthesis, Carbohydrate Synthesis, and Drug Discovery. Domino and Intramolecular Rearrangement Reactions as Advanced Synthetic Methods in Glycoscience; John Wiley & Sons, Inc., 2016; pp 121–194. doi:10.1002/9781119044222.ch7
Return to citation in text: [1] -
Hulme, C.; Chappeta, S.; Dietrich, J. Tetrahedron Lett. 2009, 50, 4054–4057. doi:10.1016/j.tetlet.2009.04.095
Return to citation in text: [1] -
Hulme, C.; Gore, V. Curr. Med. Chem. 2003, 10, 51–80. doi:10.2174/0929867033368600
Return to citation in text: [1] -
Hulme, C.; Bienaymé, H.; Nixey, T.; Chenera, B.; Jones, W.; Tempest, P.; Smith, A. L. Library Generation via Postcondensation Modifications of Isocyanide-Based Multicomponent Reactions. Methods Enzymol.; Academic Press, 2003; Vol. 369, pp 469–496. doi:10.1016/S0076-6879(03)69024-5
Return to citation in text: [1] -
Hulme, C.; Ayaz, M.; Martinez-Ariza, G.; Medda, F.; Shaw, A. Recent Advances in Multicomponent Reaction Chemistry. Small Molecule Medicinal Chemistry; John Wiley & Sons, Inc: , 2015; pp 145–187. doi:10.1002/9781118771723.ch6
Return to citation in text: [1] -
Xu, Z.; De Moliner, F.; Cappelli, A. P.; Ayaz, M.; Hulme, C. Synlett 2014, 25, 225–228. doi:10.1055/s-0033-1340219
Return to citation in text: [1] -
Alabugin, I. V.; Gilmore, K.; Manoharan, M. J. Am. Chem. Soc. 2011, 133, 12608–12623. doi:10.1021/ja203191f
Return to citation in text: [1] -
Gilmore, K.; Alabugin, I. V. Chem. Rev. 2011, 111, 6513–6556. doi:10.1021/cr200164y
Return to citation in text: [1] -
Polindara-Garcia, L. A.; Montesinos-Miguel, D.; Vazquez, A. Org. Biomol. Chem. 2015, 13, 9065–9071. doi:10.1039/C5OB01170A
Return to citation in text: [1] -
Hostetler, G.; Dunn, D.; McKenna, B. A.; Kopec, K.; Chatterjee, S. Chem. Biol. Drug Des. 2014, 83, 149–153. doi:10.1111/cbdd.12240
Return to citation in text: [1] [2]
| 52. | Polindara-Garcia, L. A.; Montesinos-Miguel, D.; Vazquez, A. Org. Biomol. Chem. 2015, 13, 9065–9071. doi:10.1039/C5OB01170A |
| 44. | Santra, S.; Andreana, T.; Bourgault, J.-P.; Andreana, P. R. Convertible Isocyanides: Application in Small Molecule Synthesis, Carbohydrate Synthesis, and Drug Discovery. Domino and Intramolecular Rearrangement Reactions as Advanced Synthetic Methods in Glycoscience; John Wiley & Sons, Inc., 2016; pp 121–194. doi:10.1002/9781119044222.ch7 |
| 45. | Hulme, C.; Chappeta, S.; Dietrich, J. Tetrahedron Lett. 2009, 50, 4054–4057. doi:10.1016/j.tetlet.2009.04.095 |
| 46. | Hulme, C.; Gore, V. Curr. Med. Chem. 2003, 10, 51–80. doi:10.2174/0929867033368600 |
| 47. | Hulme, C.; Bienaymé, H.; Nixey, T.; Chenera, B.; Jones, W.; Tempest, P.; Smith, A. L. Library Generation via Postcondensation Modifications of Isocyanide-Based Multicomponent Reactions. Methods Enzymol.; Academic Press, 2003; Vol. 369, pp 469–496. doi:10.1016/S0076-6879(03)69024-5 |
| 48. | Hulme, C.; Ayaz, M.; Martinez-Ariza, G.; Medda, F.; Shaw, A. Recent Advances in Multicomponent Reaction Chemistry. Small Molecule Medicinal Chemistry; John Wiley & Sons, Inc: , 2015; pp 145–187. doi:10.1002/9781118771723.ch6 |
| 49. | Xu, Z.; De Moliner, F.; Cappelli, A. P.; Ayaz, M.; Hulme, C. Synlett 2014, 25, 225–228. doi:10.1055/s-0033-1340219 |
| 50. | Alabugin, I. V.; Gilmore, K.; Manoharan, M. J. Am. Chem. Soc. 2011, 133, 12608–12623. doi:10.1021/ja203191f |
| 51. | Gilmore, K.; Alabugin, I. V. Chem. Rev. 2011, 111, 6513–6556. doi:10.1021/cr200164y |
| 1. | Ugi, I.; Steinbrückner, C. Angew. Chem. 1960, 72, 267–268. doi:10.1002/ange.19600720709 |
| 2. | Dömling, A.; Ugi, I. Angew. Chem., Int. Ed. 2000, 39, 3168–3210. doi:10.1002/1521-3773(20000915)39:18<3168::AID-ANIE3168>3.0.CO;2-U |
| 12. | Bourgault, J. P.; Maddirala, A. R.; Andreana, P. R. Org. Biomol. Chem. 2014, 12, 8125–8127. doi:10.1039/C4OB01148A |
| 13. | Koopmanschap, G.; Ruijter, E.; Orru, R. V. A. Beilstein J. Org. Chem. 2014, 10, 544–598. doi:10.3762/bjoc.10.50 |
| 14. | Santra, S.; Andreana, P. R. Angew. Chem., Int. Ed. 2011, 50, 9418–9422. doi:10.1002/anie.201103567 |
| 15. | Santra, S.; Andreana, P. R. J. Org. Chem. 2011, 76, 2261–2264. doi:10.1021/jo102305q |
| 40. | Maddirala, A. R.; Andreana, P. R. Eur. J. Org. Chem. 2016, 196–209. doi:10.1002/ejoc.201501273 |
| 9. | Tempest, P.; Ma, V.; Thomas, S.; Hua, Z.; Kelly, M. G.; Hulme, C. Tetrahedron Lett. 2001, 42, 4959–4962. doi:10.1016/S0040-4039(01)00919-4 |
| 10. | Dömling, A.; Wang, W.; Wang, K. Chem. Rev. 2012, 112, 3083–3135. doi:10.1021/cr100233r |
| 11. | Jiang, B.; Rajale, T.; Wever, W.; Tu, S.-J.; Li, G. Chem. – Asian J. 2010, 5, 2318–2335. doi:10.1002/asia.201000310 |
| 43. | Schuster, R. E.; Scott, J. E.; Casanova, J., Jr. Org. Synth. 1966, 46, 75. doi:10.15227/orgsyn.046.0075 |
| 4. | Hulme, C.; Peng, J.; Louridas, B.; Menard, P.; Krolikowski, P.; Kumar, N. V. Tetrahedron Lett. 1998, 39, 8047–8050. doi:10.1016/S0040-4039(98)01770-5 |
| 5. | Huang, Y.; Khoury, K.; Chanas, T.; Dömling, A. Org. Lett. 2012, 14, 5916–5919. doi:10.1021/ol302837h |
| 6. | Xu, Z.; Shaw, A. Y.; Dietrich, J.; Cappelli, A. P.; Nichol, G.; Hulme, C. Mol. Diversity 2012, 16, 73–79. doi:10.1007/s11030-011-9354-x |
| 7. | Hulme, C.; Ma, L.; Romano, J.; Morrissette, M. Tetrahedron Lett. 1999, 40, 7925–7928. doi:10.1016/S0040-4039(99)01580-4 |
| 8. | Hulme, C.; Ma, L.; Cherrier, M.-P.; Romano, J. J.; Morton, G.; Duquenne, C.; Salvino, J.; Labaudiniere, R. Tetrahedron Lett. 2000, 41, 1883–1887. doi:10.1016/S0040-4039(00)00052-6 |
| 40. | Maddirala, A. R.; Andreana, P. R. Eur. J. Org. Chem. 2016, 196–209. doi:10.1002/ejoc.201501273 |
| 3. | Sharma, U. K.; Sharma, N.; Vachhani, D. D.; Van der Eycken, E. V. Chem. Soc. Rev. 2015, 44, 1836–1860. doi:10.1039/C4CS00253A |
| 41. | Gautier, A. Justus Liebigs Ann. Chem. 1868, 146, 119–124. doi:10.1002/jlac.18681460107 |
| 42. | Eckert, H.; Nestl, A.; Ugi, I. Methyl Isocyanide. Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd, 2001. doi:10.1002/047084289X.rm198 |
| 31. | Zhu, G.; Liu, S.; Wu, S.; Peng, L.; Qu, J.; Wang, B. J. Org. Chem. 2017, 82, 4317–4327. doi:10.1021/acs.joc.7b00316 |
| 38. | Bonnaterre, F.; Bois-Choussy, M.; Zhu, J. Org. Lett. 2006, 8, 4351–4354. doi:10.1021/ol061755z |
| 24. | Bacher, N.; Tiefenthaler, M.; Sturm, S.; Stuppner, H.; Ausserlechner, M. J.; Kofler, R.; Konwalinka, G. Br. J. Haematol. 2006, 132, 615–622. doi:10.1111/j.1365-2141.2005.05907.x |
| 25. | Singh, G. S.; Desta, Z. Y. Chem. Rev. 2012, 112, 6104–6155. doi:10.1021/cr300135y |
| 26. | Zhou, B.; Yang, Y.; Shi, J.; Luo, Z.; Li, Y. J. Org. Chem. 2013, 78, 2897–2907. doi:10.1021/jo302655u |
| 27. | Lv, H.; Tiwari, B.; Mo, J.; Xing, C.; Chi, Y. R. Org. Lett. 2012, 14, 5412–5415. doi:10.1021/ol302475g |
| 28. | Zhang, B.; Feng, P.; Sun, L.-H.; Cui, Y.; Ye, S.; Jiao, N. Chem. – Eur. J. 2012, 18, 9198–9203. doi:10.1002/chem.201201375 |
| 29. | Dalpozzo, R.; Bartoli, G.; Bencivenni, G. Chem. Soc. Rev. 2012, 41, 7247–7290. doi:10.1039/c2cs35100e |
| 30. | Meloche, J. L.; Ashfeld, B. L. Angew. Chem., Int. Ed. 2017, 56, 6604–6608. doi:10.1002/anie.201701147 |
| 39. | Sharma, N.; Li, Z.; Sharma, U. K.; Van der Eycken, E. Org. Lett. 2014, 16, 3884–3887. doi:10.1021/ol5019079 |
| 21. | Dong, H.; Song, S.; Li, J.; Xu, C.; Zhang, H.; Ouyang, L. Bioorg. Med. Chem. Lett. 2015, 25, 3585–3591. doi:10.1016/j.bmcl.2015.06.076 |
| 22. | Fu, P.; Kong, F.; Li, X.; Wang, Y.; Zhu, W. Org. Lett. 2014, 16, 3708–3711. doi:10.1021/ol501523d |
| 23. | Jossang, A.; Jossang, P.; Hadi, H. A.; Sevenet, T.; Bodo, B. J. Org. Chem. 1991, 56, 6527–6530. doi:10.1021/jo00023a016 |
| 53. | Hostetler, G.; Dunn, D.; McKenna, B. A.; Kopec, K.; Chatterjee, S. Chem. Biol. Drug Des. 2014, 83, 149–153. doi:10.1111/cbdd.12240 |
| 16. | De Silva, R. A.; Santra, S.; Andreana, P. R. Org. Lett. 2008, 10, 4541–4544. doi:10.1021/ol801841m |
| 17. | Kumar, A.; Gupta, G.; Bishnoi, A. K.; Saxena, R.; Saini, K. S.; Konwar, R.; Kumar, S.; Dwivedi, A. Bioorg. Med. Chem. 2015, 23, 839–848. doi:10.1016/j.bmc.2014.12.037 |
| 18. | Santos, M. M. M. Tetrahedron 2014, 70, 9735–9757. doi:10.1016/j.tet.2014.08.005 |
| 19. | Yu, B.; Yu, D.-Q.; Liu, H.-M. Eur. J. Med. Chem. 2015, 97, 673–698. doi:10.1016/j.ejmech.2014.06.056 |
| 20. | Yu, B.; Yu, Z.; Qi, P.-P.; Yu, D.-Q.; Liu, H.-M. Eur. J. Med. Chem. 2015, 95, 35–40. doi:10.1016/j.ejmech.2015.03.020 |
| 32. | Wang, C.; Zhu, S.; Wang, G.; Li, Z.; Hui, X.-P. Eur. J. Org. Chem. 2016, 5653–5658. doi:10.1002/ejoc.201600958 |
| 33. | Liu, Y.; Wang, H.; Wan, J. Asian J. Org. Chem. 2013, 2, 374–386. doi:10.1002/ajoc.201200180 |
| 34. | Sun, Y.; Sun, J.; Yan, C.-G. Beilstein J. Org. Chem. 2013, 9, 8–14. doi:10.3762/bjoc.9.2 |
| 35. | Miyamoto, H.; Hirano, T.; Okawa, Y.; Nakazaki, A.; Kobayashi, S. Tetrahedron 2013, 69, 9481–9493. doi:10.1016/j.tet.2013.08.057 |
| 36. | Samineni, R.; Madapa, J.; Srihari, P.; Mehta, G. Org. Lett. 2017, 19, 3119–3122. doi:10.1021/acs.orglett.7b01233 |
| 37. | Yang, P.; Wang, X.; Chen, F.; Zhang, Z.-B.; Chen, C.; Peng, L.; Wang, L.-X. J. Org. Chem. 2017, 82, 3908–3916. doi:10.1021/acs.joc.6b03090 |
| 53. | Hostetler, G.; Dunn, D.; McKenna, B. A.; Kopec, K.; Chatterjee, S. Chem. Biol. Drug Des. 2014, 83, 149–153. doi:10.1111/cbdd.12240 |
© 2018 Maddirala and Andreana; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)