Abstract
Four benzothiazolium crown ether-containing styryl dyes were prepared through an optimized synthetic procedure. Two of the dyes (4b and 4d) having substituents in the 5-position of the benzothiazole ring are newly synthesized compounds. They demonstrated a higher degree of trans–cis photoisomerization and a longer life time of the higher energy forms in comparison with the known analogs. The chemical structures of all dyes in the series were characterized by NMR, UV–vis, IR spectroscopy and elemental analysis. The steady-state photophysical properties of the dyes were elucidated. The stability constants of metal complexes were determined and are in good agreement with the literature data for reference dyes. The temporal evolution of trans-to-cis isomerization was observed in a real-time regime. The dyes demonstrated a low intrinsic fluorescence of their Ba2+ complexes and high yield of E/Z photoisomerization with lifetimes of the higher energy form longer than 500 seconds. Density functional theory (DFT) calculations at the B3LYP/6-31+G(d,p) level were performed in order to predict the enthalpies (H) of the cis and trans isomers and the storage energies (ΔH) for the systems studied.
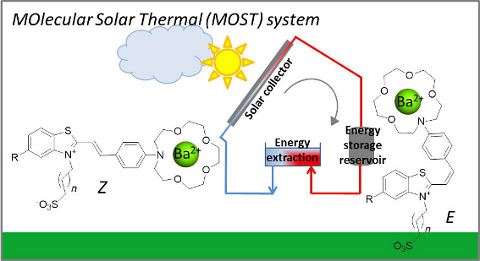
Graphical Abstract
Introduction
Molecular photoswitches permanently attract considerable interest because they hold potential for application in molecular electronic and photonic devices [1-5]. Photoswitches are a class of switches that can alternate between the thermodynamically stable forms by application of light (or change of the light intensity) as an external stimulus. If the stable forms (isomers) are not isoenergetic, they have the ability to capture and store imported (solar) energy [6]. As the stored energy is further released as thermal energy, such materials are called molecular solar thermal systems (MOST) [7]. For a pure MOST system the maximal solar energy conversion efficiency was estimated to be 10.6% [8]. It was demonstrated experimentally that by merging a MOST-system [9] with a triplet–triplet annihilation photon energy upconversion system (TTA-UC) [10,11] an effective utilization of sub-bandgap photons is possible [12,13]. Nevertheless, the identification of a precious metal-free MOST system utilizing the visible part of the sun spectrum remains a considerable challenge.
The stated requirements for such systems can be summarized as follows [7]:
(i) the potential barrier between the higher energy form and the lower energy form of the energy carriers should be as high as possible;
(ii) the higher energy form must be stable for a long time;
(iii) the higher energy form has to be with quite small molar absorptivity in comparison to the lower one;
(iv) the quantum yield of the photoisomerization has to be as high as possible, which requires the design of MOST systems with completely suppressed or minimal fluorescence, intramolecular charge transfer or other processes, quenching the photoisomerization;
(v) the energy storing MOST materials have to utilize light in the visible range of the spectrum;
(vi) it will be a crucial benefit for the MOST technology, if toxic and precious metals are avoided. Focusing on environmentally friendly MOST system is an unavoidable requirement;
(vii) the MOST materials should be of low cost and easily accessible; their synthesis and purification should be straightforward, reliable, fast, inexpensive and ideally environmentally benign.
Therefore, only MOST systems fulfilling all requirements (i)–(vii) simultaneously can be regarded as realistic technological systems for long-term solar energy storage.
However, the design and preparation of molecules matching all the above-mentioned criteria is quite difficult. Thus, the invention of new materials possessing at least parts of these requirements provides the base for further improvements and brings the scientists nearer to the identification of the “perfect” MOST system. In this connection we identified crown ether-containing styryl dyes [14,15] as promising substances due to their ability to undergo trans-to-cis photoisomerization (E/Z) with very high quantum yields. In addition, their structures predispose to an easy functionalization and low cost synthesis. Another advantage of these dyes is the very low molar absorptivity [16,17] of the higher energy cis isomer, which is a prerequisite for its photostability. The cis isomers of styryl dyes linked to crown ethers have been identified to form complexes with alkaline-earth metal cations with higher stability constant than the respective trans isomer forms [14,15,18]. Lednev et al. proposed as an explanation of the difference in the determined stability constants values for the complexes with cis/trans isomers the formation of an additional intramolecular coordination bond between the “crowned” cation and the alkylsulfonate anchoring group which is only possible for the cis form [19]. The further studies by the same group of authors indicated that benzothiazolium styryl monoazacrown ether dyes have several main advantages. First of all, the azacrown nitrogen atom is linked directly to the dye and it is part of the chromophore system, responsible for the “push–pull” effect and the photophysical properties of the dye. This provides control over the “push–pull” effect in the chromophore by switching on and off states (i.e., metal-in/metal-out from the crown ligand) [20]. Further, it was found that the azacrown benzothiazolium styryl dyes 4a and 4c exhibit ion-sensitivity in the thermal cis–trans photoisomerization [18,21], a feature, which may be used in the development of thermoreversible photoionic molecular devices [19]. Crown ether-containing styryl dyes are used as sensors for dications such as Ba2+, Ca2+ and Mg2+, or as materials for optical data storage [19]. Thus, to the best of our knowledge this kind of dyes has not been specified as potential MOST material yet.
The aim of the present study is to reveal the potential of the aza-15-crown-5-containing styryl dyes as efficient material for the TTA-UC accelerated MOST process.
Results and Discussion
Synthesis
The synthetic pathway starts with the quaternization of 2-methylbenzothiazoles 1a and 1b with 1,3-propanesultone (1c) or 1,4-butansultone (1b) in a sealed tube at 145 °C without any solvent, as it was described in the literature [17] (Scheme 1).
![[1860-5397-15-106-i1]](/bjoc/content/inline/1860-5397-15-106-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Quaternization of 2-methylbenzothiazoles with alkane sultones.
Scheme 1: Quaternization of 2-methylbenzothiazoles with alkane sultones.
A four-step synthetic pathway was used for the synthesis of 4-(aza-15-crown-5)benzocarbaldehyde 3 (Scheme 2) following the procedure described in [22].
![[1860-5397-15-106-i2]](/bjoc/content/inline/1860-5397-15-106-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 4-(aza-15-crown-5)benzocarbaldehyde (3) [22].
Scheme 2: Synthesis of 4-(aza-15-crown-5)benzocarbaldehyde (3) [22].
A series of the target dyes 4a–d was prepared using a modified literature procedure (Scheme 3) [22]. The modification consists in the addition of a 10% molar excess of 4-(aza-15-crown-5)benzocarbaldehyde (3, Scheme 3) and usage of ethyl acetate as an additional solvent for product precipitation. Because of its better solubility in ethyl acetate the unreacted excess of crown ether 3 was easily removed. In general the zwitterionic salts 2a–d reacted to complete depletion in ethanol and in the presence of piperidine as catalyst with the crown ether benzaldehyde 3 (TLC monitoring, ethyl acetate/ethanol 4.5:0.5). After the addition of ethyl acetate the precipitated target products 4a–d were isolated by filtration.
![[1860-5397-15-106-i3]](/bjoc/content/inline/1860-5397-15-106-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
It must be mentioned explicitly, that the modification of the procedure helped us to avoid cost-intensive and time-consuming purification procedures which makes the synthesis suitable for large material quantities. In this way only one further precipitation from ethanol/ethyl acetate 1:3 was needed to obtain analytically pure target dyes 4a–d.
The dyes 4a and 4c were previously described [18,21] and were used as reference compounds. To the best of our knowledge dyes 4b and 4d are new compounds. The chemical structures of all dyes from the series were proved by NMR spectroscopy, elemental analysis, IR and UV–vis spectroscopy.
Photophysical properties
Steady-state absorption and emission spectroscopy
Next we elucidated the photophysical properties of the chosen compounds and determined their suitability to be used as MOST systems. First, we defined their photophysical behavior in neat acetonitrile (ACN) solution and in the presence of barium cations. Figure 1 shows the absorption spectra of dyes 4a–d measured at different concentrations (cM) of Ba(ClO4)2. All dye solutions have similar absorption profiles with a broad long wavelength band in the vis region (Δλ = 400–600 nm) with long wavelength maxima at λmax = 520 nm indicating that their spectral properties are determined principally by the core chromophore structure. The addition of Ba2+ ions to the ACN solution of all dyes induced a decrease in the absorption maximum intensity (at about 520 nm) and to a substantial hypsochromic shift of the maximum absorption with peak at around λmax = 440 nm. The changes in the absorption spectra of the dyes upon the addition of Ba(ClO4)2 in ACN solution are characteristic for an ion complexation by the azacrown ether group of the chromoionophore and the spectra in Figure 1 were assigned to the formation of the trans-dye–Ba2+ complex [19]. For all dye solutions, a distinct isosbestic point upon titration was observed, indicating only one kind of complex formation even at the highest Ba2+ concentration.
![[1860-5397-15-106-1]](/bjoc/content/figures/1860-5397-15-106-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: a–c) Dependence of the absorption spectra of the dyes 4b, 4c and 4d, respectively (cL = 1.0 × 10−5 M in ACN) on the concentration of Ba(ClO4)2, ranging from 1 × 10−4 M up to 5 × 10−1 M. d) Absorption of dyes 4a–d at λ = 520 nm as a function of Ba(ClO4)2 concentration. Experimental conditions: rt, sample thickness d = 10 mm.
Figure 1: a–c) Dependence of the absorption spectra of the dyes 4b, 4c and 4d, respectively (cL = 1.0 × 10−5 ...
To determine the optimal Ba2+ concentration for our measurements it was necessary to define the stability constants for each dye–Ba2+ complex. The dependencies of the absorption A (Equation 1) of the dyes 4a–d at a fixed wavelength λ = 520 nm on the Ba2+ concentration in ACN is shown in Figure 1d. The curves were approximated by Equation 1, which is true for the simplest form of complexation [18]:
![[1860-5397-15-106-i4]](/bjoc/content/inline/1860-5397-15-106-i4.svg?max-width=590&scale=1.18182)
![[1860-5397-15-106-i5]](/bjoc/content/inline/1860-5397-15-106-i5.svg?max-width=590&scale=1.18182)
where A0 and A∞ are the absorptions of the chromoionophore at zero and infinite concentration of the metal ion, respectively; A is the absorption at the concentration cM of the metal ion; K is the stability constant of complex formation, and L and LM are the ligand and the metal ion complex, respectively. A∞ and K were found by approximation of Equation 1.
The stability constant for the complex formation was estimated to be K = 100 ± 15 M−1, 49.4 ± 7.6 M−1 and 44.7 ± 10 M−1 for dyes 4b, 4c and 4d, respectively. For dye 4a the stability constant was found to be K = 70 ± 15 M−1 which is in good correlation with the published data [22]. These results prompted us to identify the optimal Ba2+ concentration, necessary for a maximum degree of dye–Ba2+ inclusion complex formation. Obviously for a better complexation it is necessary to work with high Ba2+ concentrations. The explanation is related to the size of the barium cation which does not intercalate completely in the crown ether cavity. This assumption is confirmed by the slight downfield shift of the signals in the 1H NMR spectrum of dye 4b in the presence of Ba2+ cations compared to that in neat CD3CN (Supporting Information File 1, Figure S9 and Figure S10).
A common behavior demonstrated in Figure 2 was observed: the fluorescence quantum yield of all dyes (Table 1) decreases if the concentration of the Ba2+ ions increases, i.e., the formation of the trans-dye–Ba2+ complexes causes the observed fluorescence decrease.
![[1860-5397-15-106-2]](/bjoc/content/figures/1860-5397-15-106-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Dependence of the fluorescence of 4b on the concentration of the Ba2+ ions. Excitation wavelength λexc = 488 nm.
Figure 2: Dependence of the fluorescence of 4b on the concentration of the Ba2+ ions. Excitation wavelength λ...
The optical parameters of all 4 dyes, including absorption maxima (λmax,abs), fluorescence emission maxima (λmax,f), hypsochromic shift of the absorption maxima (Δλmax), and fluorescence quantum yields (Φf) for different excitation wavelengths, obtained for the dyes in ACN solution at different Ba2+ concentrations are summarized in Table 1. The emission maximum of all dyes is at about λmax,f = 595 nm and does not change significantly upon Ba2+ ion complexation. No changes in the shape of the fluorescence curves were observed, when the excitation wavelength was tuned within the region of λexc = 400–550 nm.
Table 1: Absorption maxima (λmax,abs), fluorescence emission maxima (λmax,f), shift of the absorption maxima (Δλmax), and quantum yields of fluorescence (Φf) in % (at 488 nm and 440 nm excitation) for the trans isomers of dyes 4a–d in acetonitrile solution at different Ba2+ concentrations.
| Dye | cM/cL | λmax,abs, nm | Δλabs | λmax,f, nm |
Φf %
λexc = 488 nm |
Φf %
λexc = 440 nm |
| 4a | 0 | 520 | 597.6 | 5.9 | 1.9 | |
| 1000 | 439 | 81 | 595 | 0.29 | 0.29 | |
| 10000 | 439 | 81 | 595 | 0.34 | 0.37 | |
| 4b | 0 | 523 | 594 | 6.42 | 0.83 | |
| 2000 | 438 | 85 | 588 | 1.9 | 0.38 | |
| 10000 | 438 | 85 | 580.6 | 0.91 | 0.44 | |
| 4c | 0 | 522 | 593.8 | 0.23 | 0.4 | |
| 1000 | 442 | 80 | 596 | 0.18 | 0.25 | |
| 10000 | 442 | 80 | 596 | 0.14 | 0.13 | |
| 4d | 0 | 521 | 599 | 0.56 | 0.77 | |
| 1000 | 441 | 80 | 596 | 0.31 | 0.32 | |
| 10000 | 441 | 80 | 594 | 0.49 | 0.58 | |
Generally one of the advantages of the presented Ba2+–crown ether containing styryl dye complexes as a MOST material is the extremely low intrinsic fluorescence which can be a precondition for higher quantum yields of the cis–trans photoisomerization reactions.
Real-time E/Z-photoisomerization of dyes 4a–d and their complexes
The photoisomerization of free dyes 4a–d and dye–Ba2+ inclusion complexes were investigated in real time mode upon irradiation with visible light (λ = 488 nm) close to their absorption maxima. Figure 3a illustrates the characteristic changes in the absorption spectra of dye 4b–Ba2+ complex under optical excitation with λ = 488 nm and intensity of 14 mW cm−2. The temporal evolution of the absorption at specific wavelength of λabs = 500 nm is demonstrated in Figure 3b: during the first time interval from t1 = 10 s up to t2 = 180 s the absorption is decreasing monotonically, as a result of π → π transition in the trans isomer of the free dye and its Ba2+ complex. During the next time interval for t3 > 180 s, the optical excitation was terminated and constant increase of the optical absorption was observed.
![[1860-5397-15-106-3]](/bjoc/content/figures/1860-5397-15-106-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: a) Dependence of absorption spectra of dye 4b (cL = 1.0 × 10−4 M) with Ba2+ (cM = 1 M) on the irradiation time. b) Temporal absorption evolution, measured at λabs = 500 nm, upon irradiation. Experimental conditions: Excitation wavelength λexc = 488 nm; excitation intensity 14 mW cm−2, solvent, ACN; pulse duration t = 180 s.
Figure 3: a) Dependence of absorption spectra of dye 4b (cL = 1.0 × 10−4 M) with Ba2+ (cM = 1 M) on the irrad...
The degree (R) of trans-to-cis photoisomerization at the photostationary state was evaluated from Equation 3:
![[1860-5397-15-106-i6]](/bjoc/content/inline/1860-5397-15-106-i6.svg?max-width=590&scale=1.18182)
where A0 is the absorption before irradiation and A∞ is the absorption at the photostationary state. It was found that the rate of the photoisomerization process and the degree of conversion trans-to-cis isomer depends strongly on the Ba2+ concentration. The cis isomers formed upon irradiation are thermally unstable and revert to the trans isomers in the dark [23]. The rates of trans-to-cis isomerization were determined for several Ba2+ concentrations in the range of 2 × 10−3 M up to 1 M at a fixed dye concentration (1 × 10−4 M) for all dyes. The kinetic data were found to fit well to a single exponential function (Equation 1), giving a rate constant (k) corresponding to a lifetime (1/k) [18]:
![[1860-5397-15-106-i7]](/bjoc/content/inline/1860-5397-15-106-i7.svg?max-width=590&scale=1.18182)
where A0 and A∞, are the initial and final absorptions, A is the absorption at 500 nm at a time t after termination of the irradiation. In all cases, after irradiation ceased, complete reversion toward the initial absorption spectrum was observed. As a rule, the reversion from the cis to the trans isomer leads to a gradual increase in the intensity of the absorption maximum. The photoisomerization data for all 4 dyes are summarized in Table 2.
Table 2: Degree (R) of trans-to-cis photoisomerization at the photostationary state, rate constants (k) and lifetime of trans-to-cis isomerization.
|
Dye
M |
Ba(ClO4)2
M |
R |
Lifetime
s |
k
s−1 |
|
4a
1.0 × 10−4 |
0 | 0.19 | 31 | 32.6 × 10−3 |
| 2.0 × 10−3 | 0.53 | 69 | 14.4 × 10−3 | |
| 2.0 × 10−2 | 0.52 | 264 | 3.8 × 10−3 | |
| 2.0 × 10−1 | 0.56 | 284 | 3.5 × 10−3 | |
| 1 | 0.53 | 342 | 2.9 × 10−3 | |
|
4b
1.0 × 10−4 |
0 | 0.35 | 148 | 6.0 × 10−3 |
| 2.0 × 10−3 | 0.82 | 292 | 3.4 × 10−3 | |
| 2.0 × 10−2 | 0.74 | 279 | 3.6 × 10−3 | |
| 2.0 × 10−1 | 0.54 | 481 | 2.1 × 10−3 | |
| 1 | 0.39 | 514 | 1.9 × 10−3 | |
|
4c
1.0 × 10−4 |
0 | 0.00 | 0 | 0 |
| 2.0 × 10−3 | 0.00 | 0 | 0 | |
| 2.0 × 10−2 | 0.08 | 0 | 0 | |
| 2.0 × 10−1 | 0.52 | 330 | 3.0 × 10−3 | |
| 1 | 0.44 | 390 | 2.6 × 10−3 | |
|
4d
1.0 × 10−4 |
0 | 0.21 | 18 | 55.0 × 10−3 |
| 2.0 × 10−3 | 0.23 | 20.9 | 47 × 10−3 | |
| 2.0 × 10−2 | 0.22 | 30.7 | 32 × 10−3 | |
| 2.0 × 10−1 | 0.58 | 137 | 7.3 × 10−3 | |
| 1 | 0.26 | 301 | 3.3 × 10−3 | |
Generally, the addition of Ba2+ ions was found to increase substantially the lifetime of the cis isomers. Under the abovementioned conditions the cis-4b–Ba2+ complex was detected to be most stable, while dye 4d formed stable cis-4d–Ba2+ complex only at a higher concentration of Ba2+ ions (1 M).
Figure 4 is an additional demonstration of the ability of dye 4b to undergo trans-to-cis photoisomerization in its free form and in complex with Ba2+. The absorption band corresponding to the π → π transition in the cis isomer (at around 280 nm and 320 nm, shown in Figure 4), as for the free dye 4b, increased with the irradiation time, suggesting that isomerization from trans to a cis form of the free dye or its Ba2+ complex proceeded until a photostationary state was reached. As can be seen from Figure 4 the trans-to-cis isomerization takes place to a higher extent in the free form of the dye than in the complex. However, the lifetimes of the free cis form is extremely short in comparison to that of the complex. The degree of photoisomerization of the new dye 4b is apparently higher than that of the known structure (4a). From one side it can be supposed that dye 4a aggregated much faster than its methyl-substituted analogue 4b. From another hand the substituent in the 5-position of the benzothiazole heterocycle sterically hinders the rotation of the alkylsulfo-anchoring group and thus plays the role of a controller with regard to its direction towards the crown ether. This finding is a resemblance to the results from reference [24] where the 5-methoxy-substituted benzothiazole styryl-crown ethers demonstrated higher quantum yields of trans-to-cis photoisomerization.
![[1860-5397-15-106-4]](/bjoc/content/figures/1860-5397-15-106-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: UV–vis absorption spectra of dye 4b (1.0 × 10−4 M) free and in complex with Ba2+ (1 M) before and after the end of the exposure at λ = 488 nm.
Figure 4: UV–vis absorption spectra of dye 4b (1.0 × 10−4 M) free and in complex with Ba2+ (1 M) before and a...
Insight from electronic structure calculations
To rationalize the experimental findings, we performed density functional theory (DFT) calculations at the B3LYP/6-31+G(d,p) level. The first step in the molecular modelling investigation was the optimization of the molecular structures of the cis and trans isomers of dyes 4a–d (with the –(CH2)nSO3− (n = 3, 4) tails oriented to be in proximity to the aza-15-crown-5 fragments) in the gas phase (Figure 5). The thermochemical data for these are calculated at 298.15 K and a pressure of 1 atm. In Table 3 we report the enthalpies (H) for the cis and trans isomers and the energy storage capacity (ΔH) calculated as the difference in enthalpy between the cis and trans isomers. B3LYP calculations in the gas phase predict the trans forms of the dyes 4a–d to be more stable. The cis forms are higher in energy by only 3.3 and 3.0 kJ mol−1 for 4a and 4b, respectively. The enthalpy differences calculated for dyes 4c and 4d are much higher (69.3 kJ mol−1 (4c) and 67.9 kJ mol−1 (4d), respectively).
![[1860-5397-15-106-5]](/bjoc/content/figures/1860-5397-15-106-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Optimized structures of the cis and trans isomers of dyes 4a–d.
Figure 5: Optimized structures of the cis and trans isomers of dyes 4a–d.
Compounds 4a–d can bind metal species at both isomeric forms. To determine the geometries of the 1:1 complexes with Ba2+ cations the metal cations were placed in the crown ether’s cavity and allowed to relax. In the optimized dye–Ba2+ complexes the metal cations are displaced “above” the crown ether’s cavity. Figure 6 depicts the gas-phase geometries of the Ba2+ complexes of the cis and trans isomer of 4b.
![[1860-5397-15-106-6]](/bjoc/content/figures/1860-5397-15-106-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Optimized structures of the Ba2+-complexes of cis and trans isomer of 4b.
Figure 6: Optimized structures of the Ba2+-complexes of cis and trans isomer of 4b.
The calculated enthalpies for the complex-formation reaction dye + Ba2+ → [dye–Ba]2+ in the gas phase, where dye = 4a–d, with bare metal cations are listed in Table 4. The results obtained demonstrate that all reactions in the gas phase are predicted to be favorable. The ∆∆H1 values calculated for Ba2+ complex formation with the trans isomers of 4a–d are more negative than the values obtained for the respective cis isomers.
The complex formation processes between dyes with shorter –(CH2)nSO3− tail (n = 3) are characterized by more negative ∆∆H1 values than those calculated for the dyes equipped with longer tails (n = 4), a trend corresponding to the trends in the experimentally derived stability constants. Conventional solvent treatment by methods like the polarizable continuum model (PCM) did not provide a good quantitative agreement with the experimental stability constants [23] so the data for ∆∆H in acetonitrile are not provided.
Time-dependent density functional theory (TDDFT) calculations were used to probe the electronic reorganizations upon excitation. TDPBE0 calculations with the 6–31+G(d,p) basis set for all atoms (except for Ba) and with the Stuttgart-Dresden SDD effective core potential (ECP) basis set for Ba predict one intensive band for all compounds in the range of 400–500 nm. The calculated optical parameters such as the absorption maximum (λmax), oscillator strength (f) and frontier orbital energy levels for the trans isomers of 4a–d are listed in Table 5. The positions and intensities of the bands are consistent with the experimentally observed ones. The first excited states are determined by HOMO (highest occupied molecular orbital) → LUMO (lowest unoccupied molecular orbital) transitions.
Table 5: TDDFT/PBE0 calculated absorption maxima (λmax), oscillator strength (f) HOMO and LUMO energies and energy difference (HOMO–LUMO gap, HLG) for the trans isomers of compounds 4a–d in acetonitrile.
| Compound | λmax, nm | f | HOMO, eV | LUMO, eV | HLG, eV |
| 4a | 477 | 1.58 | −5.71 | −2.98 | 2.73 |
| 4b | 481 | 1.63 | −5.64 | −2.93 | 2.71 |
| 4c | 482 | 1.63 | −5.58 | −2.90 | 2.69 |
| 4d | 483 | 1.65 | −5.56 | −2.87 | 2.69 |
The simulated spectra with spectral lifetime broadening (Gaussian function) with a full width at half-maximum (FWHM) of 0.15 eV and a height proportional to the oscillator strength for each transition spectrum for dye 4b and its Ba2+ complex (Figure 7) are consistent with the experimental ones. The experimentally measured substantial hypo- and hypsochromic shift in the absorption spectra upon Ba2+ addition are also observed in the simulated spectra of the theoretically modeled structures of the dyes and the respective complexes.
![[1860-5397-15-106-7]](/bjoc/content/figures/1860-5397-15-106-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Simulated spectra with spectral lifetime broadening TDPBE0 spectra in ACN for dye 4b and its Ba2+ complex.
Figure 7: Simulated spectra with spectral lifetime broadening TDPBE0 spectra in ACN for dye 4b and its Ba2+ c...
The simulated spectra of the metal-free and Ba2+ complexed cis form of compound 4b are also presented in Figure 7. The oscillator strengths of the cis forms, calculated at the same computational level, are found to be significantly lower than those calculated for the respective trans forms (metal-free compound trans-4b and trans-4b–Ba complex). These results correspond to the experimentally observed gradual increase in the intensity of the absorption maximum upon cis-to-trans reversion.
Conclusion
Four benzothiazolium crown ether-containing styryl dyes (two known and two novel compounds) were synthesized through an optimized synthetic procedure. The photophysical properties of the new dyes were investigated in the absence and presence of Ba2+ cations and compared with those of the known dyes. The optimal conditions for the trans-to-cis photoisomerization of the styryl-crown ether containing dyes were identified. The dyes 4b and 4d with substituents in 5-position of the benzothiazole moiety demonstrated much better photophysical properties as molecular switches and MOST materials in comparison with the unsubstituted known analogs. The calculated thermodynamic changes associated with metal-ion complexation in the gas phase match the trends in the experimental stability constants.
Experimental
General
All solvents used in the present work were commercially available (HPLC grade). The starting materials 1a, 1b, 2a, and 2b were commercially available and were used as supplied. Melting points were determined on a Kofler apparatus and are uncorrected. NMR spectra were obtained on a Bruker Avance III 500 DRX 600 MHz spectrometer in DMSO-d6. The MALDI–TOF/TOF spectra were measured at Bruker “RapifeX” at MPIP, Germany. The stepwise experimental procedures for the synthesis of compounds 2–4 and characterization data are given in Supporting Information File 1. 4-(Aza-15-crown-5)benzocarbaldehyde (3) was synthesized using a slightly modified procedure [16,17].
UV–vis spectra were measured on a Unicam 530 UV–vis spectrophotometer in conventional quartz cells of 1 cm path length. The spectral bandwidth and the scan rate were 1 nm and 140 nm min−1, respectively. Stock solutions of each compound were prepared in spectroscopic grade acetonitrile (ACN) and all experiments were carried out in red light and at room temperature. Complex formation of dyes with Ba(ClO4)2 was studied by spectrophotometric titration. In the experiment aliquots of a solution containing known concentrations of dyes and of Ba(ClO4)2 were added to a solution of dyes alone at the same concentration. So the absorption spectra were recorded for solutions with identical total dye concentration (1 × 10−5 M) and variable total Ba(ClO4)2 concentration ranging from 1 × 10−5 M to 5 × 10−1 M in ACN.
Emission spectra were recorded on FluoroLog3-22, Horiba Jobin Yvon spectrofluorometer with Quanta–φ accessory having a large 150 mm integrating sphere for the quantum yield measurements. All spectra we recorded using quartz cells of 1 cm path length. The solution concentrations were chosen to give an absorbance ≤0.05 at the excitation wavelength of 440 and 488 nm.
The photoisomerization of the dyes and their complexes was performed by irradiating the samples in quartz cells (1 cm) with 16 mW laser (Qioptiq iFLEX2000-P-2-488) at λ = 488 nm for 3 min, a time which was found to be long enough to reach a photostationary state. The kinetics of the cis–trans thermal isomerization were studied by measuring the absorbance at a fixed wavelength in the dark as a function of time after irradiation stopped until the initial absorbance value before excitation was reached. The absorbance was measured by Ocean Optic HR2000+ high resolution USB fiber optic spectrometer fitted with 500 nm interference filter in the incident beam (10 nm bandwidth). During the irradiation and kinetic studies, the solutions were intensively magnetically stirred. A cuvette holder for fluorescent measurements was used allowing recording of the absorbance spectra during the laser irradiation in the perpendicular direction and immediately after the stop of irradiation.
Computational details
Equilibrium geometries and intermolecular interaction energies for the host–guest assemblies between the dyes and metal cations were obtained by density functional theory (DFT) calculations using the B3LYP functional [25,26] (the most often used functional for organic molecules and complexes) and the 6-31+G(d,p) [27-29] basis set for the lighter atoms (C, O, S, N, H) and SDD pseudopotential for Ba atoms as implemented in the Gaussian 09 program package [30]. Frequency calculations for each optimized structure were performed at the same level of theory. No imaginary frequency was found for the lowest energy configurations of any of the optimized structures. In order to take into account the solvent effect induced by the acetonitrile solvent environment, the equilibrium geometries of the host–guest constituents and complexes were reoptimized considering the PCM (polarizable continuum model) solvent model [31]. The so-called basis set superposition error (BSSE) was not taken into account for the geometry optimization and intermolecular energy calculation. Time-dependent density functional theory calculations (TDDFT) using Perdew–Burke–Ernzerhof exchange-correlation functional (PBE0) were performed to compute the 10 lowest excited states of each structure [6–31+G(d,p) basis set for all atoms except Ba]. PyMOL molecular graphics system was used for generation of the molecular graphics images [32].
Supporting Information
| Supporting Information File 1: Experimental procedures for the synthesis of compounds 2–4 and characterization data of the new compounds. | ||
| Format: PDF | Size: 2.1 MB | Download |
References
-
Gust, D.; Andréasson, J.; Pischel, U.; Moore, T. A.; Moore, A. L. Chem. Commun. 2012, 48, 1947–1957. doi:10.1039/c1cc15329c
Return to citation in text: [1] -
Pischel, U.; Andréasson, J.; Gust, D.; Pais, V. F. ChemPhysChem 2013, 14, 28–46. doi:10.1002/cphc.201200157
Return to citation in text: [1] -
Raimondo, C.; Crivillers, N.; Reinders, F.; Sander, F.; Mayor, M.; Samorì, P. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 12375–12380. doi:10.1073/pnas.1203848109
Return to citation in text: [1] -
Orgiu, E.; Crivillers, N.; Herder, M.; Grubert, L.; Pätzel, M.; Frisch, J.; Pavlica, E.; Duong, D. T.; Bratina, G.; Salleo, A.; Koch, N.; Hecht, S.; Samorì, P. Nat. Chem. 2012, 4, 675–679. doi:10.1038/nchem.1384
Return to citation in text: [1] -
Crivillers, N.; Orgiu, E.; Reinders, F.; Mayor, M.; Samorì, P. Adv. Mater. (Weinheim, Ger.) 2011, 23, 1447–1452. doi:10.1002/adma.201003736
Return to citation in text: [1] -
Kucharski, T. J.; Tian, Y.; Akbulatov, S.; Boulatov, R. Energy Environ. Sci. 2011, 4, 4449–4472. doi:10.1039/c1ee01861b
Return to citation in text: [1] -
Lennartson, A.; Roffey, A.; Moth-Poulsen, K. Tetrahedron Lett. 2015, 56, 1457–1465. doi:10.1016/j.tetlet.2015.01.187
Return to citation in text: [1] [2] -
Börjesson, K.; Lennartson, A.; Moth-Poulsen, K. ACS Sustainable Chem. Eng. 2013, 1, 585–590. doi:10.1021/sc300107z
Return to citation in text: [1] -
Börjesson, K.; Dzebo, D.; Albinsson, B.; Moth-Poulsen, K. J. Mater. Chem. A 2013, 1, 8521–8524. doi:10.1039/c3ta12002c
Return to citation in text: [1] -
Baluschev, S.; Miteva, T.; Yakutkin, V.; Nelles, G.; Yasuda, A.; Wegner, G. Phys. Rev. Lett. 2006, 97, 143903. doi:10.1103/physrevlett.97.143903
Return to citation in text: [1] -
Baluschev, S.; Katta, K.; Avlasevich, Y.; Landfester, K. Mater. Horiz. 2016, 3, 478–486. doi:10.1039/c6mh00289g
Return to citation in text: [1] -
Dreos, A.; Wang, Z.; Udmark, J.; Ström, A.; Erhart, P.; Börjesson, K.; Nielsen, M. B.; Moth-Poulsen, K. Adv. Energy Mater. 2018, 8, 1703401. doi:10.1002/aenm.201703401
Return to citation in text: [1] -
Wang, Z.; Udmark, J.; Börjesson, K.; Rodrigues, R.; Roffey, A.; Abrahamsson, M.; Nielsen, M. B.; Moth-Poulsen, K. ChemSusChem 2017, 10, 3049–3055. doi:10.1002/cssc.201700679
Return to citation in text: [1] -
Alfimov, M. V.; Gromov, S. P.; Lednev, I. K. Chem. Phys. Lett. 1991, 185, 455–460. doi:10.1016/0009-2614(91)80242-p
Return to citation in text: [1] [2] -
Gromov, S. P.; Fedorova, O. A.; Ushakov, E. N.; Stanislavskii, O. B.; Lednev, I. K.; Alfimov, M. V. Dokl. Akad. Nauk. SSSR 1991, 317, 1134–1139.
Return to citation in text: [1] [2] -
Lednev, I. K.; Fyedorova, O. A.; Gromov, S. P.; Alfimov, M. V.; Moore, J. N.; Hester, R. E. Spectrochim. Acta, Part A 1993, 49, 1055–1063. doi:10.1016/0584-8539(93)80065-i
Return to citation in text: [1] [2] -
Lednev, I. K.; Hester, R. E.; Moore, J. N. J. Chem. Soc., Faraday Trans. 1997, 93, 1551–1558. doi:10.1039/a607389a
Return to citation in text: [1] [2] [3] -
Lednev, I. K.; Ye, T.-Q.; Hester, R. E.; Moore, J. N. J. Phys. Chem. A 1997, 101, 4966–4972. doi:10.1021/jp970685y
Return to citation in text: [1] [2] [3] [4] [5] -
Lednev, I. K.; Hester, R. E.; Moore, J. N. J. Am. Chem. Soc. 1997, 119, 3456–3461. doi:10.1021/ja964154j
Return to citation in text: [1] [2] [3] [4] -
Gromov, S. P.; Alfimov, M. V. Russ. Chem. Bull. 1997, 46, 611–636. doi:10.1007/bf02495186
Return to citation in text: [1] -
Gromov, S. P. Russ. Chem. Bull. 2008, 57, 1325–1350. doi:10.1007/s11172-008-0174-9
Return to citation in text: [1] [2] -
Li, Q.; Lin, G.-L.; Peng, B.-X.; Li, Z.-X. Dyes Pigm. 1998, 38, 211–218. doi:10.1016/s0143-7208(97)00088-0
Return to citation in text: [1] [2] [3] [4] -
Gutten, O.; Rulíšek, L. Inorg. Chem. 2013, 52, 10347–10355. doi:10.1021/ic401037x
Return to citation in text: [1] [2] -
Tulyakova, E. V.; Vermeersch, G.; Gulakova, E. N.; Fedorova, O. A.; Fedorov, Y. V.; Micheau, J. C.; Delbaere, S. Chem. – Eur. J. 2010, 16, 5661–5671. doi:10.1002/chem.200903226
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913
Return to citation in text: [1] -
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/physrevb.37.785
Return to citation in text: [1] -
Ditchfield, R.; Hehre, W. J.; Pople, J. A. J. Chem. Phys. 1971, 54, 724–728. doi:10.1063/1.1674902
Return to citation in text: [1] -
Hehre, W. J.; Ditchfield, R.; Pople, J. A. J. Chem. Phys. 1972, 56, 2257–2261. doi:10.1063/1.1677527
Return to citation in text: [1] -
Clark, T.; Chandrasekhar, J.; Spitznagel, G. W.; Schleyer, P. V. R. J. Comput. Chem. 1983, 4, 294–301. doi:10.1002/jcc.540040303
Return to citation in text: [1] -
Gaussian 09; Gaussian Inc.: Wallingford, CT, 2013.
Return to citation in text: [1] -
Miertuš, S.; Scrocco, E.; Tomasi, J. Chem. Phys. 1981, 55, 117–129. doi:10.1016/0301-0104(81)85090-2
Return to citation in text: [1] -
The PyMOL Molecular Graphics System, Version 1.7.6.6; Schrödinger, LLC.
Return to citation in text: [1]
| 23. | Gutten, O.; Rulíšek, L. Inorg. Chem. 2013, 52, 10347–10355. doi:10.1021/ic401037x |
| 16. | Lednev, I. K.; Fyedorova, O. A.; Gromov, S. P.; Alfimov, M. V.; Moore, J. N.; Hester, R. E. Spectrochim. Acta, Part A 1993, 49, 1055–1063. doi:10.1016/0584-8539(93)80065-i |
| 17. | Lednev, I. K.; Hester, R. E.; Moore, J. N. J. Chem. Soc., Faraday Trans. 1997, 93, 1551–1558. doi:10.1039/a607389a |
| 25. | Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913 |
| 26. | Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/physrevb.37.785 |
| 1. | Gust, D.; Andréasson, J.; Pischel, U.; Moore, T. A.; Moore, A. L. Chem. Commun. 2012, 48, 1947–1957. doi:10.1039/c1cc15329c |
| 2. | Pischel, U.; Andréasson, J.; Gust, D.; Pais, V. F. ChemPhysChem 2013, 14, 28–46. doi:10.1002/cphc.201200157 |
| 3. | Raimondo, C.; Crivillers, N.; Reinders, F.; Sander, F.; Mayor, M.; Samorì, P. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 12375–12380. doi:10.1073/pnas.1203848109 |
| 4. | Orgiu, E.; Crivillers, N.; Herder, M.; Grubert, L.; Pätzel, M.; Frisch, J.; Pavlica, E.; Duong, D. T.; Bratina, G.; Salleo, A.; Koch, N.; Hecht, S.; Samorì, P. Nat. Chem. 2012, 4, 675–679. doi:10.1038/nchem.1384 |
| 5. | Crivillers, N.; Orgiu, E.; Reinders, F.; Mayor, M.; Samorì, P. Adv. Mater. (Weinheim, Ger.) 2011, 23, 1447–1452. doi:10.1002/adma.201003736 |
| 9. | Börjesson, K.; Dzebo, D.; Albinsson, B.; Moth-Poulsen, K. J. Mater. Chem. A 2013, 1, 8521–8524. doi:10.1039/c3ta12002c |
| 19. | Lednev, I. K.; Hester, R. E.; Moore, J. N. J. Am. Chem. Soc. 1997, 119, 3456–3461. doi:10.1021/ja964154j |
| 8. | Börjesson, K.; Lennartson, A.; Moth-Poulsen, K. ACS Sustainable Chem. Eng. 2013, 1, 585–590. doi:10.1021/sc300107z |
| 19. | Lednev, I. K.; Hester, R. E.; Moore, J. N. J. Am. Chem. Soc. 1997, 119, 3456–3461. doi:10.1021/ja964154j |
| 7. | Lennartson, A.; Roffey, A.; Moth-Poulsen, K. Tetrahedron Lett. 2015, 56, 1457–1465. doi:10.1016/j.tetlet.2015.01.187 |
| 20. | Gromov, S. P.; Alfimov, M. V. Russ. Chem. Bull. 1997, 46, 611–636. doi:10.1007/bf02495186 |
| 6. | Kucharski, T. J.; Tian, Y.; Akbulatov, S.; Boulatov, R. Energy Environ. Sci. 2011, 4, 4449–4472. doi:10.1039/c1ee01861b |
| 18. | Lednev, I. K.; Ye, T.-Q.; Hester, R. E.; Moore, J. N. J. Phys. Chem. A 1997, 101, 4966–4972. doi:10.1021/jp970685y |
| 21. | Gromov, S. P. Russ. Chem. Bull. 2008, 57, 1325–1350. doi:10.1007/s11172-008-0174-9 |
| 14. | Alfimov, M. V.; Gromov, S. P.; Lednev, I. K. Chem. Phys. Lett. 1991, 185, 455–460. doi:10.1016/0009-2614(91)80242-p |
| 15. | Gromov, S. P.; Fedorova, O. A.; Ushakov, E. N.; Stanislavskii, O. B.; Lednev, I. K.; Alfimov, M. V. Dokl. Akad. Nauk. SSSR 1991, 317, 1134–1139. |
| 14. | Alfimov, M. V.; Gromov, S. P.; Lednev, I. K. Chem. Phys. Lett. 1991, 185, 455–460. doi:10.1016/0009-2614(91)80242-p |
| 15. | Gromov, S. P.; Fedorova, O. A.; Ushakov, E. N.; Stanislavskii, O. B.; Lednev, I. K.; Alfimov, M. V. Dokl. Akad. Nauk. SSSR 1991, 317, 1134–1139. |
| 18. | Lednev, I. K.; Ye, T.-Q.; Hester, R. E.; Moore, J. N. J. Phys. Chem. A 1997, 101, 4966–4972. doi:10.1021/jp970685y |
| 31. | Miertuš, S.; Scrocco, E.; Tomasi, J. Chem. Phys. 1981, 55, 117–129. doi:10.1016/0301-0104(81)85090-2 |
| 7. | Lennartson, A.; Roffey, A.; Moth-Poulsen, K. Tetrahedron Lett. 2015, 56, 1457–1465. doi:10.1016/j.tetlet.2015.01.187 |
| 19. | Lednev, I. K.; Hester, R. E.; Moore, J. N. J. Am. Chem. Soc. 1997, 119, 3456–3461. doi:10.1021/ja964154j |
| 12. | Dreos, A.; Wang, Z.; Udmark, J.; Ström, A.; Erhart, P.; Börjesson, K.; Nielsen, M. B.; Moth-Poulsen, K. Adv. Energy Mater. 2018, 8, 1703401. doi:10.1002/aenm.201703401 |
| 13. | Wang, Z.; Udmark, J.; Börjesson, K.; Rodrigues, R.; Roffey, A.; Abrahamsson, M.; Nielsen, M. B.; Moth-Poulsen, K. ChemSusChem 2017, 10, 3049–3055. doi:10.1002/cssc.201700679 |
| 27. | Ditchfield, R.; Hehre, W. J.; Pople, J. A. J. Chem. Phys. 1971, 54, 724–728. doi:10.1063/1.1674902 |
| 28. | Hehre, W. J.; Ditchfield, R.; Pople, J. A. J. Chem. Phys. 1972, 56, 2257–2261. doi:10.1063/1.1677527 |
| 29. | Clark, T.; Chandrasekhar, J.; Spitznagel, G. W.; Schleyer, P. V. R. J. Comput. Chem. 1983, 4, 294–301. doi:10.1002/jcc.540040303 |
| 10. | Baluschev, S.; Miteva, T.; Yakutkin, V.; Nelles, G.; Yasuda, A.; Wegner, G. Phys. Rev. Lett. 2006, 97, 143903. doi:10.1103/physrevlett.97.143903 |
| 11. | Baluschev, S.; Katta, K.; Avlasevich, Y.; Landfester, K. Mater. Horiz. 2016, 3, 478–486. doi:10.1039/c6mh00289g |
| 16. | Lednev, I. K.; Fyedorova, O. A.; Gromov, S. P.; Alfimov, M. V.; Moore, J. N.; Hester, R. E. Spectrochim. Acta, Part A 1993, 49, 1055–1063. doi:10.1016/0584-8539(93)80065-i |
| 17. | Lednev, I. K.; Hester, R. E.; Moore, J. N. J. Chem. Soc., Faraday Trans. 1997, 93, 1551–1558. doi:10.1039/a607389a |
| 22. | Li, Q.; Lin, G.-L.; Peng, B.-X.; Li, Z.-X. Dyes Pigm. 1998, 38, 211–218. doi:10.1016/s0143-7208(97)00088-0 |
| 17. | Lednev, I. K.; Hester, R. E.; Moore, J. N. J. Chem. Soc., Faraday Trans. 1997, 93, 1551–1558. doi:10.1039/a607389a |
| 22. | Li, Q.; Lin, G.-L.; Peng, B.-X.; Li, Z.-X. Dyes Pigm. 1998, 38, 211–218. doi:10.1016/s0143-7208(97)00088-0 |
| 18. | Lednev, I. K.; Ye, T.-Q.; Hester, R. E.; Moore, J. N. J. Phys. Chem. A 1997, 101, 4966–4972. doi:10.1021/jp970685y |
| 24. | Tulyakova, E. V.; Vermeersch, G.; Gulakova, E. N.; Fedorova, O. A.; Fedorov, Y. V.; Micheau, J. C.; Delbaere, S. Chem. – Eur. J. 2010, 16, 5661–5671. doi:10.1002/chem.200903226 |
| 22. | Li, Q.; Lin, G.-L.; Peng, B.-X.; Li, Z.-X. Dyes Pigm. 1998, 38, 211–218. doi:10.1016/s0143-7208(97)00088-0 |
| 23. | Gutten, O.; Rulíšek, L. Inorg. Chem. 2013, 52, 10347–10355. doi:10.1021/ic401037x |
| 19. | Lednev, I. K.; Hester, R. E.; Moore, J. N. J. Am. Chem. Soc. 1997, 119, 3456–3461. doi:10.1021/ja964154j |
| 18. | Lednev, I. K.; Ye, T.-Q.; Hester, R. E.; Moore, J. N. J. Phys. Chem. A 1997, 101, 4966–4972. doi:10.1021/jp970685y |
| 22. | Li, Q.; Lin, G.-L.; Peng, B.-X.; Li, Z.-X. Dyes Pigm. 1998, 38, 211–218. doi:10.1016/s0143-7208(97)00088-0 |
| 18. | Lednev, I. K.; Ye, T.-Q.; Hester, R. E.; Moore, J. N. J. Phys. Chem. A 1997, 101, 4966–4972. doi:10.1021/jp970685y |
| 21. | Gromov, S. P. Russ. Chem. Bull. 2008, 57, 1325–1350. doi:10.1007/s11172-008-0174-9 |
© 2019 Kandinska et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)