Abstract
The ring-opening/cyclization of cyclopropane derivatives has drawn great attention in the past several decades. In this review, recent efforts in the development of oxidative radical ring-opening/cyclization of cyclopropane derivatives, including methylenecyclopropanes, cyclopropyl olefins and cyclopropanols, are described. We hope this review will be of sufficient interest for the scientific community to further advance the application of oxidative radical strategies in the ring-opening/cyclization of cyclopropane derivatives.

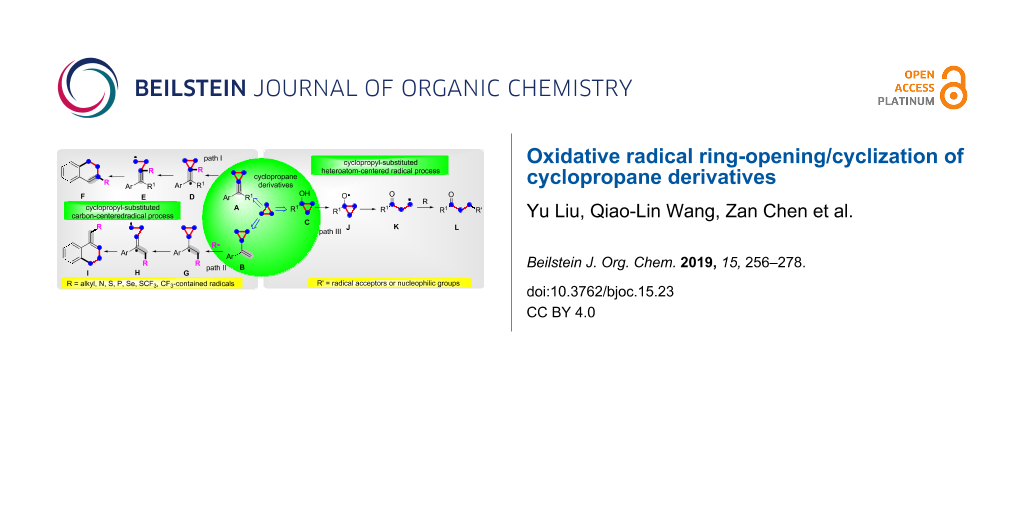
Graphical Abstract
Introduction
Cyclopropane is a cycloalkane molecule with the molecular formula C3H6, consisting of three carbon atoms linked to each other to form a ring, with each carbon atom bearing two hydrogen atoms resulting in D3h molecular symmetry. The small size of the ring creates substantial ring strain in the structure. The cyclopropane skeleton easily can take part in ring-opening reactions under certain conditions. Cyclopropane derivatives, with their three-membered carbocyclic frameworks, have spurred considerable attention especially in the domain of organic and pharmaceutical synthesis because of their highly strained three-membered carbocyclic skeletons and their easy availability [1-16]. The cyclopropane derivatives, especially methylenecyclopropanes [17-21], cyclopropyl olefins [22] and cyclopropanols [23,24] undergo ring-opening/cyclization reactions to provide a huge number of fascinating compounds with different functional groups [25-31]. However, most recently reported methods usually proceed via a radical pathway. As shown in Scheme 1 path I, the cyclopropyl-substituted carbon radical D is formed by the addition of radical R to the C–C double bond in methylenecyclopropanes (compounds A). The cyclopropyl-substituted carbon radical D easily goes through a ring-opening to generate the alkyl radical E, and then cyclizes with the phenyl ring to afford the terminal product F (path I). The cyclopropyl olefins (compounds B) also react in the same cyclopropyl-substituted carbon radical pathway to finish the ring-opening and cyclization transformation (path II). The cyclopropanols D firstly go through homolytic cleavage of the O–H bond to give the oxygen-centered radical J. The alkyl radical K, produced by ring-opening of intermediate J, reacts with a radical acceptor or a nucleophilic group to obtain the product L (path III).
![[1860-5397-15-23-i1]](/bjoc/content/inline/1860-5397-15-23-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The oxidative radical ring-opening/cyclization of cyclopropane derivatives.
Scheme 1: The oxidative radical ring-opening/cyclization of cyclopropane derivatives.
Free radical reactions have flourished and became a powerful tool in organic synthesis [32-38]. With the significant potential, this strategy has captured the human’s attention and solved considerable problems in the past several decades [39-42]. The free radical reaction was applied in a range of organic transformations because of its unique advantages such as excellent reactivity, mild conditions, functional group tolerance, and atom economy. A series of radicals, such as carbon, Se, CF3, halogen, S and N-containing radicals, were introduced into the products through oxidative radical ring-opening/cyclization of cyclopropane derivatives. In this review, we conclude recent advance in the oxidative radical ring-opening/cyclization of cyclopropane derivatives (including methylenecyclopropanes, cyclopropyl olefins and cyclopropanols) over the last 20 years.
Review
Oxidative radical ring-opening and cyclization of methylenecyclopropanes (MCPs)
In 2004, Huang and co-workers reported the first manganese(III) acetate-mediated radical ring-opening and cyclization of methylenecyclopropanes (MCPs, 1) with malonic acid diethyl esters (2, Scheme 2) [43]. This strategy provided a novel, convenient and efficient approach to construct 2-(3,4-dihydronaphthalen-2-yl)malonic acid diethyl esters 3. The MCPs 1 with the electron-deficient or electron-rich groups were all suitable for this reaction system. The mechanism for the Mn(OAc)3-mediated oxidative radical ring-opening and cyclization of MCPs with malonates is outlined in Scheme 2. Initially, the malonic acid diethyl ester (2) was transformed into radical 4 [44] under the action of Mn(OAc)3. Then, the selective addition of the radical 4 to the C–C double bond of MCPs 1 formed the more stable benzyl radical intermediate 5 [45,46], which underwent a ring-opening to generate the alkyl radical 6 [47]. Finally, the desired product 3 was generated through intramolecular cyclization of radical intermediate 6 with an aryl ring and oxidation deprotonation by another molecule Mn(OAc)3 [48].
![[1860-5397-15-23-i2]](/bjoc/content/inline/1860-5397-15-23-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Mn(OAc)3-mediated oxidative radical ring-opening and cyclization of MCPs with malonates.
Scheme 2: Mn(OAc)3-mediated oxidative radical ring-opening and cyclization of MCPs with malonates.
Later, Shi et al. demonstrated an oxidative annulation of MCPs 1 with 1,3-dicarbonyl compounds 7 using manganese(III) catalysis under room temperature conditions, which afforded 4,5-dihydrofuran derivatives 8 as [3 + 2] annulation products (cyclopropyl retained adducts) in moderate to good yields [49]. This transformation also gave another six-membered cyclic compounds 9 (cyclopropyl opened adducts) via ring-opening and cyclization process (Scheme 3). However, the [3 + 2] annulation reaction did not occur under the standard conditions when the MCPs 1 was without an aromatic group.
![[1860-5397-15-23-i3]](/bjoc/content/inline/1860-5397-15-23-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Mn(III)-mediated oxidative radical ring-opening and cyclization of MCPs with 1,3-dicarbonyl compounds.
Scheme 3: Mn(III)-mediated oxidative radical ring-opening and cyclization of MCPs with 1,3-dicarbonyl compoun...
The first method for direct [3 + 2] radical cycloaddition of MCPs 1 with elemental chalcogens 10 (S, Se, Te) was developed by Yu and co-workers. This strategy presented a simple and efficient method for the synthesis of methylene-1,2-dichalcogenolanes 11 (Scheme 4) [50]. This reaction proceeded via a radical pathway, which could take place smoothly under cartalyst- and additive-free conditions. However, the addition of the radical initiator AIBN in this reaction did not accelerate the reaction.
![[1860-5397-15-23-i4]](/bjoc/content/inline/1860-5397-15-23-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Heat-promoted ring-opening/cyclization of MCPs with elemental chalgogens.
Scheme 4: Heat-promoted ring-opening/cyclization of MCPs with elemental chalgogens.
Next, Huang’s group proposed the copper-catalyzed ring-opening and cyclization of MCPs 1 with diphenyl diselenides 12 for the synthesis of 2-phenylseleny-3,3-diarylcyclobutenes 13 under visible light irradiation (Scheme 5) [51]. The desired products 13 contained a cyclobutene group and a selenium atom, which makes the products possess unique biological and pharmaceutical activities. The mechanism of the copper(II) acetate-mediated oxidative radical ring-opening/cyclization of MCPs with diphenyl diselenides is outlined in Scheme 5. Firstly, the phenylselenyl radical 14, generated from the homolytic cleavage of diphenyl diselenide, is added to the C–C double bond of MCPs to afford the intermediate 15, which undergoes a ring-opening process to form the radical intermediate 16 [52,53]. Then, the radical 16 reacts with copper(II) acetate to produce organocopper intermediate 17. Finally, the intramolecular insertion of C–Cu in compounds 17 to the carbon–carbon double bond takes place to produce the intermediate 18 followed by β-elimination to generate the desired product 13 [54-56].
![[1860-5397-15-23-i5]](/bjoc/content/inline/1860-5397-15-23-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Copper(II) acetate-mediated oxidative radical ring-opening and cyclization of MCPs with diphenyl diselenides.
Scheme 5: Copper(II) acetate-mediated oxidative radical ring-opening and cyclization of MCPs with diphenyl di...
In 2005, Yu et al. described a novel and efficient oxidative radical ring-opening and cyclization of MCPs 1 with benzenethiol (19) for the synthesis of 3-phenylsulfanyl-1,2-dihydronaphthalenes 20 in moderate to good yields (Scheme 6) [57]. Additionally, using benzeneselenol instead of benzenethiol under the standard conditions generated the corresponding products in 31% yields.
![[1860-5397-15-23-i6]](/bjoc/content/inline/1860-5397-15-23-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: AIBN-promoted oxidative radical ring-opening and cyclization of MCPs with benzenethiol.
Scheme 6: AIBN-promoted oxidative radical ring-opening and cyclization of MCPs with benzenethiol.
In the same year, Huang’s group also reported a similar ring-opening and cyclization of MCPs 1 with diethyl phosphites 21 for building diethyl 3,4-dihydro-2-naphthylphosphonates 22 (Scheme 7) [58]. This was the first example to synthesize the diethyl 3,4-dihydro-2-naphthylphosphonates 22 that have great potential applications in organic chemistry and biochemistry.
![[1860-5397-15-23-i7]](/bjoc/content/inline/1860-5397-15-23-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: AIBN-mediated oxidative radical ring-opening and cyclization of MCPs with diethyl phosphites.
Scheme 7: AIBN-mediated oxidative radical ring-opening and cyclization of MCPs with diethyl phosphites.
In 2009, Miao’s group also discovered another method for the synthesis of 1-naphthaldehydes 25 under mild conditions via a radical-mediated ring-opening and intramolecular cyclization of MCPs 23 with organic selenium reagents 24 (Scheme 8) [59]. In this reaction, the MCPs with electron-withdrawing groups gave lower yields than that with electron-donating groups. Additionally, the use of other organoselenium reagents, such as phenylselenyl bromide or phenylselenyl chloride provided only trace amounts of the desired products. The mechanism for the organoselenium induced radical ring-opening and cyclization of MCPs derivatives is showed in Scheme 8. Firstly, phenylselenyl radical 26 was produced in the presence of free radical initiator (NH4)2S2O8 [60,61]. Next, the intermediate 26 was added to the C–C double bond of MCPs 23, and then went through a series of ring-opening, intramolecular cyclization, oxidation and dehydrogenation to generate 3-arylselanylnaphthaldehyde 25.
![[1860-5397-15-23-i8]](/bjoc/content/inline/1860-5397-15-23-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Organic-selenium induced radical ring-opening and cyclization of MCPs derivatives (cyclopropylaldehydes).
Scheme 8: Organic-selenium induced radical ring-opening and cyclization of MCPs derivatives (cyclopropylaldeh...
In 2015, Shi and co-workers reported a novel and efficient method to construct CF3-substituted dihydronaphthalene derivatives 31 in moderate to excellent yields under mild conditions through the Cu(I)-catalyzed trifluoromethylation/ring-opening/cyclization of MCPs 1 with Togni reagent II (30, Scheme 9) [62]. In this transformation, many substituted MCPs 1 with alkyl groups, Ts-protected amino groups, or halogens were tolerated well and gave the desired products 31 in good yields. Moreover, the product 31a could go through a further oxidation to afford two different products in the presence of different amount of NBS (N-bromosuccinimide). The corresponding CF3-substituted naphthalene 32 could be obtained in 69% yield when the product 31a was oxidized by 3 equiv of NBS (Scheme 9, reaction a). When the amount of NBS was increased to 6 equiv under identical conditions, the CF3-substituted naphthaldehyde 33 was obtained in 61% yield (Scheme 9, reaction b). Furthermore, the product 31a could also be transformed to the CF3-substituted epoxide 34 in the presence of 2 equiv m-CPBA (m-chloroperbenzoic acid) (Scheme 9, reaction c). A radical-trapping experiment was conducted with the addition of TEMPO or BHT under the standard conditions, and the reactions were suppressed by radical scavengers, which suggested that the reaction underwent a radical process. The proposed mechanism is depicted in Scheme 9. Initially, the CF3 radical 35 is generated from the Togni reagent II (30) under the action of Cu(I) [63,64]. Then the CF3 radical 35 adds to the C–C double bond in MCPs 1 to give the more stable benzyl radical intermediate 36 which went through a ring-opening process to provide the alkyl radical intermediate 37. The intermediate 37 undergoes intramolecular cyclization with the aromatic ring to generate intermediate 38 which is oxidized by Cu(II) to provide the CF3-substituted dihydronaphthalenes derivatives 31 along with releasing a proton [65,66].
![[1860-5397-15-23-i9]](/bjoc/content/inline/1860-5397-15-23-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Copper(I)-catalyzed oxidative radical trifluoromethylation/ring-opening/cyclization of MCPs with Togni reagent II.
Scheme 9: Copper(I)-catalyzed oxidative radical trifluoromethylation/ring-opening/cyclization of MCPs with To...
The trifluoromethylthiolation of MCPs 1 with AgSCF3 was achieved by Shi et al. which proceeds through a sequence of radical addition, ring-opening, cyclization, oxidation and dehydrogenation and successfully furnished trifluoromethylthiolated 1,2-dihydronaphthalene derivatives 39 (Scheme 10) [67]. This reaction was achieved in the presence of 3.0 equiv of Na2S2O8 as the oxidants, 0.5 equiv of HMPA (N,N,N',N',N'',N''-hexamethylphosphorotriamide) as the additive in DMSO.
![[1860-5397-15-23-i10]](/bjoc/content/inline/1860-5397-15-23-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Ag(I)-mediated trifluoromethylthiolation/ring-opening/cyclization of MCPs with AgSCF3.
Scheme 10: Ag(I)-mediated trifluoromethylthiolation/ring-opening/cyclization of MCPs with AgSCF3.
With a similar oxidative radical ring-opening and cyclization strategy, our group developed a novel method for ring-opening and cyclization of MCPs 1 with ethers 40 afforded 2-substituted 3,4-dihydronaphthalenes 41 in moderate to excellent yields (Scheme 11) [68]. This transformation just needed 2 equiv of TBHP (42), avoiding using transition metal catalysts, ligands, and bases. In the proposed mechanism (Scheme 11), the tert-butoxyl radical 43, which was formed from THBP (42) under heating conditions, attackes the ether 40 to afford the radical 44 [69-72]. Next, the addition of radical 44 to the C–C double bond of MCPs 1 generats a more stable benzyl radical 45. Final ring-opening, intramolecular cyclization, oxidation, and dehydrogenation finally delivers the desired product 41.
![[1860-5397-15-23-i11]](/bjoc/content/inline/1860-5397-15-23-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: oxidative radical ring-opening and cyclization of MCPs with α-C(sp3)-–H of ethers.
Scheme 11: oxidative radical ring-opening and cyclization of MCPs with α-C(sp3)-–H of ethers.
Next, our group reported the first oxidative ring-opening and cyclization between MCPs 1 and aldehydes 48 to provide 2-acyl-3,4-dihydronaphthalenes 49 in moderate to excellent yields via a series of radical addition, ring-opening and cyclization in the presence of DTBP (di-tert-butyl peroxide) and Lewis acids (Scheme 12) [73]. Moreover, the experimental results showed MCPs 1 with electron-rich aryl groups could deliver higher yields than that with electron-deficient ones. As outlined in Scheme 12, a tert-butoxy radical and a methyl radical were generated from cleavage of DTBP at the reaction temperature. Aldehyde 48 is easily transformed into acyl radical 50 in the presence of an alkoxy radical or a methyl radical [74-77]. The acyl radical 50 adds to the C–C double bond of MCPs giving the benzyl radical intermediate 51. The ring-opening of radical intermediate 51 occurres to form the alkyl radical intermediate 52 which intermolecularly cyclizes with the aryl ring. The following oxidation and dehydrogenation gives the target product 49.
![[1860-5397-15-23-i12]](/bjoc/content/inline/1860-5397-15-23-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Oxidative radical ring-opening and cyclization of MCPs with aldehydes.
Scheme 12: Oxidative radical ring-opening and cyclization of MCPs with aldehydes.
A new and first achievement for the synthesis of CF3-contained seven-membered ring compounds 55 and 56 through trifluoromethylation of acrylamide-tethered alkylidenecyclopropanes 54 was presented by Shi and co-workers (Scheme 13) [78]. The possible reaction pathway is outlined in Scheme 13. Initially, the Togni reagent II (30) goes through a single-electron transfer (SET) under the action of Fe2+ to generate the CF3 radical 35. The CF3 radical 35 is trapped by the C–C double bond of substrate 54 to produce the alkyl radical intermediate 57. Then, the intramolecular addition of an alkyl radical to the less hindered central carbon of MCPs 54 gives the benzyl radical intermediate 58, which undergoes a ring-opening process to provide the alkyl radical intermediate 59 [79,80]. Because of the different substituent groups on the MCPs 54 (whether R1 was a para-methoxy substituent or not), this reaction proceeds through two different pathways. When R1 is not a para-methoxy group, the intermediate 59 undergoes a conventional cyclization with aromatic ring to afford the radical intermediate 60. After oxidation and aromatization, the corresponding product 55 is formed. An ipso-cyclization with aromatic ring occurres and gives the intermediate 61 when R1 is a para-methoxy group. The oxonium ion 62 is produced by the oxidation of the intermediate 61 under the action of Fe3+ [81]. Lastly, the oxonium ion 62 is transformed into the desired product 56 in the presence of 2-indobenzoic acid anion.
![[1860-5397-15-23-i13]](/bjoc/content/inline/1860-5397-15-23-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Cu(I) or Fe(II)-catalyzed oxidative radical trifluoromethylation/ring-opening/cyclization of MCPs derivatives (acrylamide-tethered alkylidenecyclopropanes).
Scheme 13: Cu(I) or Fe(II)-catalyzed oxidative radical trifluoromethylation/ring-opening/cyclization of MCPs d...
Recently, Shi’s group developed the first ring expansion of MCPs 63 with a nitrogen atom to furnish azetidines 64 (Scheme 14) [82]. The author proposed that Rh(II) had an effective impact on the reactions and could improve the reaction yields. Unfortunately, the MCPs 63 with the groups R1 and R2 = H were not suitable for this transformation. The reason was because the formed intermediate was unstable under this conditions. A possible mechanism is outlined in Scheme 14. Initially, the Rh-nitrene intermediate 65 [83-86] is generated from the coordination of azide to Rh2(esp)2 complex (bis[rhodium-(α,α,α’,α’-tetramethyl-1,3-benzenedipropionic acid)]) and extrusion of N2. Then, the Rh-nitrene intermediate 65 goes through an intramolecular single electron transfer (SET) to give the nitrogen-centered radical intermediate 66 [87-90]. Next, the radical addition of intermediate 66 to the C–C double bond in MCPs moiety furnishes the more stable benzyl radical intermediate 67, which is ring-opened to give alkyl radical 68. Finally, intermediate 68 goes through SET with the Rh(III) species and intramolecular cyclization with the 2-position of the indole moiety to afford the target product 64 along with the regenerated Rh(III) catalyst.
![[1860-5397-15-23-i14]](/bjoc/content/inline/1860-5397-15-23-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Rh(II)-catalyzed oxidative radical ring-opening and cyclization of MCPs.
Scheme 14: Rh(II)-catalyzed oxidative radical ring-opening and cyclization of MCPs.
A silver-catalyzed intramolecular cascade amination/ring-opening/cyclization of a variety of substituted MCPs 69 was proposed by Fan and co-workers, which provided a simple and efficient way for the building of [2,3-c]dihydrocarbazoles 70 and [2,3-c]carbazoles 71 (Scheme 15) [91]. This process permitted the use of readily available and cheap AgOAc as the catalyst and oxidant, and DMF as the solvent. Notably, the product 70 was easily transformed into 71 in the presence of chloranil (1.4 equiv) at 120 °C under Ar atmosphere for 5 h. In this transformation, substrates with electron-donating groups showed higher yields than the ones with electron-withdrawing groups.
![[1860-5397-15-23-i15]](/bjoc/content/inline/1860-5397-15-23-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Ag(I)-catalyzed oxidative radical amination/ring-opening/cyclization of MCPs derivatives.
Scheme 15: Ag(I)-catalyzed oxidative radical amination/ring-opening/cyclization of MCPs derivatives.
In the same year, Shi et al. reported an effective ring-opening and cyclization of arylvinylidenecyclopropanes 72 with diaryl diselenides 73 for the synthesis of 1,2-diarylselenocyclopentene 74 in moderate to good yields at 150 °C for 1.5 h (Scheme 16) [92]. The electron-rich, electron-neutral and electron-poor arylvinylidenecyclopropanes were tolerated well in this transformation. The detailed mechanism is outlined in Scheme 16. Initially, the homolysis of diphenyldiselenide 73 under heating conditions produces the phenylseleno radical 26 [93]. Then, the additon of radical 26 to the C–C double bond of MCPs derivatives 72 affords the radical intermediate 75 [94]. Next, the radical 75 goes through a ring-opening process to give the radical intermediate 76. The intermediate 77, produced by the intramolecular cyclization of intermediate 76, reactes with diphenyl diselenide 73 to form the target product 74 via homolytic substitution (SH).
![[1860-5397-15-23-i16]](/bjoc/content/inline/1860-5397-15-23-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Heating-promoted radical ring-opening and cyclization of MCP derivatives (arylvinylidenecyclopropanes) with diaryl diselenides.
Scheme 16: Heating-promoted radical ring-opening and cyclization of MCP derivatives (arylvinylidenecyclopropan...
In 2013, Ryu and co-workers developed the bromine radical-mediated ring-opening and alkylation of alkylidenecyclopropanes 1 with allylic bromides 78 for the synthesis of 2-bromo-1,6-dienes 79 via radical ring-opening and SH2’ reactions (path V in Scheme 17) [95]. The experimental results suggested that radical carbonylation could also be incorporated in the reaction sequence, leading to 2-bromo-1,7-dien-5-ones 80 (path IV in Scheme 17).
![[1860-5397-15-23-i17]](/bjoc/content/inline/1860-5397-15-23-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Bromine radical-mediated ring-opening of alkylidenecyclopropanes.
Scheme 17: Bromine radical-mediated ring-opening of alkylidenecyclopropanes.
In 2016, Xu’s group exploited the fluoroalkyl (RF) radical-mediated ring-opening of MCPs 1 for the synthesis of fluorinated homoallylic compounds (80 and 81, Scheme 18) [96]. In this reaction system, the radical reaction of MCPs 1 with RF-X (X = Br, I) furnished homoallylic halides in excellent yields (path VII in Scheme 18). Similarly, the radical reaction of MCPs 1 with the RFTMS/CsF/PhI(OAc)2 gave homoallylic acohol esters in moderate to good yields (path VI in Scheme 18).
![[1860-5397-15-23-i18]](/bjoc/content/inline/1860-5397-15-23-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Fluoroalkyl (Rf) radical-mediated ring-opening of MCPs.
Scheme 18: Fluoroalkyl (Rf) radical-mediated ring-opening of MCPs.
Oxidative radical ring-opening and cyclization of cyclopropyl olefins
In 2016, Li’s group reported a photoredox catalysis oxidative radical ring-opening and cyclization of cyclopropyl olefins 83 with bromides 84 for the synthesis of partially saturated naphthalenes 85 in moderate to excellent yields (Scheme 19) [97]. It was the first example for alkylation, ring-opening and cyclization cascade reaction of the cyclopropyl olefins under photoredox catalysis. The alkylation reagents could be extended to other bromides, such as monofluoro-substituted bromides, trifluoro-substituted bromides, bromoacetonitrile and bromomalonate. This alkylation/ring-opening/cyclization was carried out by using Ir(ppy)2(dtbbpy)PF4 as photocatalyst, and K2HPO4 as base in MeCN under the irradiation of 24 W blue LED light at room temperature for 12–36 h. A plausible mechanism is shown in Scheme 19. Firstly, the substrate 84a underwent oxidative quenching under the action of an iridium photoredox catalyst to afford the alkyl radical 86, which adds to the C–C double bond of MCPs 83 to deliver the benzyl radical 87. Then, it undergoes a ring-opening process to afford the terminal alkyl radical 88. Next, the alkyl radical 88 intramolecular cyclizes with the phenyl ring to give intermediate 89. Finally, the resulting aryl radical intermediate 89 is oxidized and deprotonated to provide the target product 85. In the process, two new C–C bonds and a new ring are formed.
![[1860-5397-15-23-i19]](/bjoc/content/inline/1860-5397-15-23-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Visible-light-induced alkylation/ring-opening/cyclization of cyclopropyl olefins with bromides.
Scheme 19: Visible-light-induced alkylation/ring-opening/cyclization of cyclopropyl olefins with bromides.
Oxidative radical ring-opening of cyclopropanols
In 2011, Chiba’s group presented Mn(III)-mediated ring-opening and [3 + 3]-annulation of cyclopropanols 91 and vinyl azides 92 for the synthesis of azaheterocyles 93 (Scheme 20) [98]. This strategy could also be applied to the synthesis of the quaternary indole alkaloid and melinonine-E.
![[1860-5397-15-23-i20]](/bjoc/content/inline/1860-5397-15-23-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Mn(III)-mediated ring-opening and [3 + 3]-annulation of cyclopropanols and vinyl azides.
Scheme 20: Mn(III)-mediated ring-opening and [3 + 3]-annulation of cyclopropanols and vinyl azides.
Quinones play an important role in organic chemistry because of their unique structure. In 2013, Malayappasamy and co-workers reported an efficient and convenient method for the synthesis of γ-carbonyl quinones 95 via ring-opening and functionalization of cyclopropanols 91 with quinones 94 (Scheme 21) [99]. In this transformation, both AgNO3 and FeSO4 were all efficient catalysts for the ring-opening and functionalization reaction. However, AgNO3 was superior than FeSO4 according to the reaction yields and time. Interestingly, aromatic cyclopropanols delivered higher yields than aliphatic ones. The mechanism for the Ag(I)-catalyzed oxidative ring-opening and functionalization of cyclopropanols with quinones is outlined in Scheme 21. Firstly, the sulfate radical anion 97 is generated from persulfate 96 under the action of Ag(I). Next, the radical 97 reacts with cyclopropanol 91 to give the cyclopropoxy radical 98, which undergoes a ring-opening process to produce β-keto radical 99. The radical 100 is formed through the addition of radical 99 to the quinones 94. Finally, the intermediate 100 occurres reoxidation with Ag(II) to provide the final product 95 along with regenerated Ag(I).
![[1860-5397-15-23-i21]](/bjoc/content/inline/1860-5397-15-23-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Ag(I)-catalyzed oxidative ring-opening of cyclopropanols with quinones.
Scheme 21: Ag(I)-catalyzed oxidative ring-opening of cyclopropanols with quinones.
In 2015, Duan et al. developed a Ag(I)-catalyzed oxidative ring-opening of cyclopropanols 91 with heteroarenes 101 or 103 for the synthesis of carbonyl-containing alkyl-substituted heteroarenes 102 or 104 under mild conditions in moderate to good yields with good functional group tolerance (Scheme 22) [100]. This reaction went through a selective C(sp3)–C(sp3) bond cleavage, C–H activation and C(sp3)–C(sp2) bond formation. Notably, this finding was the first example for silver-catalyzed regioselective C2-alkylation of heterorarenes with primary alkyl radicals, generated from cyclopropanols through a radical ring-opening process.
![[1860-5397-15-23-i22]](/bjoc/content/inline/1860-5397-15-23-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Ag(I)-catalyzed oxidative ring-opening of cyclopropanols with heteroarenes.
Scheme 22: Ag(I)-catalyzed oxidative ring-opening of cyclopropanols with heteroarenes.
Lopp’s group also reported an efficient approach for copper-catalyzed ring-opening and trifluoromethylation of cyclopropanols 91 to construct β-trifluoromethyl-substituted ketones 106 (Scheme 23) [101]. Additionally, a series of cyclopropanols with different functional R groups were successfully scaled up to 1 mmol. In this transformation, there exist two possible pathways to produce the target product 106. The Togni reagent (105) reacts with CuCl to generate Cu(III) complex 108. Then, the intermediated 109 is generated from the electrophilic attack of copper(III) 108 with cyclopropanol 91. Finally, the desired product 106 is formed through reductive elimination of CuCl in intermediated 109. On the other hand, the intermediated 108 can lose the CF3 radical to generate the Cu(II) complex 110. Next, the complex 110 reacts with 91 to give the radical 99. The desired product 106 was produced by the interception of the CF3 radical, which came from CuCF3Cl.
![[1860-5397-15-23-i23]](/bjoc/content/inline/1860-5397-15-23-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Cu(I)-catalyzed oxidative ring-opening/trifluoromethylation of cyclopropanols.
Scheme 23: Cu(I)-catalyzed oxidative ring-opening/trifluoromethylation of cyclopropanols.
In the same year, Dai’s group also reported a copper-catalyzed ring-opening and trifluoromethylation or trifluoromethylthiolation of cyclopropanols 91 for the synthesis of β-CF3/SCF3-substituted ketones 113 (Scheme 24) [102]. This strategy was also applied to the synthesis of LY2409021. The LY2409021 was a glucagon receptor antagonist and used in clinical trials for type 2 diabetes mellitus. Xu et al. also presented the similar ring-opening/trifluoromethylation of cyclopropanols for the synthesis of various β-trifluoromethyl ketones [103].
![[1860-5397-15-23-i24]](/bjoc/content/inline/1860-5397-15-23-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Cu(I)-catalyzed oxidative ring-opening and trifluoromethylation/trifluoromethylthiolation of cyclopropanols.
Scheme 24: Cu(I)-catalyzed oxidative ring-opening and trifluoromethylation/trifluoromethylthiolation of cyclop...
In this year, Loh et al. [104] and Zhu et al. [105] proposed a oxidative ring-opening and fluorination of cyclopropanols 91 with Selectfluor to construct β-fluorinated ketones 114 (Scheme 25). In Loh’s work, the Fe(III)- or Ag(I)-catalyzed oxidative ring-opening and fluorination of cyclopropanols 91 via radical rearrangement is disclosed. Notably, this reaction proceeds at room temperature and tolerates a diverse array of cyclopropanols. In Zhu’s work, the fluorination of 91a was notable because the seven-membered cyclic product 114a and five-membered cyclic product 114b were formed.
![[1860-5397-15-23-i25]](/bjoc/content/inline/1860-5397-15-23-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Ag(I)-mediated oxidative ring-opening/fluorination of cyclopropanols with Selectfluor.
Scheme 25: Ag(I)-mediated oxidative ring-opening/fluorination of cyclopropanols with Selectfluor.
Lectka’s group also presented a new approach to β-fluorinated ketones 114 via photocatalyzed ring-opening and fluorination of cyclopropanols 91 with Selectfluor under mild and simple conditions (Scheme 26) [106]. It is worth mentioning that a number of electronically and sterically diverse β-fluorinated carbonyl-containing compounds 114 and γ-fluoro alcohols 115 could be prepared through this method.
![[1860-5397-15-23-i26]](/bjoc/content/inline/1860-5397-15-23-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Photocatalyzed ring-opening/fluorination of cyclopropanols with Selectfluor.
Scheme 26: Photocatalyzed ring-opening/fluorination of cyclopropanols with Selectfluor.
In 2015, Duan and co-workers introduced the Na2S2O8-promoted ring-opening/alkynylation of cyclopropanols 91 with ethynylbenziodoxolones (EBX) 116 for the synthesis of the alkynylated ketones 117 (Scheme 27) [107]. This reaction involved a C–C bond cleavage, radical rearrangement, and C–C bond formation, and showed a wide substrates scope under mild conditions. Surprisingly, four- and five-membered cycloalkanols were suitable in this system.
![[1860-5397-15-23-i27]](/bjoc/content/inline/1860-5397-15-23-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Na2S2O8-promoted ring-opening/alkynylation of cyclopropanols with EBX.
Scheme 27: Na2S2O8-promoted ring-opening/alkynylation of cyclopropanols with EBX.
In 2015, Zhu’s group developed the silver-catalyzed ring-opening of cycloalkanols 91 with NCS 118 for the synthesis of distally chlorinated ketones 119 (Scheme 28) [108]. The reaction was carried out with inexpensive reagents and can also be applied to the distal bromination of cycloalkanols. The possible mechanism is outlined in Scheme 28. The cycloalkoxy radical 98 is generated from cyclopropanol 91 under the action of the metastable Ag(II) species, which is formed by the interaction of AgNO3 and K2S2O8. The radical 98 undergoes a ring-opening to give the alkyl radical 99. Finally, the radical 99 is intercepted by NCS 118 to furnish the chlorinated ketone 119. The generated imidyl radical 120 can also participate in hydrogen abstraction of cyclopropanol 91 to form the radical 98.
![[1860-5397-15-23-i28]](/bjoc/content/inline/1860-5397-15-23-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: Ag(I)-catalyzed ring-opening and chlorination of cyclopropanols with aldehydes.
Scheme 28: Ag(I)-catalyzed ring-opening and chlorination of cyclopropanols with aldehydes.
In 2016, the silver-promoted oxidative ring-opening/alkynylation of cyclopropanols 91 with ethynylbenziodoxolones (EBX) 116 had been presented by Li and co-workers (Scheme 29) [109]. Both silver(I) nitrate and potassium persulfate played an important role in this transformation.
![[1860-5397-15-23-i29]](/bjoc/content/inline/1860-5397-15-23-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: Ag(I)-catalyzed ring-opening/alkynylation of cyclopropanols with EBX.
Scheme 29: Ag(I)-catalyzed ring-opening/alkynylation of cyclopropanols with EBX.
In 2016, Hu and co-workers developed a novel ring-opening of cyclopropanols 91 with acrylamides 122 for the synthesis of oxindoles 123 (Scheme 30) [110]. A series of desired γ-carbonylalkyl-substituted oxindoles 123 were synthesized between N-phenyl acrylamides 122 and tertiary cyclopropanols 91 through Na2S2O8-promoted radical cyclization under transition-metal free conditions. With the addition of a radical scavenger such as TEMPO or BHT, the reaction was suppressed remarkably.
![[1860-5397-15-23-i30]](/bjoc/content/inline/1860-5397-15-23-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 30: Na2S2O8-promoted ring-opening/alkylation of cyclopropanols with acrylamides.
Scheme 30: Na2S2O8-promoted ring-opening/alkylation of cyclopropanols with acrylamides.
In the same year, Dai’s group also reported the ring-opening-initiated tandem cyclization of cyclopropanols 91 with acrylamides 122 or 2-isocyanobiphenyls 124 (Scheme 31) [111]. This transformation involved a C–C bond cleavage and two C–C bond formations, and showed excellent functional group tolerance, satisfactory yields and operational simplicity.
![[1860-5397-15-23-i31]](/bjoc/content/inline/1860-5397-15-23-i31.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 31: Cyclopropanol ring-opening initiated tandem cyclization with acrylamides or 2-isocyanobiphenyls.
Scheme 31: Cyclopropanol ring-opening initiated tandem cyclization with acrylamides or 2-isocyanobiphenyls.
In 2017, Mohr’s group proposed a straightforward approach to synthesize β-fluorinated ketones 114 by using AgF2 as both oxidant and fluorine atom source via the silver(II)-mediated ring-opening and fluorination of cyclopropanols 91 (Scheme 32) [112]. Through this method, a fluorine atom could easily be introduced in the β-position of a ketone. The mechanism is outlined in Scheme 32, the Ag-alkoxide complex 126 is initially formed from the process of ligand exchange between the substrate and AgF2. The alkoxy radical 98 is produced via a single-electron oxidation by Ag–O bond homolysis. As a feature of the cyclopropane system, the radical 98 goes through a ring fission to form the alkyl radical 99. Finally, the radical 99 abstracted an F-atom from another molecule of AgF2 to produce the target product 114.
![[1860-5397-15-23-i32]](/bjoc/content/inline/1860-5397-15-23-i32.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 32: Ag(II)-mediated oxidative ring-opening/fluorination of cyclopropanols with AgF2.
Scheme 32: Ag(II)-mediated oxidative ring-opening/fluorination of cyclopropanols with AgF2.
Kananovich and co-workers demonstrated the copper-catalyzed ring-opening and trifluoromethylation of teriary cyclopropanols 91 with fluorinated sulfinate salts 127 for the synthesis of β-trifluoromethyl ketones 128 at room temperature and in an open flask (Scheme 33) [113]. The presented results provided an efficient and convenient method for the synthesis of diverse fulorinated ketones from cyclopropanols.
![[1860-5397-15-23-i33]](/bjoc/content/inline/1860-5397-15-23-i33.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 33: Cu(II)-catalyzed ring-opening/fluoromethylation of cyclopropanols with sulfinate salts.
Scheme 33: Cu(II)-catalyzed ring-opening/fluoromethylation of cyclopropanols with sulfinate salts.
In the same year, this group developed a similar copper-catalyzed ring-opening and sulfonylation of teriary cyclopropanols 91 with sodium sulfinates 129 for the synthesis of γ-keto sulfones 130 in excellent yields (Scheme 34) [114]. The reaction was compatible with a series of fluoroalkyl, aryl and alkyl sulfinate salts. Notably, oxygen instead of THBP as oxidation was viable in this transformation.
![[1860-5397-15-23-i34]](/bjoc/content/inline/1860-5397-15-23-i34.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 34: Cu(II)-catalyzed ring-opening/sulfonylation of cyclopropanols with sulfinate salts.
Scheme 34: Cu(II)-catalyzed ring-opening/sulfonylation of cyclopropanols with sulfinate salts.
In 2017, Reddy and co-workers reported the first radical cyclization of propiolamides (131 and 133) with cyclopropanols 91 for the synthesis of azaspiro[4.5]deca-3,6,9-triene-2,8-diones 132 and 6,7-dihydro-3H-pyrrolo[2,1-j]quinoline-3,9(5H)-diones 134 (Scheme 35) [115]. Interestingly, this transformation proceeded under transition-metal-free conditions with high selectivity and yields. A series of substituents such as methoxy, dimethoxy, trimethoxy, methyl, chloro, bromo, and fluoro on the aromatic ring of cyclopropanols were tolerated well. The mechanism is outlined in Scheme 35. A β-carbonylalkyl radical 99 is produced from cyclopropanol 91 through a SET process. Then, addition of the radical 99 at the α-position of carbonyl in the substrate 131 furnishes the vinyl radical 135. Next, the vinyl radical 135 occurred 5-exo cyclization with the phenyl ring to generate the intermediate 136. Finally, the intermediate 136 underwent oxidation and deprotonation to give the desired product 132.
![[1860-5397-15-23-i35]](/bjoc/content/inline/1860-5397-15-23-i35.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 35: Na2S2O8-promoted ring-opening/arylation of cyclopropanols with propiolamides.
Scheme 35: Na2S2O8-promoted ring-opening/arylation of cyclopropanols with propiolamides.
In this year, Melchiorre’s group reported the ring-opening and [3 + 2]-annulation of cyclopropanols 91 with α,β-unsaturated aldehydes 138 for the synthesis of stereochemically dense cyclopentanols 139 with excellent enantioselectivity (Scheme 36) [116]. This transformation merged a stereocontrolled radical pattern with a classical ionic process in a cascade sequence.
![[1860-5397-15-23-i36]](/bjoc/content/inline/1860-5397-15-23-i36.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 36: The ring-opening and [3 + 2]-annulation of cyclopropanols with α,β-unsaturated aldehydes.
Scheme 36: The ring-opening and [3 + 2]-annulation of cyclopropanols with α,β-unsaturated aldehydes.
In 2018, Orellana et al. developed the Ag(II)-catalyzed ring-opening and functionalization of cyclopropanols 91 with electron-poor aromatic nitrogen heterocyles 140 under acid-free conditions and used a well-defined catalyst [Ag(II)(bipy)2S2O8] at low loadings (Scheme 37) [117]. This finding indicated that the silver pyridine complex plays an important role in single electron oxidants of cyclopropanols.
![[1860-5397-15-23-i37]](/bjoc/content/inline/1860-5397-15-23-i37.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 37: Cu(II)-catalyzed ring-opening/arylation of cyclopropanols with aromatic nitrogen heterocyles.
Scheme 37: Cu(II)-catalyzed ring-opening/arylation of cyclopropanols with aromatic nitrogen heterocyles.
In the same year, a silver-catalyzed ring-opening and difluoromethylthiolation of cyclopropanols 91 with PhSO2SCF2H 142 for the synthesis of difluoromethylthioethers 143 was reported by Shen and co-workers (Scheme 38) [118]. AgNO3 was utilized as catalyst, K2S2O8 as oxidant, and SDS (sodium dodecyl sulfate) as addictive in water. The SDS plays a key role in this transformation, and it enhances the solubility of both reactants in water. The cycloalkanol derivatives with electron-rich substituents on the phenyl rings deliver the corresponding products in higher yields than that with electron-deficient substituents.
![[1860-5397-15-23-i38]](/bjoc/content/inline/1860-5397-15-23-i38.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 38: Ag(I)-catalyzed ring-opening and difluoromethylthiolation of cyclopropanols with PhSO2SCF2H.
Scheme 38: Ag(I)-catalyzed ring-opening and difluoromethylthiolation of cyclopropanols with PhSO2SCF2H.
In 2018, Zhu and co-workers also reported the first silver-catalyzed ring-opening and acylation of cyclopropanols 91 with aldehydes 48 for the synthesis of 1,4-diketones 144 (Scheme 39) [119]. They proposed that the involvement of an uncommon water-assisted 1,2-HAT process was strongly exothermic and it promoted the addition of alkyl radicals to C=O bonds in aldehydes. The electronic effect of the phenyl rings in the aldehydes showed important influence on the reaction yields.
![[1860-5397-15-23-i39]](/bjoc/content/inline/1860-5397-15-23-i39.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 39: Ag(I)-catalyzed ring-opening and acylation of cyclopropanols with aldehydes.
Scheme 39: Ag(I)-catalyzed ring-opening and acylation of cyclopropanols with aldehydes.
In 2017, Kananovich developed a simple and efficient one-pot method for the preparation of enantiomerically enriched 2-oxyranyl ketones 146 by aerobic oxidation of easily available cyclopropanols 91 via intermediate formation of peroxyketone intermediates 145, followed by enantioselective epoxide formation in the presence of a poly-L-leucine catalyst and DBU (Scheme 40) [120].
![[1860-5397-15-23-i40]](/bjoc/content/inline/1860-5397-15-23-i40.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 40: Aerobic oxidation ring-opening of cyclopropanols for the synthesis of 2-oxyranyl ketones.
Scheme 40: Aerobic oxidation ring-opening of cyclopropanols for the synthesis of 2-oxyranyl ketones.
In 2014, a practical method for the conversion of 1,2-disubstituted cyclopropanols 91 derived from Kulinkovich cyclopropanation into linear enones 147 was developed by Wu and co-workers [121]. The approach features the regioselective cleavage of the cyclopropane rings in EtOH at room temperature with cheap and readily available Co(acac)2 as the catalyst and air as the reagent (Scheme 41).
![[1860-5397-15-23-i41]](/bjoc/content/inline/1860-5397-15-23-i41.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 41: Aerobic oxidation ring-opening of cyclopropanols for the synthesis of linear enones.
Scheme 41: Aerobic oxidation ring-opening of cyclopropanols for the synthesis of linear enones.
In 2015, Tyagi’s group presented a biomimetic synthesis of metabolite 149 from intermediate 148 by using catalytic vanadyl acetylacetonate and molecular O2 (Scheme 42) [122]. The transformation went through aerobic oxidation ring-opening of cyclopropanols. The results showed that the oxygen atom of newly-formed hydroxy group came from molecular O2.
![[1860-5397-15-23-i42]](/bjoc/content/inline/1860-5397-15-23-i42.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 42: Aerobic oxidation ring-opening of cyclopropanols for the synthesis of metabolite.
Scheme 42: Aerobic oxidation ring-opening of cyclopropanols for the synthesis of metabolite.
Conclusion
In the past 20 years, the field of oxidative radical ring-opening/cyclization of cyclopropane derivatives (including methylenecyclopropanes, cyclopropyl olefins and cyclopropanols) has experienced significant advances. This utility has been highlighted in a number of complex natural product syntheses. In this review, we have systematically summarized various oxidative radical strategies developed for the ring-opening and cyclization of cyclopropane derivatives. Despite these advances, there still exist opportunities for exploration and many questions to be addressed. Although oxidative radical ring-opening/cyclization of functionalized cyclopropane derivatives has been well developed, the ring-opening/cyclization of common cyclopropane derivatives is conspicuously absent. On the other hand, green and environmentally friendly strategies, such as photocatalysis or electrocatalysis, can be another orientation for further developments. This review opens the scope for future developments in new methodologies which promise the synthesis of novel fused cyclic systems with a wide range of medicinal and synthetic applications.
Acknowledgements
The work was supported by the National Nature Science Foundation of China (Grant No. 21602056), Scientific Research Fund of Hunan Provincial Science and Technology Department (Grant No. 2018JJ3208), Scientific Research Fund of Hunan Provincial Education Department (Grant No. 16A087).
References
-
Brandi, A.; Goti, A. Chem. Rev. 1998, 98, 589–636. doi:10.1021/cr940341t
Return to citation in text: [1] -
Schneider, T. F.; Kaschel, J.; Werz, D. B. Angew. Chem., Int. Ed. 2014, 53, 5504–5523. doi:10.1002/anie.201309886
Return to citation in text: [1] -
Rubin, M.; Rubina, M.; Gevorgyan, V. Chem. Rev. 2007, 107, 3117–3179. doi:10.1021/cr050988l
Return to citation in text: [1] -
de Meijere, A. Angew. Chem., Int. Ed. Engl. 1979, 18, 809–826. doi:10.1002/anie.197908093
Return to citation in text: [1] -
Wu, W.; Lin, Z.; Jiang, H. Org. Biomol. Chem. 2018, 16, 7315–7329. doi:10.1039/c8ob01187g
Return to citation in text: [1] -
Agrawal, D.; Yadav, V. K. Chem. Commun. 2008, 6471–6488. doi:10.1039/b812285g
Return to citation in text: [1] -
Carson, C. A.; Kerr, M. A. Chem. Soc. Rev. 2009, 38, 3051–3060. doi:10.1039/b901245c
Return to citation in text: [1] -
Cavitt, M. A.; Phun, L. H.; France, S. Chem. Soc. Rev. 2014, 43, 804–818. doi:10.1039/c3cs60238a
Return to citation in text: [1] -
Chen, D. Y. K.; Pouwerb, R. H.; Richardc, J. A. Chem. Soc. Rev. 2012, 41, 4631–4642. doi:10.1039/c2cs35067j
Return to citation in text: [1] -
Gnad, F.; Reiser, O. Chem. Rev. 2003, 103, 1603–1624. doi:10.1021/cr010015v
Return to citation in text: [1] -
Maas, G. Chem. Soc. Rev. 2004, 33, 183–190. doi:10.1039/b309046a
Return to citation in text: [1] -
Qian, D.; Zhang, J. Chem. Soc. Rev. 2015, 44, 677–698. doi:10.1039/c4cs00304g
Return to citation in text: [1] -
Rademacher, P. Chem. Rev. 2003, 103, 933–976. doi:10.1021/cr0100143
Return to citation in text: [1] -
Walsh, R. Chem. Soc. Rev. 2005, 34, 714–732. doi:10.1039/b310975p
Return to citation in text: [1] -
Wessjohann, L. A.; Brandt, W.; Thiemann, T. Chem. Rev. 2003, 103, 1625–1648. doi:10.1021/cr0100188
Return to citation in text: [1] -
Fumagalli, G.; Stanton, S.; Bower, J. F. Chem. Rev. 2017, 117, 9404–9432. doi:10.1021/acs.chemrev.6b00599
Return to citation in text: [1] -
Shi, M.; Shao, L.-X.; Lu, J.-M.; Wei, Y.; Mizuno, K.; Maeda, H. Chem. Rev. 2010, 110, 5883–5913. doi:10.1021/cr900381k
Return to citation in text: [1] -
Yu, L.-Z.; Chen, K.; Zhu, Z.-Z.; Shi, M. Chem. Commun. 2017, 53, 5935–5945. doi:10.1039/c7cc02596c
Return to citation in text: [1] -
Wong, H. N. C.; Hon, M. Y.; Tse, C. W.; Yip, Y. C.; Tanko, J.; Hudlicky, T. Chem. Rev. 1989, 89, 165–198. doi:10.1021/cr00091a005
Return to citation in text: [1] -
Pellissier, H. Tetrahedron 2010, 66, 8341–8375. doi:10.1016/j.tet.2010.08.031
Return to citation in text: [1] -
Goldschmidt, Z.; Crammer, B. Chem. Soc. Rev. 1988, 17, 229–267. doi:10.1039/cs9881700229
Return to citation in text: [1] -
Meazza, M.; Guo, H.; Rios, R. Org. Biomol. Chem. 2017, 15, 2479–2490. doi:10.1039/c6ob02647h
Return to citation in text: [1] -
Gibson, D. H.; DePuy, C. H. Chem. Rev. 1974, 74, 605–623. doi:10.1021/cr60292a001
Return to citation in text: [1] -
Kulinkovich, O. G. Chem. Rev. 2003, 103, 2597–2632. doi:10.1021/cr010012i
Return to citation in text: [1] -
Wu, X.; Zhu, C. Chem. Rec. 2018, 18, 587–598. doi:10.1002/tcr.201700090
Return to citation in text: [1] -
Nikolaev, A.; Orellana, A. Synthesis 2016, 1741–1768. doi:10.1055/s-0035-1560442
Return to citation in text: [1] -
Brandi, A.; Cicchi, S.; Cordero, F. M.; Goti, A. Chem. Rev. 2014, 114, 7317–7420. doi:10.1021/cr400686j
Return to citation in text: [1] -
Grover, H. K.; Emmett, M. R.; Kerr, M. A. Org. Biomol. Chem. 2015, 13, 655–671. doi:10.1039/c4ob02117g
Return to citation in text: [1] -
Nonhebel, D. C. Chem. Soc. Rev. 1993, 22, 347–359. doi:10.1039/cs9932200347
Return to citation in text: [1] -
Reissig, H.-U.; Zimmer, R. Chem. Rev. 2003, 103, 1151–1196. doi:10.1021/cr010016n
Return to citation in text: [1] -
Thankachan, A. P.; Sindhu, K. S.; Krishnan, K. K.; Anilkumar, G. Org. Biomol. Chem. 2015, 13, 8780–8802. doi:10.1039/c5ob01088h
Return to citation in text: [1] -
McCarroll, A. J.; Walton, J. C. J. Chem. Soc., Perkin Trans. 1 2001, 3215–3229. doi:10.1039/b105234a
Return to citation in text: [1] -
Dowd, P.; Zhang, W. Chem. Rev. 1993, 93, 2091–2115. doi:10.1021/cr00022a007
Return to citation in text: [1] -
Easton, C. J. Chem. Rev. 1997, 97, 53–82. doi:10.1021/cr9402844
Return to citation in text: [1] -
Frey, P. A.; Hegeman, A. D.; Reed, G. H. Chem. Rev. 2006, 106, 3302–3316. doi:10.1021/cr050292s
Return to citation in text: [1] -
Snider, B. B. Chem. Rev. 1996, 96, 339–364. doi:10.1021/cr950026m
Return to citation in text: [1] -
Studer, A. Chem. Soc. Rev. 2004, 33, 267–273. doi:10.1039/b307652k
Return to citation in text: [1] -
Penkett, C. S.; Sanderson, J. T. Annu. Rep. Prog. Chem., Sect. B: Org. Chem. 2001, 97, 3–20. doi:10.1039/b102954c
Return to citation in text: [1] -
Recupero, F.; Punta, C. Chem. Rev. 2007, 107, 3800–3842. doi:10.1021/cr040170k
Return to citation in text: [1] -
Rowlands, G. J. Annu. Rep. Prog. Chem., Sect. B: Org. Chem. 2012, 108, 15–28. doi:10.1039/c2oc90010f
Return to citation in text: [1] -
Shang, X.; Liu, Z.-Q. Org. Biomol. Chem. 2016, 14, 7829–7831. doi:10.1039/c6ob00797j
Return to citation in text: [1] -
Litwinienko, G.; Beckwith, A. L. J.; Ingold, K. U. Chem. Soc. Rev. 2011, 40, 2157–2163. doi:10.1039/c1cs15007c
Return to citation in text: [1] -
Zhou, H.; Huang, X.; Chen, W. J. Org. Chem. 2004, 69, 5471–5472. doi:10.1021/jo0496289
Return to citation in text: [1] -
Snider, B. B.; Patricia, J. J.; Kates, S. A. J. Org. Chem. 1988, 53, 2137–2143. doi:10.1021/jo00245a001
Return to citation in text: [1] -
Heiba, E.-A. I.; Dessau, R. M. J. Org. Chem. 1974, 39, 3456–3457. doi:10.1021/jo00937a052
Return to citation in text: [1] -
Heiba, E. I.; Dessau, R. M.; Koehl, W. J., Jr. J. Am. Chem. Soc. 1968, 90, 5905–5906. doi:10.1021/ja01023a049
Return to citation in text: [1] -
Back, T. G.; Muralidharan, K. R. J. Org. Chem. 1989, 54, 121–125. doi:10.1021/jo00262a028
Return to citation in text: [1] -
Citterio, A.; Fancelli, D.; Finzi, C.; Pesce, L.; Santi, R. J. Org. Chem. 1989, 54, 2713–2718. doi:10.1021/jo00272a047
Return to citation in text: [1] -
Huang, J.-W.; Shi, M. J. Org. Chem. 2005, 70, 3859–3863. doi:10.1021/jo050181t
Return to citation in text: [1] -
Yu, L.; Wu, Y.; Chen, T.; Pan, Y.; Xu, Q. Org. Lett. 2013, 15, 144–147. doi:10.1021/ol3031846
Return to citation in text: [1] -
Yu, L.; Huang, X. Synlett 2007, 1371–1374. doi:10.1055/s-2007-980361
Return to citation in text: [1] -
Xu, B.; Chen, Y.; Shi, M. Tetrahedron Lett. 2002, 43, 2781–2784. doi:10.1016/s0040-4039(02)00384-2
Return to citation in text: [1] -
Liu, L.-P.; Shi, M. Chem. Commun. 2004, 2878–2879. doi:10.1039/b412823k
Return to citation in text: [1] -
Kochi, J. K. J. Am. Chem. Soc. 1963, 85, 1958–1968. doi:10.1021/ja00896a014
Return to citation in text: [1] -
Kochi, J. K.; Bemis, A. J. Am. Chem. Soc. 1968, 90, 4038–4051. doi:10.1021/ja01017a022
Return to citation in text: [1] -
Kochi, J. K. Free Radicals; Wiley: New York, NY, U.S.A., 1968; Vol. I, p 591.
Return to citation in text: [1] -
Yu, L.; Huang, X.; Xie, M. H. Synlett 2006, 423–426. doi:10.1055/s-2006-926268
Return to citation in text: [1] -
Huang, X.; Yu, L. Synlett 2005, 2953–2957. doi:10.1055/s-2005-918947
Return to citation in text: [1] -
Miao, M.; Huang, X. J. Org. Chem. 2009, 74, 5636–5639. doi:10.1021/jo900805a
Return to citation in text: [1] -
Ma, S.; Lu, L.; Zhang, J. J. Am. Chem. Soc. 2004, 126, 9645–9660. doi:10.1021/ja0494860
Return to citation in text: [1] -
Liu, Z.-Q.; Sun, L.; Wang, J.-G.; Han, J.; Zhao, Y.-K.; Zhou, B. Org. Lett. 2009, 11, 1437–1439. doi:10.1021/ol900145u
Return to citation in text: [1] -
Zhu, Z.-Z.; Chen, K.; Yu, L.-Z.; Tang, X.-Y.; Shi, M. Org. Lett. 2015, 17, 5994–5997. doi:10.1021/acs.orglett.5b02940
Return to citation in text: [1] -
Wang, X.; Ye, Y.; Zhang, S.; Feng, J.; Xu, Y.; Zhang, Y.; Wang, J. J. Am. Chem. Soc. 2011, 133, 16410–16413. doi:10.1021/ja207775a
Return to citation in text: [1] -
Dong, X.; Sang, R.; Wang, Q.; Tang, X.-Y.; Shi, M. Chem. – Eur. J. 2013, 19, 16910–16915. doi:10.1002/chem.201303623
Return to citation in text: [1] -
Li, Y.; Lu, Y.; Qiu, G.; Ding, Q. Org. Lett. 2014, 16, 4240–4243. doi:10.1021/ol501939m
Return to citation in text: [1] -
Xu, J.; Wang, Y.-L.; Gong, T.-J.; Xiao, B.; Fu, Y. Chem. Commun. 2014, 50, 12915–12918. doi:10.1039/c4cc05692b
Return to citation in text: [1] -
Chen, M.-T.; Tang, X.-Y.; Shi, M. Org. Chem. Front. 2017, 4, 86–90. doi:10.1039/c6qo00536e
Return to citation in text: [1] -
Liu, Y.; Wang, Q.-L.; Zhou, C.-S.; Xiong, B.-Q.; Zhang, P.-L.; Yang, C.-a.; Tang, K.-W. J. Org. Chem. 2017, 82, 7394–7401. doi:10.1021/acs.joc.7b00970
Return to citation in text: [1] -
Pan, C.; Zhang, H.; Zhu, C. Tetrahedron Lett. 2016, 57, 595–598. doi:10.1016/j.tetlet.2015.12.092
Return to citation in text: [1] -
Feng, S.; Xie, X.; Zhang, W.; Liu, L.; Zhong, Z.; Xu, D.; She, X. Org. Lett. 2016, 18, 3846–3849. doi:10.1021/acs.orglett.6b01857
Return to citation in text: [1] -
Wei, W.-T.; Zhou, M.-B.; Fan, J.-H.; Liu, W.; Song, R.-J.; Liu, Y.; Hu, M.; Xie, P.; Li, J.-H. Angew. Chem., Int. Ed. 2013, 52, 3638–3641. doi:10.1002/anie.201210029
Return to citation in text: [1] -
Zhang, X.; Wang, M.; Li, P.; Wang, L. Chem. Commun. 2014, 50, 8006–8009. doi:10.1039/c4cc01189a
Return to citation in text: [1] -
Liu, Y.; Wang, Q.-L.; Zhou, C.-S.; Xiong, B.-Q.; Zhang, P.-L.; Yang, C.-a.; Tang, K.-W. J. Org. Chem. 2018, 83, 4657–4664. doi:10.1021/acs.joc.8b00427
Return to citation in text: [1] -
Lv, L.; Xi, H.; Bai, X.; Li, Z. Org. Lett. 2015, 17, 4324–4327. doi:10.1021/acs.orglett.5b02138
Return to citation in text: [1] -
Jia, F.; Liu, K.; Xi, H.; Lu, S.; Li, Z. Tetrahedron Lett. 2013, 54, 6337–6340. doi:10.1016/j.tetlet.2013.09.048
Return to citation in text: [1] -
Chatgilialoglu, C.; Crich, D.; Komatsu, M.; Ryu, I. Chem. Rev. 1999, 99, 1991–2070. doi:10.1021/cr9601425
Return to citation in text: [1] -
Pan, C.; Ni, Q.; Fu, Y.; Yu, J.-T. J. Org. Chem. 2017, 82, 7683–7688. doi:10.1021/acs.joc.7b01255
Return to citation in text: [1] -
Yu, L.-Z.; Xu, Q.; Tang, X.-Y.; Shi, M. ACS Catal. 2016, 6, 526–531. doi:10.1021/acscatal.5b02400
Return to citation in text: [1] -
Horner, J. H.; Tanaka, N.; Newcomb, M. J. Am. Chem. Soc. 1998, 120, 10379–10390. doi:10.1021/ja9819460
Return to citation in text: [1] -
Hollis, R.; Hughes, L.; Bowry, V. W.; Ingold, K. U. J. Org. Chem. 1992, 57, 4284–4287. doi:10.1021/jo00041a040
Return to citation in text: [1] -
Wang, L.-J.; Wang, A.-Q.; Xia, Y.; Wu, X.-X.; Liu, X.-Y.; Liang, Y.-M. Chem. Commun. 2014, 50, 13998–14001. doi:10.1039/c4cc06923d
Return to citation in text: [1] -
Chen, K.; Zhu, Z.-Z.; Liu, J.-X.; Tang, X.-Y.; Wei, Y.; Shi, M. Chem. Commun. 2016, 52, 350–353. doi:10.1039/c5cc07292a
Return to citation in text: [1] -
Stokes, B. J.; Richert, K. J.; Driver, T. G. J. Org. Chem. 2009, 74, 6442–6451. doi:10.1021/jo901224k
Return to citation in text: [1] -
Driver, T. G. Org. Biomol. Chem. 2010, 8, 3831–3846. doi:10.1039/c005219c
Return to citation in text: [1] -
Sun, K.; Liu, S.; Bec, P. M.; Driver, T. G. Angew. Chem., Int. Ed. 2011, 50, 1702–1706. doi:10.1002/anie.201006917
Return to citation in text: [1] -
Stokes, B. J.; Liu, S.; Driver, T. G. J. Am. Chem. Soc. 2011, 133, 4702–4705. doi:10.1021/ja111060q
Return to citation in text: [1] -
Fiori, K. W.; Du Bois, J. J. Am. Chem. Soc. 2007, 129, 562–568. doi:10.1021/ja0650450
Return to citation in text: [1] -
Zalatan, D. N.; Du Bois, J. J. Am. Chem. Soc. 2009, 131, 7558–7559. doi:10.1021/ja902893u
Return to citation in text: [1] -
Zhang, X.; Ke, Z.; DeYonker, N. J.; Xu, H.; Li, Z.-F.; Xu, X.; Zhang, X.; Su, C.-Y.; Phillips, D. L.; Zhao, C. J. Org. Chem. 2013, 78, 12460–12468. doi:10.1021/jo402101h
Return to citation in text: [1] -
Zhang, J.; Jiang, J.; Xu, D.; Luo, Q.; Wang, H.; Chen, J.; Li, H.; Wang, Y.; Wan, X. Angew. Chem., Int. Ed. 2015, 54, 1231–1235. doi:10.1002/anie.201408874
Return to citation in text: [1] -
Fan, X.; Yu, L.-Z.; Wei, Y.; Shi, M. Org. Lett. 2017, 19, 4476–4479. doi:10.1021/acs.orglett.7b01957
Return to citation in text: [1] -
Shi, M.; Lu, J.-M.; Xu, G.-C. Tetrahedron Lett. 2005, 46, 4745–4748. doi:10.1016/j.tetlet.2005.05.029
Return to citation in text: [1] -
Mathew, L.; Warkentin, J. J. Am. Chem. Soc. 1986, 108, 7981–7984. doi:10.1021/ja00285a016
Return to citation in text: [1] -
Chu, J. Y. C.; Lewicki, J. W. J. Org. Chem. 1977, 42, 2491–2493. doi:10.1021/jo00434a031
Return to citation in text: [1] -
Kippo, T.; Hamaoka, K.; Ryu, I. J. Am. Chem. Soc. 2013, 135, 632–635. doi:10.1021/ja311821h
Return to citation in text: [1] -
Xie, H.; Xu, B. Eur. J. Org. Chem. 2016, 2594–2598. doi:10.1002/ejoc.201600380
Return to citation in text: [1] -
Li, J.; Chen, J.; Jiao, W.; Wang, G.; Li, Y.; Cheng, X.; Li, G. J. Org. Chem. 2016, 81, 9992–10001. doi:10.1021/acs.joc.6b01825
Return to citation in text: [1] -
Wang, Y.-F.; Toh, K. K.; Ng, E. P. J.; Chiba, S. J. Am. Chem. Soc. 2011, 133, 6411–6421. doi:10.1021/ja200879w
Return to citation in text: [1] -
Ilangovan, A.; Saravanakumar, S.; Malayappasamy, S. Org. Lett. 2013, 15, 4968–4971. doi:10.1021/ol402229m
Return to citation in text: [1] -
Lu, S.-C.; Li, H.-S.; Xu, S.; Duan, G.-Y. Org. Biomol. Chem. 2017, 15, 324–327. doi:10.1039/c6ob02330d
Return to citation in text: [1] -
Kananovich, D. G.; Konik, Y. A.; Zubrytski, D. M.; Järving, I.; Lopp, M. Chem. Commun. 2015, 51, 8349–8352. doi:10.1039/c5cc02386f
Return to citation in text: [1] -
Li, Y.; Ye, Z.; Bellman, T. M.; Chi, T.; Dai, M. Org. Lett. 2015, 17, 2186–2189. doi:10.1021/acs.orglett.5b00782
Return to citation in text: [1] -
He, X.-P.; Shu, Y.-J.; Dai, J.-J.; Zhang, W.-M.; Feng, Y.-S.; Xu, H.-J. Org. Biomol. Chem. 2015, 13, 7159–7163. doi:10.1039/c5ob00808e
Return to citation in text: [1] -
Ren, S.; Feng, C.; Loh, T.-P. Org. Biomol. Chem. 2015, 13, 5105–5109. doi:10.1039/c5ob00632e
Return to citation in text: [1] -
Zhao, H.; Fan, X.; Yu, J.; Zhu, C. J. Am. Chem. Soc. 2015, 137, 3490–3493. doi:10.1021/jacs.5b00939
Return to citation in text: [1] -
Bloom, S.; Bume, D. D.; Pitts, C. R.; Lectka, T. Chem. – Eur. J. 2015, 21, 8060–8063. doi:10.1002/chem.201501081
Return to citation in text: [1] -
Wang, S.; Guo, L.; Wang, H.; Duan, X.-H. Org. Lett. 2015, 17, 4798–4801. doi:10.1021/acs.orglett.5b02353
Return to citation in text: [1] -
Fan, X.; Zhao, H.; Yu, J.; Bao, X.; Zhu, C. Org. Chem. Front. 2016, 3, 227–232. doi:10.1039/c5qo00368g
Return to citation in text: [1] -
Wang, C. Y.; Song, R. J.; Xie, Y. X.; Li, J. H. Synthesis 2016, 223–230. doi:10.1055/s-0035-1560374
Return to citation in text: [1] -
Guo, L.-N.; Deng, Z.-Q.; Wu, Y.; Hu, J. RSC Adv. 2016, 6, 27000–27003. doi:10.1039/c6ra03431d
Return to citation in text: [1] -
Davis, D. C.; Haskins, C. W.; Dai, M. G. Synlett 2017, 28, 913–918. doi:10.1055/s-0036-1588929
Return to citation in text: [1] -
Deng, Y.; Kauser, N. I.; Islam, S. M.; Mohr, J. T. Eur. J. Org. Chem. 2017, 5872–5879. doi:10.1002/ejoc.201700899
Return to citation in text: [1] -
Konik, Y. A.; Kudrjashova, M.; Konrad, N.; Kaabel, S.; Järving, I.; Lopp, M.; Kananovich, D. G. Org. Biomol. Chem. 2017, 15, 4635–4643. doi:10.1039/c7ob00680b
Return to citation in text: [1] -
Konik, Y. A.; Elek, G. Z.; Kaabel, S.; Järving, I.; Lopp, M.; Kananovich, D. G. Org. Biomol. Chem. 2017, 15, 8334–8340. doi:10.1039/c7ob01605k
Return to citation in text: [1] -
Reddy, C. R.; Yarlagadda, S.; Ramesh, B.; Reddy, M. R.; Sridhar, B.; Reddy, B. V. S. Eur. J. Org. Chem. 2017, 2332–2337. doi:10.1002/ejoc.201700058
Return to citation in text: [1] -
Woźniak, Ł.; Magagnano, G.; Melchiorre, P. Angew. Chem., Int. Ed. 2018, 57, 1068–1072. doi:10.1002/anie.201711397
Return to citation in text: [1] -
Nikolaev, A.; Legault, C. Y.; Zhang, M.; Orellana, A. Org. Lett. 2018, 20, 796–799. doi:10.1021/acs.orglett.7b03938
Return to citation in text: [1] -
Xu, B.; Wang, D.; Hu, Y.; Shen, Q. Org. Chem. Front. 2018, 5, 1462–1465. doi:10.1039/c8qo00115d
Return to citation in text: [1] -
Che, C.; Qian, Z.; Wu, M.; Zhao, Y.; Zhu, G. J. Org. Chem. 2018, 83, 5665–5673. doi:10.1021/acs.joc.8b00666
Return to citation in text: [1] -
Elek, G. Z.; Borovkov, V.; Lopp, M.; Kananovich, D. G. Org. Lett. 2017, 19, 3544–3547. doi:10.1021/acs.orglett.7b01519
Return to citation in text: [1] -
Han, W.-B.; Li, S.-G.; Lu, X.-W.; Wu, Y. Eur. J. Org. Chem. 2014, 3841–3846. doi:10.1002/ejoc.201402175
Return to citation in text: [1] -
Tyagi, S.; Cook, C. D.; DiDonato, D. A.; Key, J. A.; McKillican, B. P.; Eberle, W. J.; Carlin, T. J.; Hunt, D. A.; Marshall, S. J.; Bow, N. L. J. Org. Chem. 2015, 80, 11941–11947. doi:10.1021/acs.joc.5b01700
Return to citation in text: [1]
| 73. | Liu, Y.; Wang, Q.-L.; Zhou, C.-S.; Xiong, B.-Q.; Zhang, P.-L.; Yang, C.-a.; Tang, K.-W. J. Org. Chem. 2018, 83, 4657–4664. doi:10.1021/acs.joc.8b00427 |
| 74. | Lv, L.; Xi, H.; Bai, X.; Li, Z. Org. Lett. 2015, 17, 4324–4327. doi:10.1021/acs.orglett.5b02138 |
| 75. | Jia, F.; Liu, K.; Xi, H.; Lu, S.; Li, Z. Tetrahedron Lett. 2013, 54, 6337–6340. doi:10.1016/j.tetlet.2013.09.048 |
| 76. | Chatgilialoglu, C.; Crich, D.; Komatsu, M.; Ryu, I. Chem. Rev. 1999, 99, 1991–2070. doi:10.1021/cr9601425 |
| 77. | Pan, C.; Ni, Q.; Fu, Y.; Yu, J.-T. J. Org. Chem. 2017, 82, 7683–7688. doi:10.1021/acs.joc.7b01255 |
| 78. | Yu, L.-Z.; Xu, Q.; Tang, X.-Y.; Shi, M. ACS Catal. 2016, 6, 526–531. doi:10.1021/acscatal.5b02400 |
| 92. | Shi, M.; Lu, J.-M.; Xu, G.-C. Tetrahedron Lett. 2005, 46, 4745–4748. doi:10.1016/j.tetlet.2005.05.029 |
| 93. | Mathew, L.; Warkentin, J. J. Am. Chem. Soc. 1986, 108, 7981–7984. doi:10.1021/ja00285a016 |
| 87. | Fiori, K. W.; Du Bois, J. J. Am. Chem. Soc. 2007, 129, 562–568. doi:10.1021/ja0650450 |
| 88. | Zalatan, D. N.; Du Bois, J. J. Am. Chem. Soc. 2009, 131, 7558–7559. doi:10.1021/ja902893u |
| 89. | Zhang, X.; Ke, Z.; DeYonker, N. J.; Xu, H.; Li, Z.-F.; Xu, X.; Zhang, X.; Su, C.-Y.; Phillips, D. L.; Zhao, C. J. Org. Chem. 2013, 78, 12460–12468. doi:10.1021/jo402101h |
| 90. | Zhang, J.; Jiang, J.; Xu, D.; Luo, Q.; Wang, H.; Chen, J.; Li, H.; Wang, Y.; Wan, X. Angew. Chem., Int. Ed. 2015, 54, 1231–1235. doi:10.1002/anie.201408874 |
| 91. | Fan, X.; Yu, L.-Z.; Wei, Y.; Shi, M. Org. Lett. 2017, 19, 4476–4479. doi:10.1021/acs.orglett.7b01957 |
| 82. | Chen, K.; Zhu, Z.-Z.; Liu, J.-X.; Tang, X.-Y.; Wei, Y.; Shi, M. Chem. Commun. 2016, 52, 350–353. doi:10.1039/c5cc07292a |
| 83. | Stokes, B. J.; Richert, K. J.; Driver, T. G. J. Org. Chem. 2009, 74, 6442–6451. doi:10.1021/jo901224k |
| 84. | Driver, T. G. Org. Biomol. Chem. 2010, 8, 3831–3846. doi:10.1039/c005219c |
| 85. | Sun, K.; Liu, S.; Bec, P. M.; Driver, T. G. Angew. Chem., Int. Ed. 2011, 50, 1702–1706. doi:10.1002/anie.201006917 |
| 86. | Stokes, B. J.; Liu, S.; Driver, T. G. J. Am. Chem. Soc. 2011, 133, 4702–4705. doi:10.1021/ja111060q |
| 79. | Horner, J. H.; Tanaka, N.; Newcomb, M. J. Am. Chem. Soc. 1998, 120, 10379–10390. doi:10.1021/ja9819460 |
| 80. | Hollis, R.; Hughes, L.; Bowry, V. W.; Ingold, K. U. J. Org. Chem. 1992, 57, 4284–4287. doi:10.1021/jo00041a040 |
| 81. | Wang, L.-J.; Wang, A.-Q.; Xia, Y.; Wu, X.-X.; Liu, X.-Y.; Liang, Y.-M. Chem. Commun. 2014, 50, 13998–14001. doi:10.1039/c4cc06923d |
| 94. | Chu, J. Y. C.; Lewicki, J. W. J. Org. Chem. 1977, 42, 2491–2493. doi:10.1021/jo00434a031 |
| 95. | Kippo, T.; Hamaoka, K.; Ryu, I. J. Am. Chem. Soc. 2013, 135, 632–635. doi:10.1021/ja311821h |
| 96. | Xie, H.; Xu, B. Eur. J. Org. Chem. 2016, 2594–2598. doi:10.1002/ejoc.201600380 |
| 103. | He, X.-P.; Shu, Y.-J.; Dai, J.-J.; Zhang, W.-M.; Feng, Y.-S.; Xu, H.-J. Org. Biomol. Chem. 2015, 13, 7159–7163. doi:10.1039/c5ob00808e |
| 104. | Ren, S.; Feng, C.; Loh, T.-P. Org. Biomol. Chem. 2015, 13, 5105–5109. doi:10.1039/c5ob00632e |
| 101. | Kananovich, D. G.; Konik, Y. A.; Zubrytski, D. M.; Järving, I.; Lopp, M. Chem. Commun. 2015, 51, 8349–8352. doi:10.1039/c5cc02386f |
| 102. | Li, Y.; Ye, Z.; Bellman, T. M.; Chi, T.; Dai, M. Org. Lett. 2015, 17, 2186–2189. doi:10.1021/acs.orglett.5b00782 |
| 99. | Ilangovan, A.; Saravanakumar, S.; Malayappasamy, S. Org. Lett. 2013, 15, 4968–4971. doi:10.1021/ol402229m |
| 100. | Lu, S.-C.; Li, H.-S.; Xu, S.; Duan, G.-Y. Org. Biomol. Chem. 2017, 15, 324–327. doi:10.1039/c6ob02330d |
| 97. | Li, J.; Chen, J.; Jiao, W.; Wang, G.; Li, Y.; Cheng, X.; Li, G. J. Org. Chem. 2016, 81, 9992–10001. doi:10.1021/acs.joc.6b01825 |
| 98. | Wang, Y.-F.; Toh, K. K.; Ng, E. P. J.; Chiba, S. J. Am. Chem. Soc. 2011, 133, 6411–6421. doi:10.1021/ja200879w |
| 106. | Bloom, S.; Bume, D. D.; Pitts, C. R.; Lectka, T. Chem. – Eur. J. 2015, 21, 8060–8063. doi:10.1002/chem.201501081 |
| 107. | Wang, S.; Guo, L.; Wang, H.; Duan, X.-H. Org. Lett. 2015, 17, 4798–4801. doi:10.1021/acs.orglett.5b02353 |
| 105. | Zhao, H.; Fan, X.; Yu, J.; Zhu, C. J. Am. Chem. Soc. 2015, 137, 3490–3493. doi:10.1021/jacs.5b00939 |
| 1. | Brandi, A.; Goti, A. Chem. Rev. 1998, 98, 589–636. doi:10.1021/cr940341t |
| 2. | Schneider, T. F.; Kaschel, J.; Werz, D. B. Angew. Chem., Int. Ed. 2014, 53, 5504–5523. doi:10.1002/anie.201309886 |
| 3. | Rubin, M.; Rubina, M.; Gevorgyan, V. Chem. Rev. 2007, 107, 3117–3179. doi:10.1021/cr050988l |
| 4. | de Meijere, A. Angew. Chem., Int. Ed. Engl. 1979, 18, 809–826. doi:10.1002/anie.197908093 |
| 5. | Wu, W.; Lin, Z.; Jiang, H. Org. Biomol. Chem. 2018, 16, 7315–7329. doi:10.1039/c8ob01187g |
| 6. | Agrawal, D.; Yadav, V. K. Chem. Commun. 2008, 6471–6488. doi:10.1039/b812285g |
| 7. | Carson, C. A.; Kerr, M. A. Chem. Soc. Rev. 2009, 38, 3051–3060. doi:10.1039/b901245c |
| 8. | Cavitt, M. A.; Phun, L. H.; France, S. Chem. Soc. Rev. 2014, 43, 804–818. doi:10.1039/c3cs60238a |
| 9. | Chen, D. Y. K.; Pouwerb, R. H.; Richardc, J. A. Chem. Soc. Rev. 2012, 41, 4631–4642. doi:10.1039/c2cs35067j |
| 10. | Gnad, F.; Reiser, O. Chem. Rev. 2003, 103, 1603–1624. doi:10.1021/cr010015v |
| 11. | Maas, G. Chem. Soc. Rev. 2004, 33, 183–190. doi:10.1039/b309046a |
| 12. | Qian, D.; Zhang, J. Chem. Soc. Rev. 2015, 44, 677–698. doi:10.1039/c4cs00304g |
| 13. | Rademacher, P. Chem. Rev. 2003, 103, 933–976. doi:10.1021/cr0100143 |
| 14. | Walsh, R. Chem. Soc. Rev. 2005, 34, 714–732. doi:10.1039/b310975p |
| 15. | Wessjohann, L. A.; Brandt, W.; Thiemann, T. Chem. Rev. 2003, 103, 1625–1648. doi:10.1021/cr0100188 |
| 16. | Fumagalli, G.; Stanton, S.; Bower, J. F. Chem. Rev. 2017, 117, 9404–9432. doi:10.1021/acs.chemrev.6b00599 |
| 25. | Wu, X.; Zhu, C. Chem. Rec. 2018, 18, 587–598. doi:10.1002/tcr.201700090 |
| 26. | Nikolaev, A.; Orellana, A. Synthesis 2016, 1741–1768. doi:10.1055/s-0035-1560442 |
| 27. | Brandi, A.; Cicchi, S.; Cordero, F. M.; Goti, A. Chem. Rev. 2014, 114, 7317–7420. doi:10.1021/cr400686j |
| 28. | Grover, H. K.; Emmett, M. R.; Kerr, M. A. Org. Biomol. Chem. 2015, 13, 655–671. doi:10.1039/c4ob02117g |
| 29. | Nonhebel, D. C. Chem. Soc. Rev. 1993, 22, 347–359. doi:10.1039/cs9932200347 |
| 30. | Reissig, H.-U.; Zimmer, R. Chem. Rev. 2003, 103, 1151–1196. doi:10.1021/cr010016n |
| 31. | Thankachan, A. P.; Sindhu, K. S.; Krishnan, K. K.; Anilkumar, G. Org. Biomol. Chem. 2015, 13, 8780–8802. doi:10.1039/c5ob01088h |
| 114. | Konik, Y. A.; Elek, G. Z.; Kaabel, S.; Järving, I.; Lopp, M.; Kananovich, D. G. Org. Biomol. Chem. 2017, 15, 8334–8340. doi:10.1039/c7ob01605k |
| 23. | Gibson, D. H.; DePuy, C. H. Chem. Rev. 1974, 74, 605–623. doi:10.1021/cr60292a001 |
| 24. | Kulinkovich, O. G. Chem. Rev. 2003, 103, 2597–2632. doi:10.1021/cr010012i |
| 52. | Xu, B.; Chen, Y.; Shi, M. Tetrahedron Lett. 2002, 43, 2781–2784. doi:10.1016/s0040-4039(02)00384-2 |
| 53. | Liu, L.-P.; Shi, M. Chem. Commun. 2004, 2878–2879. doi:10.1039/b412823k |
| 22. | Meazza, M.; Guo, H.; Rios, R. Org. Biomol. Chem. 2017, 15, 2479–2490. doi:10.1039/c6ob02647h |
| 49. | Huang, J.-W.; Shi, M. J. Org. Chem. 2005, 70, 3859–3863. doi:10.1021/jo050181t |
| 112. | Deng, Y.; Kauser, N. I.; Islam, S. M.; Mohr, J. T. Eur. J. Org. Chem. 2017, 5872–5879. doi:10.1002/ejoc.201700899 |
| 17. | Shi, M.; Shao, L.-X.; Lu, J.-M.; Wei, Y.; Mizuno, K.; Maeda, H. Chem. Rev. 2010, 110, 5883–5913. doi:10.1021/cr900381k |
| 18. | Yu, L.-Z.; Chen, K.; Zhu, Z.-Z.; Shi, M. Chem. Commun. 2017, 53, 5935–5945. doi:10.1039/c7cc02596c |
| 19. | Wong, H. N. C.; Hon, M. Y.; Tse, C. W.; Yip, Y. C.; Tanko, J.; Hudlicky, T. Chem. Rev. 1989, 89, 165–198. doi:10.1021/cr00091a005 |
| 20. | Pellissier, H. Tetrahedron 2010, 66, 8341–8375. doi:10.1016/j.tet.2010.08.031 |
| 21. | Goldschmidt, Z.; Crammer, B. Chem. Soc. Rev. 1988, 17, 229–267. doi:10.1039/cs9881700229 |
| 50. | Yu, L.; Wu, Y.; Chen, T.; Pan, Y.; Xu, Q. Org. Lett. 2013, 15, 144–147. doi:10.1021/ol3031846 |
| 113. | Konik, Y. A.; Kudrjashova, M.; Konrad, N.; Kaabel, S.; Järving, I.; Lopp, M.; Kananovich, D. G. Org. Biomol. Chem. 2017, 15, 4635–4643. doi:10.1039/c7ob00680b |
| 44. | Snider, B. B.; Patricia, J. J.; Kates, S. A. J. Org. Chem. 1988, 53, 2137–2143. doi:10.1021/jo00245a001 |
| 47. | Back, T. G.; Muralidharan, K. R. J. Org. Chem. 1989, 54, 121–125. doi:10.1021/jo00262a028 |
| 110. | Guo, L.-N.; Deng, Z.-Q.; Wu, Y.; Hu, J. RSC Adv. 2016, 6, 27000–27003. doi:10.1039/c6ra03431d |
| 43. | Zhou, H.; Huang, X.; Chen, W. J. Org. Chem. 2004, 69, 5471–5472. doi:10.1021/jo0496289 |
| 48. | Citterio, A.; Fancelli, D.; Finzi, C.; Pesce, L.; Santi, R. J. Org. Chem. 1989, 54, 2713–2718. doi:10.1021/jo00272a047 |
| 111. | Davis, D. C.; Haskins, C. W.; Dai, M. G. Synlett 2017, 28, 913–918. doi:10.1055/s-0036-1588929 |
| 39. | Recupero, F.; Punta, C. Chem. Rev. 2007, 107, 3800–3842. doi:10.1021/cr040170k |
| 40. | Rowlands, G. J. Annu. Rep. Prog. Chem., Sect. B: Org. Chem. 2012, 108, 15–28. doi:10.1039/c2oc90010f |
| 41. | Shang, X.; Liu, Z.-Q. Org. Biomol. Chem. 2016, 14, 7829–7831. doi:10.1039/c6ob00797j |
| 42. | Litwinienko, G.; Beckwith, A. L. J.; Ingold, K. U. Chem. Soc. Rev. 2011, 40, 2157–2163. doi:10.1039/c1cs15007c |
| 108. | Fan, X.; Zhao, H.; Yu, J.; Bao, X.; Zhu, C. Org. Chem. Front. 2016, 3, 227–232. doi:10.1039/c5qo00368g |
| 32. | McCarroll, A. J.; Walton, J. C. J. Chem. Soc., Perkin Trans. 1 2001, 3215–3229. doi:10.1039/b105234a |
| 33. | Dowd, P.; Zhang, W. Chem. Rev. 1993, 93, 2091–2115. doi:10.1021/cr00022a007 |
| 34. | Easton, C. J. Chem. Rev. 1997, 97, 53–82. doi:10.1021/cr9402844 |
| 35. | Frey, P. A.; Hegeman, A. D.; Reed, G. H. Chem. Rev. 2006, 106, 3302–3316. doi:10.1021/cr050292s |
| 36. | Snider, B. B. Chem. Rev. 1996, 96, 339–364. doi:10.1021/cr950026m |
| 37. | Studer, A. Chem. Soc. Rev. 2004, 33, 267–273. doi:10.1039/b307652k |
| 38. | Penkett, C. S.; Sanderson, J. T. Annu. Rep. Prog. Chem., Sect. B: Org. Chem. 2001, 97, 3–20. doi:10.1039/b102954c |
| 45. | Heiba, E.-A. I.; Dessau, R. M. J. Org. Chem. 1974, 39, 3456–3457. doi:10.1021/jo00937a052 |
| 46. | Heiba, E. I.; Dessau, R. M.; Koehl, W. J., Jr. J. Am. Chem. Soc. 1968, 90, 5905–5906. doi:10.1021/ja01023a049 |
| 109. | Wang, C. Y.; Song, R. J.; Xie, Y. X.; Li, J. H. Synthesis 2016, 223–230. doi:10.1055/s-0035-1560374 |
| 54. | Kochi, J. K. J. Am. Chem. Soc. 1963, 85, 1958–1968. doi:10.1021/ja00896a014 |
| 55. | Kochi, J. K.; Bemis, A. J. Am. Chem. Soc. 1968, 90, 4038–4051. doi:10.1021/ja01017a022 |
| 56. | Kochi, J. K. Free Radicals; Wiley: New York, NY, U.S.A., 1968; Vol. I, p 591. |
| 57. | Yu, L.; Huang, X.; Xie, M. H. Synlett 2006, 423–426. doi:10.1055/s-2006-926268 |
| 117. | Nikolaev, A.; Legault, C. Y.; Zhang, M.; Orellana, A. Org. Lett. 2018, 20, 796–799. doi:10.1021/acs.orglett.7b03938 |
| 118. | Xu, B.; Wang, D.; Hu, Y.; Shen, Q. Org. Chem. Front. 2018, 5, 1462–1465. doi:10.1039/c8qo00115d |
| 115. | Reddy, C. R.; Yarlagadda, S.; Ramesh, B.; Reddy, M. R.; Sridhar, B.; Reddy, B. V. S. Eur. J. Org. Chem. 2017, 2332–2337. doi:10.1002/ejoc.201700058 |
| 116. | Woźniak, Ł.; Magagnano, G.; Melchiorre, P. Angew. Chem., Int. Ed. 2018, 57, 1068–1072. doi:10.1002/anie.201711397 |
| 68. | Liu, Y.; Wang, Q.-L.; Zhou, C.-S.; Xiong, B.-Q.; Zhang, P.-L.; Yang, C.-a.; Tang, K.-W. J. Org. Chem. 2017, 82, 7394–7401. doi:10.1021/acs.joc.7b00970 |
| 69. | Pan, C.; Zhang, H.; Zhu, C. Tetrahedron Lett. 2016, 57, 595–598. doi:10.1016/j.tetlet.2015.12.092 |
| 70. | Feng, S.; Xie, X.; Zhang, W.; Liu, L.; Zhong, Z.; Xu, D.; She, X. Org. Lett. 2016, 18, 3846–3849. doi:10.1021/acs.orglett.6b01857 |
| 71. | Wei, W.-T.; Zhou, M.-B.; Fan, J.-H.; Liu, W.; Song, R.-J.; Liu, Y.; Hu, M.; Xie, P.; Li, J.-H. Angew. Chem., Int. Ed. 2013, 52, 3638–3641. doi:10.1002/anie.201210029 |
| 72. | Zhang, X.; Wang, M.; Li, P.; Wang, L. Chem. Commun. 2014, 50, 8006–8009. doi:10.1039/c4cc01189a |
| 65. | Li, Y.; Lu, Y.; Qiu, G.; Ding, Q. Org. Lett. 2014, 16, 4240–4243. doi:10.1021/ol501939m |
| 66. | Xu, J.; Wang, Y.-L.; Gong, T.-J.; Xiao, B.; Fu, Y. Chem. Commun. 2014, 50, 12915–12918. doi:10.1039/c4cc05692b |
| 67. | Chen, M.-T.; Tang, X.-Y.; Shi, M. Org. Chem. Front. 2017, 4, 86–90. doi:10.1039/c6qo00536e |
| 62. | Zhu, Z.-Z.; Chen, K.; Yu, L.-Z.; Tang, X.-Y.; Shi, M. Org. Lett. 2015, 17, 5994–5997. doi:10.1021/acs.orglett.5b02940 |
| 121. | Han, W.-B.; Li, S.-G.; Lu, X.-W.; Wu, Y. Eur. J. Org. Chem. 2014, 3841–3846. doi:10.1002/ejoc.201402175 |
| 63. | Wang, X.; Ye, Y.; Zhang, S.; Feng, J.; Xu, Y.; Zhang, Y.; Wang, J. J. Am. Chem. Soc. 2011, 133, 16410–16413. doi:10.1021/ja207775a |
| 64. | Dong, X.; Sang, R.; Wang, Q.; Tang, X.-Y.; Shi, M. Chem. – Eur. J. 2013, 19, 16910–16915. doi:10.1002/chem.201303623 |
| 122. | Tyagi, S.; Cook, C. D.; DiDonato, D. A.; Key, J. A.; McKillican, B. P.; Eberle, W. J.; Carlin, T. J.; Hunt, D. A.; Marshall, S. J.; Bow, N. L. J. Org. Chem. 2015, 80, 11941–11947. doi:10.1021/acs.joc.5b01700 |
| 119. | Che, C.; Qian, Z.; Wu, M.; Zhao, Y.; Zhu, G. J. Org. Chem. 2018, 83, 5665–5673. doi:10.1021/acs.joc.8b00666 |
| 60. | Ma, S.; Lu, L.; Zhang, J. J. Am. Chem. Soc. 2004, 126, 9645–9660. doi:10.1021/ja0494860 |
| 61. | Liu, Z.-Q.; Sun, L.; Wang, J.-G.; Han, J.; Zhao, Y.-K.; Zhou, B. Org. Lett. 2009, 11, 1437–1439. doi:10.1021/ol900145u |
| 120. | Elek, G. Z.; Borovkov, V.; Lopp, M.; Kananovich, D. G. Org. Lett. 2017, 19, 3544–3547. doi:10.1021/acs.orglett.7b01519 |
© 2019 Liu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)