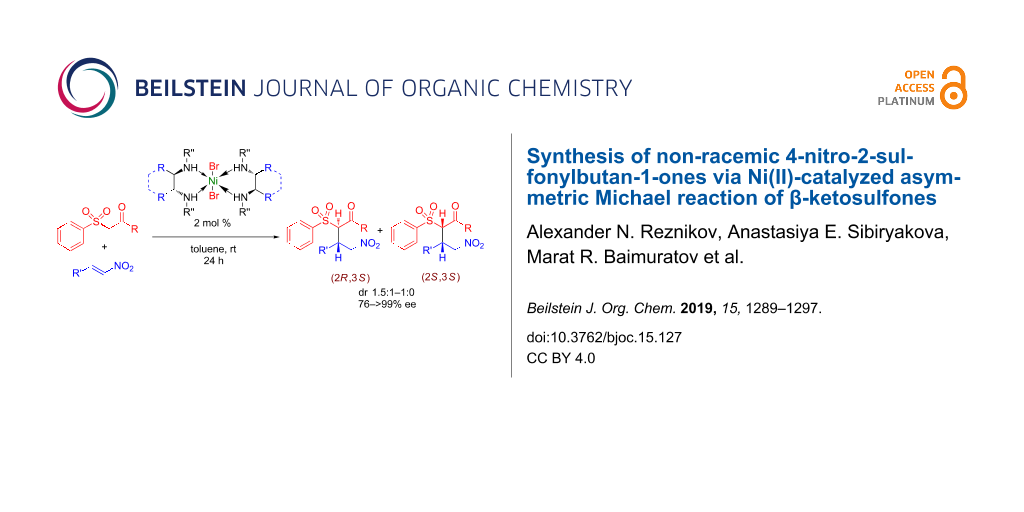
Abstract
Functionally substituted sulfones with stereogenic centers are valuable reagents in organic synthesis and key motifs in some bioactive compounds. The asymmetric Michael addition of β-ketosulfones to conjugated nitroalkenes in the presence of Ni(II) complexes with various chiral vicinal diamines was studied. This reaction provides convenient access to non-racemic 4-nitro-2-sulfonylbutan-1-ones with two stereocenters with high yield and excellent enantioselectivity (up to 99%). It has been established that the catalytic Michael reaction itself was carried out with high diastereoselectivity, but the Michael adducts may epimerize at the C-2 position at a significant rate. Conditions for the preparation of individual diastereomers were found.

Graphical Abstract
Introduction
Sulfones are widely used in organic synthesis, particularly, in various reactions of C–C and C=C-bond formation [1-4]. The use of sulfones in Julia–Kocienski [1] and Ramberg–Bäcklund reactions [2] made this class of compounds frequently used in the synthesis of organic fine chemicals and natural compounds. In addition to using the sulfonyl group as an auxiliary, it is also included in some chiral bioactive molecules, such as remikiren (1, renin inhibitor for the treatment of hypertension) [5,6], eletriptan (2, Relpax®, serotonin 5-HT1 receptor agonist for the treatment of migraine) [7], and apremilast (3, Otezla®, inhibitor of the PDE4 for the treatment of certain types of psoriasis and psoriatic arthritis) [8] (Figure 1). Recently we have shown that racemic sulfone 4 exhibits high antiviral activity against BVDV with low cytotoxicity [9]. However, it is of great importance to obtain all stereoisomers for the study of biological activity.
![[1860-5397-15-127-1]](/bjoc/content/figures/1860-5397-15-127-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Рharmacologically active sulfones.
Figure 1: Рharmacologically active sulfones.
Therefore, the development of methods for the asymmetric synthesis of polyfunctional sulfones is valuable. The most notable of them are Ag- and Cu-catalyzed 1,3-dipolar cycloaddition reactions, which allows to obtain chiral cyclic sulfones with high enantioselectivity [10-12]. Also non-racemic cyclic sulfones can be obtained by the Diels–Alder reaction, catalyzed by chiral Lewis acids or organocatalysts. Rh- and Cu-catalyzed CH-insertion reactions occurring at moderate or high enantioselectivity are also known [13-18]. The studied methods for obtaining acyclic sulfones with stereogenic centers in the side chain are more limited. One of the most significant approaches to obtaining both cyclic and acyclic chiral sulfones is asymmetric hydrogenation in the presence of transition metal complexes. The preparation of hydroxy sulfones from β-ketosulfones in the presence of Ru [19,20], Ir [21] and Rh [22] complexes was described. Chiral sulfones were also obtained by hydrogenation of the C=C bond with α,β-unsaturated sulfones in the presence of Ir(I) complexes with P,N-ligands [23].
The asymmetric addition of various nucleophiles to unsaturated sulfones is also considered as an effective route to chiral sulfones. The conjugated addition of arylboronic acids to unsaturated sulfones under catalysis of Rh complexes was reported [24-26]. It was shown that arylboronic acids are attached to 1,2-disubstituted α,β-unsaturated sulfones in the presence of the Rh/(S,S)-chiraphos catalytic system. Modern methods for the synthesis of functionalized sulfones, with stereocenters in the side chain, by Michael addition are based, mainly, on the use of vinyl sulfones as Michael acceptors and aldehydes [27-32], ketones [33], α-cyano esters [34,35], β-keto esters [35], β-keto acids [36], thiomalonates [37], nitroalkanes [38], oxindoles [39] and thiols [40] as Michael donors. There are only few examples of the use of sulfones as Michael donors in asymmetric addition reactions. Thus, ketonitrosulfones were obtained with good enantiomeric excesses via asymmetric addition of α-nitrosulfones to enones in the presence of organocatalysts [41]. Asymmetric addition of β-ketosulfones to nitroalkenes was implemented using various organocatalysts [42]. The reaction of ketosulfones with nitroalkenes in the presence of organocatalysts shows high enantioselectivity, however, leads to a mixture of diastereomers.
For the catalytic activation of β-ketosulfones by metal complexes chiral Lewis acids may be considered as an alternative way to carry out the asymmetric Michael reaction with their participation. Asymmetric conjugate addition of activated methylene compounds (such as diketones, keto esters and malonates) to nitroalkenes in the presence of Mg [43], Co [44,45], Mn [45] and Ru [46] complexes was performed. The most remarkable results were obtained with Ni(II) complexes as catalyst for the Michael addition of 1,3-dicarbonyl compounds to nitroalkenes [47-54]. This reaction was used as a key stage in the synthesis of non-racemic analogues of GABA and substituted pyrrolidinones with neurotropic activity [47,51,52,54].
It should be noted that the reaction of β-keto phosphonates with nitroalkenes in the presence of Ni(II) complexes with chiral vicinal diamines was carried out not only with excellent enantioselectivity, but also diastereoselectivity [55]. Moreover, β-keto sulfoxides react with nitroalkenes under catalysis by Ni(II) complexes [56].
The above considerations lead to the use of β-ketosulfones in the Ni(II)-catalyzed reaction, since the proposed mechanism [47], that involves the formation of cyclic Ni enolate, and the high CH acidity of ketosulfones (pKa 9.8–10.5 [4]). The formation of the key intermediate can be provided by the coordination of β-ketosulfones through the oxygen atom of the sulfonyl group with Ni. For example, sulfoxide complexes with O-coordination of the corresponding ligands are widely known [57]. Although the donor properties of sulfones are lower than those of sulfoxides.
Results and Discussion
Initially we carried out a screening of catalysts using the Ni(II) complexes 7a–h with chiral vicinal diamines L1–L8 (Figure 2) in a model reaction of 1-phenyl-2-(phenylsulfonyl)ethan-1-one (5a) with ω-nitrostyrene (6a). The results of the study are shown in Table 1.
![[1860-5397-15-127-2]](/bjoc/content/figures/1860-5397-15-127-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structures of the ligands L1–L8.
Figure 2: Structures of the ligands L1–L8.
Table 1: Screening of Ni(II) complexes with chiral diamines in the asymmetric addition of 1-phenyl-2-(phenylsulfonyl)ethan-1-one (5) to ω-nitrostyrene (6a)a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-127-i3.svg?max-width=637&scale=1.0)
|
||||
| Entry | Catalyst | Conversion, %b |
drb,c
8a:9a |
ee, %d |
| 1 |
[NiBr2L12]
7a |
86 | 2:1 | 91:82 |
| 2 |
[NiBr2L22]
7b |
82 | 7.3/1 | 89:80 |
| 3 |
[NiBr2L32]
7c |
80 | 2.2:1 | 91:82 |
| 4 |
[NiBr2L42]
7d |
40 | 6.5:1 | 70:65 |
| 5 |
[NiBr2L52]
7e |
42 | 1.4:1 | 92:71 |
| 6 |
[NiBr2L62]
7f |
40 | 2.4:1 | 96:51 |
| 7 |
[NiBr2L72]
7g |
39 | 1:1.4 | >99:87 |
| 8 |
[NiBr2L82]
7h |
86 | 1.4:1 | 87:70 |
| 9 | no cat. | – | – | – |
aReaction conditions: 5a 1.00 mmol, 6a 1.05 mmol, THF 1.5 mL, catalysts 7a–h 0.02 mmol, 20 °C, 24 h; bdetermined by 1H NMR; cthe absolute configuration of compound 8a was assumed by analogy with 8d; the relative configurations of the compounds 8a and 9a were assigned by studying their 1H NMR spectra in comparison with compound 8d, for which X-ray diffraction data were obtained (see below); ddetermined by chiral HPLC.
The Michael addition of the β-ketosulfone 5a was carried out with moderate to high enantioselectivity but low diastereoselectivity and led to the formation of two diastereomers (2R,3S)-8a and (2S,3S)-9a. Since we did not succeed in growing a crystal that was suitable for X-ray determination of the absolute configuration of 8а, we defined the absolute configuration as compared to the analogous adamantyl derivative, which will be discussed below. It should be noted that low diastereoselectivity was previously observed in the asymmetric addition of various Michael donors to nitroalkenes in the presence of both metal complexes [47] and organocatalysts [58-61]. This was explained by the authors as a result of the high CH acidity of the corresponding Michael adducts. The highest reaction rate with good enantioselectivity is achieved using catalyst 7a (Table 1, entry 1). For this reason, further studies were carried out with catalyst 7a. The study of the solvent effect on the reaction are summarized in Table 2.
Table 2: Solvent effect on the reaction of 1-phenyl-2-(phenylsulfonyl)ethan-1-one (5a) with ω-nitrostyrene (6a)a.
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-127-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Solvent | Conversion, %b |
drb
8a:9a |
eec
8a:9a |
| 1 | toluene | 98 | 1:– | 94 |
| 2 | EtOAc | 82 | 1.6:1 | 87:57 |
| 3 | THF | 86 | 2:1 | 91:82 |
| 4 | CH2Cl2 | 89 | 2.2:1 | 81:63 |
| 5 | CHF2CF2CH2OH | 40 | 1.8:1 | 83:75 |
| 6 | MeOH | 47 | 1.8:1 | 83:35 |
| 7 | CH3NO2 | 42 | 1:1 | 80:75 |
| 8 | DMF | 43 | 1:1 | 0:0 |
| 9 | ethane-1,2-diol | 18 | 1:1.9 | NDd |
| 10 | MeCN | 50 | 1.2:1 | 89:75 |
aReaction conditions: 5a 1.00 mmol, 6a 1.05 mmol, solvent 1.5 mL, catalyst 7a 0.02 mmol, 20 °C, 24 h; bdetermined by 1H NMR; cdetermined by chiral HPLC; dnot determined.
As can be seen from Table 2, the dr value decreases when using more polar solvents. An individual diastereomer 8a is formed when toluene is used as a solvent (Table 2, entry 1). The highest reaction rate and enantioselectivity are also achieved in toluene. Considering these factors, toluene was chosen as the best solvent for this reaction.
The obtained experimental data show that in some cases the formation of diastereomer (2R,3S)-8a or diastereomer (2S,3S)-9a as the major one is observed depending on the type of catalyst, and, on the other hand, the ratio of diastereomers in the presence of the same catalyst strongly depends on the solvent used. This suggests that the dr is determined by the rates ratio of the catalytic reaction (which can occur with high or low diastereoselectivity) and the epimerization of products 8a or 9a. Оne of the stereoisomers can be formed directly during the reaction. To test this hypothesis, we decided to study the evolution of dr on the course of reaction by 1H NMR spectroscopy (Figure 3). The reaction of sulfone 5a with ω-nitrostyrene (6a) was chosen as the model reaction.
![[1860-5397-15-127-3]](/bjoc/content/figures/1860-5397-15-127-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Evolution of the conversion of 5 and diastereomeric composition of the products of reaction of 5a with 6a in the presence of catalyst 7a (2 mol %) in chloroform-d.
Figure 3: Evolution of the conversion of 5 and diastereomeric composition of the products of reaction of 5a w...
Conversion was determined by decrease of the integral intensity of the sulfone 5a methylene group signal at 4.75 ppm. The diastereomers 8a:9a ratio was determined by the ratio of the integral intensities of signals of the methine groups of 8a and 9a at 4.62–4.57 and 4.53–4.47 ppm, respectively. As the studies have shown, during the first 12 hours, one diastereomer (2R,3S)-8a was formed predominantly (dr 8a:9a of more than 14:1). During this time, the conversion reached 71%. Only after this period, the appearance of the significant amount of second diastereomer (2S,3S)-9a was recorded. After 24 hours, the dr (8a:9a) reached 6.3:1, while the conversion is 82%. After 50 hours, the dr reached 3:1, while the reaction practically stopped.
Further, we carried out a control experiment to evaluate the epimerization rate of product 8a in solution under the same conditions. For this purpose the individual diastereomer 8a was dissolved in chloroform-d and the formation of the second diastereomer 9a was monitored by 1H NMR. Surprisingly, we found that, along with the epimerization of (2R,3S)-8a to (2S,3S)-9a, retro-Michael reaction occurred (Figure 4).
![[1860-5397-15-127-4]](/bjoc/content/figures/1860-5397-15-127-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Time profile of epimerization and retro-Michael reaction of (2R,3S)-8a in chloroform-d solution.
Figure 4: Time profile of epimerization and retro-Michael reaction of (2R,3S)-8a in chloroform-d solution.
It is noteworthy that firstly the formation of sulfone 5a was more rapid than the epimerization of product 8a. After about 60 hours the content of sulfone 5a in solution became almost constant, while the amount of epimer 9a continued to increase. This fact may indicate that the formation of compound 9a may occur not only as a result of keto–enol tautomerism in solution, but also as a result of dynamic equilibrium between the Michael/retro-Michael products. This assumption is confirmed by a partial racemization of sulfones 8 at a prolonged storage, passing both the stereocenter at position 2 and the stereocenter at the position 3 (according to the HPLC data).
This study shows that the asymmetric addition of sulfone 5a to ω-nitrostyrene (6a) occurs enantio- and diastereoselectively and leads to isomer 8a, and the formation of 9a is explained by subsequent epimerization of 8a. The study of the reaction of various β-ketosulfones with nitroalkenes in the presence of complex 7a was carried out under the optimized conditions (Table 3).
Table 3: Asymmetric addition of β-ketosulfones to nitroalkenes in the presence of complex 7aa.
![[Graphic 3]](/bjoc/content/inline/1860-5397-15-127-i5.svg?max-width=637&scale=1.0)
|
|||||||||
| Entry | R | R' | Compd | Conv, %b |
drc,
8:9 |
eec, %
for 8:for 9 |
Yieldd, %
8:9 |
dre,
8:9 |
eee, %
for 8: for 9 |
| 1 | Ph | Ph | 8a/9a | 98 | 1:– | 94:– | 77:– | 1:– | >99:– |
| 2 | 4-ClC6H4 | Ph | 8b/9b | 90 | 1:– | 92:– | 72:– | 1:– | >99:– |
| 3 | 3-MeOC6H4 | Ph | 8c/9c | 80 | 1.9:1 | 88:82 | –:46 | 1: 29 | –:>99 |
| 4 | 1-Ad | Ph | 8d/9d | 87 | 1:– | 93:– | 67:– | 1:– | >99:– |
| 5 | Ph | 4-FC6H4 | 8e/9e | 88 | 1:– | 96:– | 74:– | 1:– | >99:– |
| 6 | Ph | 4-ClC6H4 | 8f/9f | 93 | 1:– | >99:– | 71 | 1:– | >99:– |
| 7 | Ph | 2-ClC6H4 | 8g/9g | 98 | 2.8:1 | 92:93 | 93f | 2.8:1 | 92:93 |
| 8 | Ph | 4-NO2C6H4 | 8h/9h | 95 | 1:– | 76:– | 65:– | 1:– | >99:– |
| 9 | Ph | 3-MeO C6H4 | 8i/9i | 93 | 1:1.13 | 78:89 | 52f | 1:2 | >99:>99 |
aReaction conditions: β-ketosulfone 5a–d 1.00 mmol, nitroalkene 6a–f 1.05 mmol, toluene 1.5 mL, catalyst 7a 0.02 mmol, 20 °C, 48 h; bdetermined by 1H NMR; cdr (by 1H NMR) and ee (by chiral HPLC) in reaction mixture; the absolute configuration of compounds 8a–i was assumed by analogy with 8d; the relative configurations of the compounds 8a–i and 9a–i were assigned by studying their 1H NMR spectra in comparison with compound 8d, for which X-ray diffraction data were obtained (see below); disolated yields; edr and ee after crystallization (for entries 1–6, 8, 9) or column chromatography (for entry 7); fisolated yields for a mixtures of diastereomers.
The dr values for 8:9 are ranged from 1:– to 1:1.13. The formation of а mixture of diastereomers is observed in the presence of substituents in the 2- or 3-position of the aryl ring of sulfones or nitroalkenes.
Crystals suitable for X-ray analysis were obtained for compound 8d (which is formed as an individual diastereomer). This made it possible to determine its absolute (2R,3S)-configuration, as well as to assign the absolute configuration for the other compounds obtained. The molecular structure of compound 8d is shown in Figure 5.
![[1860-5397-15-127-5]](/bjoc/content/figures/1860-5397-15-127-5.png?scale=2.0&max-width=1024&background=FFFFFF)
The relative configurations of other Michael adducts 8 and 9 were determined by comparing their NMR data with those of compound 8d. For (2R,3S)-isomers the value of 3JHH for proton at 2-C was 5 Hz, while for (2S,3S)-isomers this value was 11 Hz.
It is possible to use the previously proposed mechanism for 1,3-dicarbonyl compounds [47] to explain how Ni catalysts are able to activate the substrates. The postulated catalytic cycle is summarized in Scheme 1.
![[1860-5397-15-127-i1]](/bjoc/content/inline/1860-5397-15-127-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The proposed mechanism of asymmetric addition of β-ketosulfones to nitroalkenes.
Scheme 1: The proposed mechanism of asymmetric addition of β-ketosulfones to nitroalkenes.
We assume that the β-ketosulfone coordinates to the Ni complex generating Ni-enolate B. The nitroalkene is activated by coordination to Ni. The complex B regenerates after the conjugate addition via transition state C and coordination of a new β-ketosulfone molecule to Ni. TS1 and TS2 are proposed by analogy with 1,3-dicarbonyl compounds [47] to rationalize the asymmetric induction. As illustrated in Scheme 2, β-ketosulfone is coordinated to Ni in more Lewis acidic equatorial position, whereas the nitroalkene is positioned in apical by avoiding the steric repulsion of benzyl groups (TS2-I and 2-II vs TS1-I and 1-II, Scheme 2). Additional hydrogen bonding between the hydrogen atom of the amino group and the oxygen atom of the nitroalkene in TS2-I and 2-II may also help to rigidify the transition state and improve the stereoselectivities.
![[1860-5397-15-127-i2]](/bjoc/content/inline/1860-5397-15-127-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Transition state models for asymmetric addition of β-ketosulfones to nitroalkenes.
Scheme 2: Transition state models for asymmetric addition of β-ketosulfones to nitroalkenes.
The observed (2R,3S)-diastereoselectivity in the presence of catalyst 7a stems from the addition of the Re face of the β-ketosulfone to the Si face of the nitroalkene in TS2-I. We suppose that the CO group of sulfone is placed on the side of the nitro group, whereas the bulkier sulfonyl group is oriented opposed to the nitro group to minimize steric interactions.
Conclusion
In summary, a convenient synthetic protocol for the preparation of valuable non-racemic 4-nitro-2-sulfonylbutan-1-ones via Ni(II)-catalyzed Michael addition was developed. Corresponding sulfones were obtained with high enantiomeric excesses (up to 99%) by asymmetric addition of β-ketosulfones to nitroalkenes in the presence of Ni(II) complexes with chiral vicinal diamines. In some cases, a high diastereoselectivity of the reaction was observed.
Supporting Information
| Supporting Information File 1: Experimental procedures, copies of NMR, FTIR, mass spectra, HPLC and X-ray diffraction data. | ||
| Format: PDF | Size: 6.4 MB | Download |
References
-
Blakemore, P. R.; Sephton, S. M.; Ciganek, E. Org. React. 2018, 95, 1–422. doi:10.1002/0471264180.or095.01
Return to citation in text: [1] [2] -
Taylor, R. J. K.; Casy, G. Org. React. 2003, 62, 359–475. doi:10.1002/0471264180.or062.02
Return to citation in text: [1] [2] -
Carretero, J. C.; Arrayás, R. G.; Adrio, J. Sulfones in Asymmetric Catalysis. In Organosulfur Chemistry in Asymmetric Synthesis; Toru, T.; Bolm, C., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; pp 291–320. doi:10.1002/9783527623235.ch9
Return to citation in text: [1] -
Markitanov, Y. M.; Timoshenko, V. M.; Shermolovich, Y. G. J. Sulfur Chem. 2014, 35, 188–236. doi:10.1080/17415993.2013.815749
Return to citation in text: [1] [2] -
Richter, W. F.; Whitby, B. R.; Chou, R. C. Xenobiotica 1996, 26, 243–254. doi:10.3109/00498259609046705
Return to citation in text: [1] -
Himmelmann, A.; Bergbrant, A.; Svensson, A.; Hansson, L.; Aurell, M. Am. J. Hypertens. 1996, 9, 517–522. doi:10.1016/0895-7061(95)00340-1
Return to citation in text: [1] -
Capi, M.; Curto, M.; Lionetto, L.; de Andrés, F.; Gentile, G.; Negro, A.; Martelletti, P. Ther. Adv. Neurol. Disord. 2016, 9, 414–423. doi:10.1177/1756285616650619
Return to citation in text: [1] -
Keating, G. M. Drugs 2017, 77, 459–472. doi:10.1007/s40265-017-0709-1
Return to citation in text: [1] -
Shiryaev, V. A.; Radchenko, E. V.; Palyulin, V. A.; Zefirov, N. S.; Bormotov, N. I.; Serova, O. A.; Shishkina, L. N.; Baimuratov, M. R.; Bormasheva, K. M.; Gruzd, Y. A.; Ivleva, E. A.; Leonova, M. V.; Lukashenko, A. V.; Osipov, D. V.; Osyanin, V. A.; Reznikov, A. N.; Shadrikova, V. A.; Sibiryakova, A. E.; Tkachenko, I. M.; Klimochkin, Y. N. Eur. J. Med. Chem. 2018, 158, 214–235. doi:10.1016/j.ejmech.2018.08.009
Return to citation in text: [1] -
Liang, G.; Tong, M.-C.; Wang, C.-J. Adv. Synth. Catal. 2009, 351, 3101–3106. doi:10.1002/adsc.200900552
Return to citation in text: [1] -
Llamas, T.; Arrayás, R. G.; Carretero, J. C. Org. Lett. 2006, 8, 1795–1798. doi:10.1021/ol060314c
Return to citation in text: [1] -
Harada, M.; Kato, S.; Haraguchi, R.; Fukuzawa, S.-i. Chem. – Eur. J. 2018, 24, 2580–2583. doi:10.1002/chem.201706033
Return to citation in text: [1] -
Kennedy, M.; McKervey, M. A.; Maguire, A. R.; Roos, G. H. P. J. Chem. Soc., Chem. Commun. 1990, 361–362. doi:10.1039/c39900000361
Return to citation in text: [1] -
Honma, M.; Sawada, T.; Fujisawa, Y.; Utsugi, M.; Watanabe, H.; Umino, A.; Matsumura, T.; Hagihara, T.; Takano, M.; Nakada, M. J. Am. Chem. Soc. 2003, 125, 2860–2861. doi:10.1021/ja029534l
Return to citation in text: [1] -
Honma, M.; Nakada, M. Tetrahedron Lett. 2003, 44, 9007–9011. doi:10.1016/j.tetlet.2003.09.215
Return to citation in text: [1] -
Sawada, T.; Nakada, M. Adv. Synth. Catal. 2005, 347, 1527–1532. doi:10.1002/adsc.200505173
Return to citation in text: [1] -
Takeda, H.; Watanabe, H.; Nakada, M. Tetrahedron 2006, 62, 8054–8063. doi:10.1016/j.tet.2006.06.022
Return to citation in text: [1] -
Shiely, A. E.; Clarke, L.-A.; Flynn, C. J.; Buckley, A. M.; Ford, A.; Lawrence, S. E.; Maguire, A. R. Eur. J. Org. Chem. 2018, 2277–2289. doi:10.1002/ejoc.201800077
Return to citation in text: [1] -
Bertus, P.; Phansavath, P.; Ratovelomanana-Vidal, V.; Genêt, J.-P.; Touati, A. R.; Homri, T.; Ben Hassine, B. Tetrahedron Lett. 1999, 40, 3175–3178. doi:10.1016/s0040-4039(99)00453-0
Return to citation in text: [1] -
Genêt, J. P.; Pinel, C.; Ratovelomanana-Vidal, V.; Mallart, S.; Pfister, X.; Bischoff, L.; De Andrade, M. C. C.; Darses, S.; Galopin, C.; Laffitte, J. A. Tetrahedron: Asymmetry 1994, 5, 675–690. doi:10.1016/0957-4166(94)80030-8
Return to citation in text: [1] -
Tao, L.; Yin, C.; Dong, X.-Q.; Zhang, X. Org. Biomol. Chem. 2019, 17, 785–788. doi:10.1039/c8ob02923g
Return to citation in text: [1] -
Zhang, H.-L.; Hou, X.-L.; Dai, L.-X.; Luo, Z.-B. Tetrahedron: Asymmetry 2007, 18, 224–228. doi:10.1016/j.tetasy.2007.01.009
Return to citation in text: [1] -
Peters, B. K.; Zhou, T.; Rujirawanich, J.; Cadu, A.; Singh, T.; Rabten, W.; Kerdphon, S.; Andersson, P. G. J. Am. Chem. Soc. 2014, 136, 16557–16562. doi:10.1021/ja5079877
Return to citation in text: [1] -
Mauleón, P.; Carretero, J. C. Org. Lett. 2004, 6, 3195–3198. doi:10.1021/ol048690p
Return to citation in text: [1] -
Mauleón, P.; Alonso, I.; Rodriguez Rivero, M.; Carretero, J. C. J. Org. Chem. 2007, 72, 9924–9935. doi:10.1021/jo7016197
Return to citation in text: [1] -
Mauleón, P.; Carretero, J. C. Chem. Commun. 2005, 4961–4963. doi:10.1039/b508142d
Return to citation in text: [1] -
Kanada, Y.; Yuasa, H.; Nakashima, K.; Murahashi, M.; Tada, N.; Itoh, A.; Koseki, Y.; Miura, T. Tetrahedron Lett. 2013, 54, 4896–4899. doi:10.1016/j.tetlet.2013.06.141
Return to citation in text: [1] -
Nakashima, K.; Murahashi, M.; Yuasa, H.; Ina, M.; Tada, N.; Itoh, A.; Hirashima, S.-i.; Koseki, Y.; Miura, T. Molecules 2013, 18, 14529–14542. doi:10.3390/molecules181214529
Return to citation in text: [1] -
Miura, T.; Yuasa, H.; Murahashi, M.; Ina, M.; Nakashima, K.; Tada, N.; Itoh, A. Synlett 2012, 23, 2385–2388. doi:10.1055/s-0032-1317137
Return to citation in text: [1] -
Murphy, J. J.; Quintard, A.; McArdle, P.; Alexakis, A.; Stephens, J. C. Angew. Chem., Int. Ed. 2011, 50, 5095–5098. doi:10.1002/anie.201100804
Return to citation in text: [1] -
Zhu, Q.; Lu, Y. Chem. Commun. 2010, 46, 2235–2237. doi:10.1039/b919549a
Return to citation in text: [1] -
Mossé, S.; Andrey, O.; Alexakis, A. Chimia 2006, 60, 216–219. doi:10.2533/000942906777674778
Return to citation in text: [1] -
Zhu, Q.; Cheng, L.; Lu, Y. Chem. Commun. 2008, 6315–6317. doi:10.1039/b816307c
Return to citation in text: [1] -
Li, H.; Song, J.; Liu, X.; Deng, L. J. Am. Chem. Soc. 2005, 127, 8948–8949. doi:10.1021/ja0511063
Return to citation in text: [1] -
Li, H.; Song, J.; Deng, L. Tetrahedron 2009, 65, 3139–3148. doi:10.1016/j.tet.2008.11.054
Return to citation in text: [1] [2] -
Wei, Y.; Guo, R.; Dang, Y.; Nie, J.; Ma, J.-A. Adv. Synth. Catal. 2016, 358, 2721–2726. doi:10.1002/adsc.201600485
Return to citation in text: [1] -
Qiao, B.; Liu, Q.; Liu, H.; Yan, L.; Jiang, Z. Chem. – Asian J. 2014, 9, 1252–1256. doi:10.1002/asia.201400049
Return to citation in text: [1] -
Zhu, Q.; Lu, Y. Org. Lett. 2009, 11, 1721–1724. doi:10.1021/ol9003349
Return to citation in text: [1] -
Zong, L.; Du, S.; Chin, K. F.; Wang, C.; Tan, C.-H. Angew. Chem., Int. Ed. 2015, 54, 9390–9393. doi:10.1002/anie.201503844
Return to citation in text: [1] -
Fang, X.; Dong, X.-Q.; Liu, Y.-Y.; Wang, C.-J. Tetrahedron Lett. 2013, 54, 4509–4511. doi:10.1016/j.tetlet.2013.06.059
Return to citation in text: [1] -
Bera, K.; Namboothiri, I. N. N. Chem. Commun. 2013, 49, 10632–10634. doi:10.1039/c3cc45985c
Return to citation in text: [1] -
García Mancheño, O.; Tangen, P.; Rohlmann, R.; Fröhlich, R.; Alemán, J. Chem. – Eur. J. 2011, 17, 984–992. doi:10.1002/chem.201001914
Return to citation in text: [1] -
Barnes, D. M.; Ji, J.; Fickes, M. G.; Fitzgerald, M. A.; King, S. A.; Morton, H. E.; Plagge, F. A.; Preskill, M.; Wagaw, S. H.; Wittenberger, S. J.; Zhang, J. J. Am. Chem. Soc. 2002, 124, 13097–13105. doi:10.1021/ja026788y
Return to citation in text: [1] -
Lewis, K. G.; Ghosh, S. K.; Bhuvanesh, N.; Gladysz, J. A. ACS Cent. Sci. 2015, 1, 50–56. doi:10.1021/acscentsci.5b00035
Return to citation in text: [1] -
Reznikov, A. N.; Golovin, E. V.; Klimochkin, Y. N. Russ. J. Gen. Chem. 2012, 82, 1827–1829. doi:10.1134/s1070363212110163
Return to citation in text: [1] [2] -
Watanabe, M.; Ikagawa, A.; Wang, H.; Murata, K.; Ikariya, T. J. Am. Chem. Soc. 2004, 126, 11148–11149. doi:10.1021/ja046296g
Return to citation in text: [1] -
Evans, D. A.; Mito, S.; Seidel, D. J. Am. Chem. Soc. 2007, 129, 11583–11592. doi:10.1021/ja0735913
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Zhu, Q.; Huang, H.; Shi, D.; Shen, Z.; Xia, C. Org. Lett. 2009, 11, 4536–4539. doi:10.1021/ol901776n
Return to citation in text: [1] -
Wilckens, K.; Duhs, M.-A.; Lentz, D.; Czekelius, C. Eur. J. Org. Chem. 2011, 5441–5446. doi:10.1002/ejoc.201100488
Return to citation in text: [1] -
Jin, R.; Liu, K.; Xia, D.; Qian, Q.; Liu, G.; Li, H. Adv. Synth. Catal. 2012, 354, 3265–3274. doi:10.1002/adsc.201200222
Return to citation in text: [1] -
Reznikov, A. N.; Kapranov, L. E.; Ivankina, V. V.; Sibiryakova, A. E.; Rybakov, V. B.; Klimochkin, Y. N. Helv. Chim. Acta 2018, 101, e1800170. doi:10.1002/hlca.201800170
Return to citation in text: [1] [2] -
Reznikov, A. N.; Golovin, E. V.; Klimochkin, Y. N. Russ. J. Org. Chem. 2013, 49, 663–668. doi:10.1134/s1070428013050047
Return to citation in text: [1] [2] -
Reznikov, A. N.; Sidnin, E. A.; Klimochkin, Y. N. Russ. J. Org. Chem. 2013, 49, 1600–1604. doi:10.1134/s1070428013110067
Return to citation in text: [1] -
Sibiryakova, A. E.; Shiryaev, V. A.; Reznikov, A. N.; Kabanova, A. A.; Klimochkin, Y. N. Synthesis 2019, 51, 463–469. doi:10.1055/s-0037-1610824
Return to citation in text: [1] [2] -
Reznikov, A. N.; Sibiryakova, A. E.; Rybakov, V. B.; Klimochkin, Y. N. Tetrahedron: Asymmetry 2015, 26, 1050–1057. doi:10.1016/j.tetasy.2015.08.003
Return to citation in text: [1] -
Reznikov, A. N.; Sibiryakova, A. E.; Klimochkin, Yu. N. Russ. J. Org. Chem. 2014, 50, 1695–1696. doi:10.1134/s107042801411027x
Return to citation in text: [1] -
Calligaris, M. Coord. Chem. Rev. 2004, 248, 351–375. doi:10.1016/j.ccr.2004.02.005
Return to citation in text: [1] -
Okino, T.; Hoashi, Y.; Furukawa, T.; Xu, X.; Takemoto, Y. J. Am. Chem. Soc. 2005, 127, 119–125. doi:10.1021/ja044370p
Return to citation in text: [1] -
Almaşi, D.; Alonso, D. A.; Gómez-Bengoa, A. E.; Nájera, C. J. Org. Chem. 2009, 74, 6163–6168. doi:10.1021/jo9010552
Return to citation in text: [1] -
Manzano, R.; Andrés, J.; Pedrosa, R. Synlett 2011, 2203–2205. doi:10.1055/s-0030-1261139
Return to citation in text: [1] -
Andrés, J. M.; Losada, J.; Maestro, A.; Rodríguez-Ferrer, P.; Pedrosa, R. J. Org. Chem. 2017, 82, 8444–8454. doi:10.1021/acs.joc.7b01177
Return to citation in text: [1]
| 45. | Reznikov, A. N.; Golovin, E. V.; Klimochkin, Y. N. Russ. J. Gen. Chem. 2012, 82, 1827–1829. doi:10.1134/s1070363212110163 |
| 46. | Watanabe, M.; Ikagawa, A.; Wang, H.; Murata, K.; Ikariya, T. J. Am. Chem. Soc. 2004, 126, 11148–11149. doi:10.1021/ja046296g |
| 47. | Evans, D. A.; Mito, S.; Seidel, D. J. Am. Chem. Soc. 2007, 129, 11583–11592. doi:10.1021/ja0735913 |
| 48. | Zhu, Q.; Huang, H.; Shi, D.; Shen, Z.; Xia, C. Org. Lett. 2009, 11, 4536–4539. doi:10.1021/ol901776n |
| 49. | Wilckens, K.; Duhs, M.-A.; Lentz, D.; Czekelius, C. Eur. J. Org. Chem. 2011, 5441–5446. doi:10.1002/ejoc.201100488 |
| 50. | Jin, R.; Liu, K.; Xia, D.; Qian, Q.; Liu, G.; Li, H. Adv. Synth. Catal. 2012, 354, 3265–3274. doi:10.1002/adsc.201200222 |
| 51. | Reznikov, A. N.; Kapranov, L. E.; Ivankina, V. V.; Sibiryakova, A. E.; Rybakov, V. B.; Klimochkin, Y. N. Helv. Chim. Acta 2018, 101, e1800170. doi:10.1002/hlca.201800170 |
| 52. | Reznikov, A. N.; Golovin, E. V.; Klimochkin, Y. N. Russ. J. Org. Chem. 2013, 49, 663–668. doi:10.1134/s1070428013050047 |
| 53. | Reznikov, A. N.; Sidnin, E. A.; Klimochkin, Y. N. Russ. J. Org. Chem. 2013, 49, 1600–1604. doi:10.1134/s1070428013110067 |
| 54. | Sibiryakova, A. E.; Shiryaev, V. A.; Reznikov, A. N.; Kabanova, A. A.; Klimochkin, Y. N. Synthesis 2019, 51, 463–469. doi:10.1055/s-0037-1610824 |
| 1. | Blakemore, P. R.; Sephton, S. M.; Ciganek, E. Org. React. 2018, 95, 1–422. doi:10.1002/0471264180.or095.01 |
| 2. | Taylor, R. J. K.; Casy, G. Org. React. 2003, 62, 359–475. doi:10.1002/0471264180.or062.02 |
| 3. | Carretero, J. C.; Arrayás, R. G.; Adrio, J. Sulfones in Asymmetric Catalysis. In Organosulfur Chemistry in Asymmetric Synthesis; Toru, T.; Bolm, C., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; pp 291–320. doi:10.1002/9783527623235.ch9 |
| 4. | Markitanov, Y. M.; Timoshenko, V. M.; Shermolovich, Y. G. J. Sulfur Chem. 2014, 35, 188–236. doi:10.1080/17415993.2013.815749 |
| 7. | Capi, M.; Curto, M.; Lionetto, L.; de Andrés, F.; Gentile, G.; Negro, A.; Martelletti, P. Ther. Adv. Neurol. Disord. 2016, 9, 414–423. doi:10.1177/1756285616650619 |
| 27. | Kanada, Y.; Yuasa, H.; Nakashima, K.; Murahashi, M.; Tada, N.; Itoh, A.; Koseki, Y.; Miura, T. Tetrahedron Lett. 2013, 54, 4896–4899. doi:10.1016/j.tetlet.2013.06.141 |
| 28. | Nakashima, K.; Murahashi, M.; Yuasa, H.; Ina, M.; Tada, N.; Itoh, A.; Hirashima, S.-i.; Koseki, Y.; Miura, T. Molecules 2013, 18, 14529–14542. doi:10.3390/molecules181214529 |
| 29. | Miura, T.; Yuasa, H.; Murahashi, M.; Ina, M.; Nakashima, K.; Tada, N.; Itoh, A. Synlett 2012, 23, 2385–2388. doi:10.1055/s-0032-1317137 |
| 30. | Murphy, J. J.; Quintard, A.; McArdle, P.; Alexakis, A.; Stephens, J. C. Angew. Chem., Int. Ed. 2011, 50, 5095–5098. doi:10.1002/anie.201100804 |
| 31. | Zhu, Q.; Lu, Y. Chem. Commun. 2010, 46, 2235–2237. doi:10.1039/b919549a |
| 32. | Mossé, S.; Andrey, O.; Alexakis, A. Chimia 2006, 60, 216–219. doi:10.2533/000942906777674778 |
| 47. | Evans, D. A.; Mito, S.; Seidel, D. J. Am. Chem. Soc. 2007, 129, 11583–11592. doi:10.1021/ja0735913 |
| 5. | Richter, W. F.; Whitby, B. R.; Chou, R. C. Xenobiotica 1996, 26, 243–254. doi:10.3109/00498259609046705 |
| 6. | Himmelmann, A.; Bergbrant, A.; Svensson, A.; Hansson, L.; Aurell, M. Am. J. Hypertens. 1996, 9, 517–522. doi:10.1016/0895-7061(95)00340-1 |
| 33. | Zhu, Q.; Cheng, L.; Lu, Y. Chem. Commun. 2008, 6315–6317. doi:10.1039/b816307c |
| 58. | Okino, T.; Hoashi, Y.; Furukawa, T.; Xu, X.; Takemoto, Y. J. Am. Chem. Soc. 2005, 127, 119–125. doi:10.1021/ja044370p |
| 59. | Almaşi, D.; Alonso, D. A.; Gómez-Bengoa, A. E.; Nájera, C. J. Org. Chem. 2009, 74, 6163–6168. doi:10.1021/jo9010552 |
| 60. | Manzano, R.; Andrés, J.; Pedrosa, R. Synlett 2011, 2203–2205. doi:10.1055/s-0030-1261139 |
| 61. | Andrés, J. M.; Losada, J.; Maestro, A.; Rodríguez-Ferrer, P.; Pedrosa, R. J. Org. Chem. 2017, 82, 8444–8454. doi:10.1021/acs.joc.7b01177 |
| 2. | Taylor, R. J. K.; Casy, G. Org. React. 2003, 62, 359–475. doi:10.1002/0471264180.or062.02 |
| 23. | Peters, B. K.; Zhou, T.; Rujirawanich, J.; Cadu, A.; Singh, T.; Rabten, W.; Kerdphon, S.; Andersson, P. G. J. Am. Chem. Soc. 2014, 136, 16557–16562. doi:10.1021/ja5079877 |
| 4. | Markitanov, Y. M.; Timoshenko, V. M.; Shermolovich, Y. G. J. Sulfur Chem. 2014, 35, 188–236. doi:10.1080/17415993.2013.815749 |
| 1. | Blakemore, P. R.; Sephton, S. M.; Ciganek, E. Org. React. 2018, 95, 1–422. doi:10.1002/0471264180.or095.01 |
| 24. | Mauleón, P.; Carretero, J. C. Org. Lett. 2004, 6, 3195–3198. doi:10.1021/ol048690p |
| 25. | Mauleón, P.; Alonso, I.; Rodriguez Rivero, M.; Carretero, J. C. J. Org. Chem. 2007, 72, 9924–9935. doi:10.1021/jo7016197 |
| 26. | Mauleón, P.; Carretero, J. C. Chem. Commun. 2005, 4961–4963. doi:10.1039/b508142d |
| 57. | Calligaris, M. Coord. Chem. Rev. 2004, 248, 351–375. doi:10.1016/j.ccr.2004.02.005 |
| 13. | Kennedy, M.; McKervey, M. A.; Maguire, A. R.; Roos, G. H. P. J. Chem. Soc., Chem. Commun. 1990, 361–362. doi:10.1039/c39900000361 |
| 14. | Honma, M.; Sawada, T.; Fujisawa, Y.; Utsugi, M.; Watanabe, H.; Umino, A.; Matsumura, T.; Hagihara, T.; Takano, M.; Nakada, M. J. Am. Chem. Soc. 2003, 125, 2860–2861. doi:10.1021/ja029534l |
| 15. | Honma, M.; Nakada, M. Tetrahedron Lett. 2003, 44, 9007–9011. doi:10.1016/j.tetlet.2003.09.215 |
| 16. | Sawada, T.; Nakada, M. Adv. Synth. Catal. 2005, 347, 1527–1532. doi:10.1002/adsc.200505173 |
| 17. | Takeda, H.; Watanabe, H.; Nakada, M. Tetrahedron 2006, 62, 8054–8063. doi:10.1016/j.tet.2006.06.022 |
| 18. | Shiely, A. E.; Clarke, L.-A.; Flynn, C. J.; Buckley, A. M.; Ford, A.; Lawrence, S. E.; Maguire, A. R. Eur. J. Org. Chem. 2018, 2277–2289. doi:10.1002/ejoc.201800077 |
| 21. | Tao, L.; Yin, C.; Dong, X.-Q.; Zhang, X. Org. Biomol. Chem. 2019, 17, 785–788. doi:10.1039/c8ob02923g |
| 56. | Reznikov, A. N.; Sibiryakova, A. E.; Klimochkin, Yu. N. Russ. J. Org. Chem. 2014, 50, 1695–1696. doi:10.1134/s107042801411027x |
| 10. | Liang, G.; Tong, M.-C.; Wang, C.-J. Adv. Synth. Catal. 2009, 351, 3101–3106. doi:10.1002/adsc.200900552 |
| 11. | Llamas, T.; Arrayás, R. G.; Carretero, J. C. Org. Lett. 2006, 8, 1795–1798. doi:10.1021/ol060314c |
| 12. | Harada, M.; Kato, S.; Haraguchi, R.; Fukuzawa, S.-i. Chem. – Eur. J. 2018, 24, 2580–2583. doi:10.1002/chem.201706033 |
| 22. | Zhang, H.-L.; Hou, X.-L.; Dai, L.-X.; Luo, Z.-B. Tetrahedron: Asymmetry 2007, 18, 224–228. doi:10.1016/j.tetasy.2007.01.009 |
| 47. | Evans, D. A.; Mito, S.; Seidel, D. J. Am. Chem. Soc. 2007, 129, 11583–11592. doi:10.1021/ja0735913 |
| 9. | Shiryaev, V. A.; Radchenko, E. V.; Palyulin, V. A.; Zefirov, N. S.; Bormotov, N. I.; Serova, O. A.; Shishkina, L. N.; Baimuratov, M. R.; Bormasheva, K. M.; Gruzd, Y. A.; Ivleva, E. A.; Leonova, M. V.; Lukashenko, A. V.; Osipov, D. V.; Osyanin, V. A.; Reznikov, A. N.; Shadrikova, V. A.; Sibiryakova, A. E.; Tkachenko, I. M.; Klimochkin, Y. N. Eur. J. Med. Chem. 2018, 158, 214–235. doi:10.1016/j.ejmech.2018.08.009 |
| 47. | Evans, D. A.; Mito, S.; Seidel, D. J. Am. Chem. Soc. 2007, 129, 11583–11592. doi:10.1021/ja0735913 |
| 51. | Reznikov, A. N.; Kapranov, L. E.; Ivankina, V. V.; Sibiryakova, A. E.; Rybakov, V. B.; Klimochkin, Y. N. Helv. Chim. Acta 2018, 101, e1800170. doi:10.1002/hlca.201800170 |
| 52. | Reznikov, A. N.; Golovin, E. V.; Klimochkin, Y. N. Russ. J. Org. Chem. 2013, 49, 663–668. doi:10.1134/s1070428013050047 |
| 54. | Sibiryakova, A. E.; Shiryaev, V. A.; Reznikov, A. N.; Kabanova, A. A.; Klimochkin, Y. N. Synthesis 2019, 51, 463–469. doi:10.1055/s-0037-1610824 |
| 19. | Bertus, P.; Phansavath, P.; Ratovelomanana-Vidal, V.; Genêt, J.-P.; Touati, A. R.; Homri, T.; Ben Hassine, B. Tetrahedron Lett. 1999, 40, 3175–3178. doi:10.1016/s0040-4039(99)00453-0 |
| 20. | Genêt, J. P.; Pinel, C.; Ratovelomanana-Vidal, V.; Mallart, S.; Pfister, X.; Bischoff, L.; De Andrade, M. C. C.; Darses, S.; Galopin, C.; Laffitte, J. A. Tetrahedron: Asymmetry 1994, 5, 675–690. doi:10.1016/0957-4166(94)80030-8 |
| 55. | Reznikov, A. N.; Sibiryakova, A. E.; Rybakov, V. B.; Klimochkin, Y. N. Tetrahedron: Asymmetry 2015, 26, 1050–1057. doi:10.1016/j.tetasy.2015.08.003 |
| 36. | Wei, Y.; Guo, R.; Dang, Y.; Nie, J.; Ma, J.-A. Adv. Synth. Catal. 2016, 358, 2721–2726. doi:10.1002/adsc.201600485 |
| 34. | Li, H.; Song, J.; Liu, X.; Deng, L. J. Am. Chem. Soc. 2005, 127, 8948–8949. doi:10.1021/ja0511063 |
| 35. | Li, H.; Song, J.; Deng, L. Tetrahedron 2009, 65, 3139–3148. doi:10.1016/j.tet.2008.11.054 |
| 47. | Evans, D. A.; Mito, S.; Seidel, D. J. Am. Chem. Soc. 2007, 129, 11583–11592. doi:10.1021/ja0735913 |
| 35. | Li, H.; Song, J.; Deng, L. Tetrahedron 2009, 65, 3139–3148. doi:10.1016/j.tet.2008.11.054 |
| 47. | Evans, D. A.; Mito, S.; Seidel, D. J. Am. Chem. Soc. 2007, 129, 11583–11592. doi:10.1021/ja0735913 |
| 43. | Barnes, D. M.; Ji, J.; Fickes, M. G.; Fitzgerald, M. A.; King, S. A.; Morton, H. E.; Plagge, F. A.; Preskill, M.; Wagaw, S. H.; Wittenberger, S. J.; Zhang, J. J. Am. Chem. Soc. 2002, 124, 13097–13105. doi:10.1021/ja026788y |
| 44. | Lewis, K. G.; Ghosh, S. K.; Bhuvanesh, N.; Gladysz, J. A. ACS Cent. Sci. 2015, 1, 50–56. doi:10.1021/acscentsci.5b00035 |
| 45. | Reznikov, A. N.; Golovin, E. V.; Klimochkin, Y. N. Russ. J. Gen. Chem. 2012, 82, 1827–1829. doi:10.1134/s1070363212110163 |
| 41. | Bera, K.; Namboothiri, I. N. N. Chem. Commun. 2013, 49, 10632–10634. doi:10.1039/c3cc45985c |
| 42. | García Mancheño, O.; Tangen, P.; Rohlmann, R.; Fröhlich, R.; Alemán, J. Chem. – Eur. J. 2011, 17, 984–992. doi:10.1002/chem.201001914 |
| 39. | Zong, L.; Du, S.; Chin, K. F.; Wang, C.; Tan, C.-H. Angew. Chem., Int. Ed. 2015, 54, 9390–9393. doi:10.1002/anie.201503844 |
| 40. | Fang, X.; Dong, X.-Q.; Liu, Y.-Y.; Wang, C.-J. Tetrahedron Lett. 2013, 54, 4509–4511. doi:10.1016/j.tetlet.2013.06.059 |
| 37. | Qiao, B.; Liu, Q.; Liu, H.; Yan, L.; Jiang, Z. Chem. – Asian J. 2014, 9, 1252–1256. doi:10.1002/asia.201400049 |
© 2019 Reznikov et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)