Abstract
We report a new synthetic approach to assemble spirothiazolidinediones via a [2 + 2 + 2] cyclotrimerization reaction and the derivatives were further functionalized through DA chemistry and click reaction. Using flow cytometry, it was shown for the first time that the new benzyl alcohol derivatives of thiazolidine-2,4-dione generated here are efficient apoptosis inducers in the HeLa, Hek293, U937, Jurkat, and K562 cell lines.
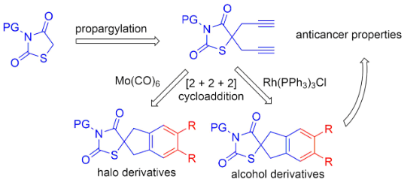
Graphical Abstract
Introduction
Heterocyclic compounds play a vital role in the metabolism of all living cells. Thus, most of the biologically active and pharmaceutically important compounds consist of heterocycles [1-4]. Especially, thiazolidinediones (TZDs) are important heterocyclic compounds. In 1954, Visentini reported the first ever pharmacological evaluation of a thiazolidinedione derivative, i.e., its anti-TB activity [3]. The promising activity shown by the compounds containing a thiazolidinedione nucleus cover numerous categories such as antihyperglycaemics [5], aldose reductase inhibitors (ARI) [6,7], anti-inflammatory [8,9], anti-arthritics [10], anticancer [11-13] and antimicrobial [14-18], etc., has made them an indispensable anchor for the development of new therapeutic agents [19-24]. Thiazolidinedione derivatives, such as rosiglitazone, pioglitazone and troglitazone, etc., are members of the glitazone family of drugs, used for the treatment of diabetes as potent agonists of the γ-peroxisome proliferator activated receptor (PPARγ) [23,24]. The discovery of biological activities of TZDs and the development of medicinal and pharmaceutical chemistry has led to an enhanced interest in the design of new synthetic methods for the preparation of various TZDs. The most conventional method of synthesis of thiazolidinedione derivatives is refluxing chloroacetic acid (2) with thiourea (1), followed by a Knoevenagel condensation with an aldehyde (Scheme 1) [25].
![[1860-5397-15-269-i1]](/bjoc/content/inline/1860-5397-15-269-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Conventional method of synthesis of thiazolidine-2,4-dione derivatives.
Scheme 1: Conventional method of synthesis of thiazolidine-2,4-dione derivatives.
Results and Discussion
Limited reports are available dealing with the synthesis of spiro derivatives of thiazolidine-2,4-diones [26-28]. In view of the importance of thiazolidine-2,4-dione derivatives, we conceived a new synthetic strategy to diverse spirocyclic thiazolidinediones based on a [2 + 2 + 2] cyclotrimerization [29-51] as a key step.
In this regard, the required diyne precursor of thiazolidinedione was prepared from N-methylthiazolidine-2,4-dione (5a) and propargyl bromide (6a) in the presence of K2CO3 in DMF to afford the dipropargylated intermediate thiazolidinedione 7a in 85% yield. Diyne 7a was then reacted with propargyl bromide (6a) in the presence of Mo(CO)6 in acetonitrile at 90 °C under microwave irradiation (MWI) conditions to give the co-trimerized spiro derivative 8a (Scheme 2).
![[1860-5397-15-269-i2]](/bjoc/content/inline/1860-5397-15-269-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: [2 + 2 + 2] Cyclotrimerization of N-methylthiazolidinedione.
Scheme 2: [2 + 2 + 2] Cyclotrimerization of N-methylthiazolidinedione.
The free NH moiety of thiazolidinedione 3 was alkylated using alkyl or aryl halides in the presence of Et3N using DCM as solvent. To our surprise, during an attempted protection with (Boc)2O, we obtained N-tert-butylthiazolidinedione 5b rather than the expected N-Boc-protected thiazolidinedione 9 as reported by Paladhi et al. [52] (Scheme 3). The formation of compound 5b was confirmed on the basis of spectral data. We observed only two peaks in the carbonyl region of the 13C NMR spectrum and no third carbonyl peak (Figure 1). Finally, mass spectral (HRMS) data confirmed the molecular formula. Next, we prepared the diyne precursors and examined the [2 + 2 + 2] cyclotrimerization strategy with different propargyl halides 6 to obtain spirothiazolidinedione derivatives (Scheme 4).
![[1860-5397-15-269-i3]](/bjoc/content/inline/1860-5397-15-269-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Unexpected product 5b obtained in the attempted NH-protection of thiazolidinedione with (Boc)2O.
Scheme 3: Unexpected product 5b obtained in the attempted NH-protection of thiazolidinedione with (Boc)2O.
![[1860-5397-15-269-1]](/bjoc/content/figures/1860-5397-15-269-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Comparison of 13C NMR values of 9 and 5b.
Figure 1: Comparison of 13C NMR values of 9 and 5b.
![[1860-5397-15-269-i4]](/bjoc/content/inline/1860-5397-15-269-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: [2 + 2 + 2] Cyclotrimerization of dipropargylthiazolidinediones with propargyl halides.
Scheme 4: [2 + 2 + 2] Cyclotrimerization of dipropargylthiazolidinediones with propargyl halides.
Further, the dibromo N-methyl derivative of thiazolidinedione 8b was converted to sultine 13 using rongalite and tetra-n-butylammonium bromide (TBAB) in DMF. Then, sultine 13 was treated with different dienophiles 14 in a DA fashion and linearly fused spirocyclic derivatives 15 were isolated in good yields (Scheme 5).
![[1860-5397-15-269-i5]](/bjoc/content/inline/1860-5397-15-269-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Formation of sultine 13 from compound 8b followed by DA reaction.
Scheme 5: Formation of sultine 13 from compound 8b followed by DA reaction.
Since TZDs are pharmaceutically important targets, we intended to prepare spirocyclic derivatives of thiazolidinedione. In this context, N-carboxy ester and N-propargyl derivatives of thiazolidinedione were prepared, which on further modification, can generate new derivatives suitable for the multiple interaction sites. Further, the antitumour activity of these compounds were studied.
Therefore, we synthesized the required diyne derivatives of thiazolidinedione for [2 + 2 + 2] cycloaddition, in the presence of propargyl bromide and K2CO3 in DMF. The dipropargyl derivatives 7e and 7f were then treated with different propargyl alcohols 16 in the presence of Wilkinson’s catalyst/Ti(OiPr)4 under ethanol reflux conditions [53] to obtain the corresponding [2 + 2 + 2] cyclotrimerized alcohols 17–19 and 21 in good yields. During the [2 + 2 + 2] cycloaddition of compound 7e with 3-butyn-1-ol (16b) we were able to isolate the self trimerized derivative 20 as a minor product (Scheme 7).
![[1860-5397-15-269-i6]](/bjoc/content/inline/1860-5397-15-269-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Dipropargylation of 2,4-thiazolidinedione derivatives.
Scheme 6: Dipropargylation of 2,4-thiazolidinedione derivatives.
![[1860-5397-15-269-i7]](/bjoc/content/inline/1860-5397-15-269-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: [2 + 2 + 2] Cycloaddition in the presence of Wilkinson’s catalyst.
Scheme 7: [2 + 2 + 2] Cycloaddition in the presence of Wilkinson’s catalyst.
The ester derivative of thiazolidinedione 18 was hydrolyzed under AcOH, HCl reflux conditions to obtain N-acid derivative 22 in 90% yield (Scheme 8) [54].
![[1860-5397-15-269-i8]](/bjoc/content/inline/1860-5397-15-269-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: N-Ester derivative 18 hydrolysis to N-acid derivative 22.
Scheme 8: N-Ester derivative 18 hydrolysis to N-acid derivative 22.
Fused 1,2,3-triazoles represent an important class of nitrogen-containing biologically active compounds which exhibit various biological properties, such as antiviral, antibacterial and anticancer, etc. [55-58]. Recently, the use of 1,2,3-triazole derivatives as drug candidates has been increased for clinical therapy of various diseases. Hence, we also modified the N-propargyl alcohol derivative 21 to 1,2,3-triazolo alcohol derivative 24 by click chemistry using 4-nitrophenyl azide (23) and the corresponding triazolo alcohol derivative 24 was obtained as yellow solid in 80% yield (Scheme 9) [58].
![[1860-5397-15-269-i9]](/bjoc/content/inline/1860-5397-15-269-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Synthesis of triazolo derivative 24 via click reaction.
Scheme 9: Synthesis of triazolo derivative 24 via click reaction.
All alcohol derivatives of thiazolidinedione were used to study the cytotoxic activity in comparison with camptothecin and etoposide on Jurkat, K562, HEK293, HELA, A549 and U937 cell lines. Compounds 20 and 17 showed the highest activity (IC50 = 0.29 and 0.36 nM, respectively) for leukemic monocytic lymphoma cells (U937), and 22 was active against T-cell leukemia (Jurkat) with an IC50 value of 0.34 nM. Further, these three compounds (22, 20 and 17) were used to study apoptosis and the cell cycle in cells of the corresponding cell cultures. The highest percentage of apoptosis was observed when compound 22 was added to the culture of Jurkat cells at a concentration of 0.75 nM and was 68.65%. Under similar conditions, compound 20 showed about 95% in K562 cells of late apoptosis and compound 17 about 30.28% for cell line U937. The results of flow cytometry showed that in all three cell lines, Jurkat, K562 and U937, a hypodiploidal DNA peak appears 24 hours after the action of compounds 22, 20 and 17, which shows the inability to stop the fission cycle at the checkpoints, and ultimately, leads to cell death.
Conclusion
In summary, we have synthesized spirothiazolidinedione derivatives via a [2 + 2 + 2] cyclotrimerization strategy with propargyl halides. We have also shown the synthesis of linearly fused spirocyclic alcohol derivatives of thiazolidinedione using Wilkinson’s catalyst and these compounds were tested for their anticancer activity. We have shown for the first time that the new benzyl alcohol derivatives of thiazolidinedione generated here are efficient apoptosis inducers in the HeLa, Hek293, U937, Jurkat, and K562 cell lines.
Experimental
General: All commercially available products were used as received without further purification and moisture-sensitive reagents were transferred by using syringe–septum techniques. All the solvents used as reaction media were dried over oven-dried molecular sieves (4 Å). Column chromatography was performed with silica gel (100–200 mesh) using mixtures of petroleum ether and EtOAc as eluent. 1H NMR and 13C NMR spectral data were recorded with 400 MHz and 100 MHz or 500 MHz and 125 MHz spectrometers using tetramethylsilane (TMS) as an internal standard and chloroform-d as a solvent. High resolution mass spectrometry (HRMS) was performed using a Bruker (Maxis Impact) or a Micromass Q-ToF spectrometer. The melting points recorded are uncorrected. The microwave used here was a Discover® SP by CEM Corporation and all the microwave reactions were performed under the standard method, where time and temperature can be monitored manually.
Cell culture: Cells (Jurkat, K562, U937) were purchased from the Russian Cell Culture Collection (Institute of Cytology of the Russian Academy of Sciences) and cultured according to standard mammalian tissue culture protocols and sterile technique. Human cancer cell lines HEK293 and HeLa were obtained from the HPA Culture Collections (UK). All cell lines used in the study were tested and shown to be free of mycoplasma and viral contamination.
Cell culture was performed in a similar manner as described in [59]. HEK293 and HeLa cell lines were cultured as monolayers and maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco BRL) supplemented with 10% foetal bovine serum and 1% penicillin streptomycin solution at 37 °C in a humidified incubator under a 5% CO2 atmosphere.
Cells were maintained in RPMI 1640 (Jurkat, K562, U937) (Gibco) supplemented with 4 mM glutamine, 10% FBS (Sigma) and 100 units/mL penicillin streptomycin (Sigma). All types of cells were grown in an atmosphere of 5% CO2 at 37 °C. The cells were subcultured at 2–3 days intervals. Adherent cells (HEK293, HeLa) were suspended using trypsin/EDTA and counted after they have reached 80% confluency. Cells were then seeded in 24 well plates at 5 × 104 cells per well and incubated overnight. Jurkat, K562, U937 cells were subcultured at 2 day intervals with a seeding density of 1 × 105 cells per 24 well plates in RPMI with 10% FBS.
MTT Assay: The MTT assay is a colorimetric assay for evaluation of cell metabolic activity. The NADPH-dependent cellular oxidoreductases present in the living cell can reflect, under certain conditions, the cell viability. These enzymes can reduce the tetrazolium dye, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (МТТ) or yellow tetrazole, to give insoluble formazan, which develops purple color particularly in living cells. Thus, the color gradient can serve to determine the degree of cytostatic activity (shift from proliferation to dormancy) of drug candidates and toxic compounds. In vitro cytotoxicity was assessed using a standard MTT colorimetric assay in a similar manner as described in [60,61]. HEK293 cells were plated in 96-well microassay culture plates (1 × 104 cells per well) and grown overnight at 37 °C in a 5% CO2 incubator. The cells were then incubated with the test compounds at different concentrations for an additional 24 h. Control wells were prepared by the addition of dimethyl sulfoxide (DMSO, 1%). At the end of this incubation, 20 μL of MTT (5 mg/mL in PBS) was added to each well. After incubation for 4 h, the formazan crystals were dissolved in 150 μL of DMSO, and the absorbance was determined at 595 nm using a microplate spectrophotometer. The IC50 value was determined from plots of percent viability against the dose of the compound added.
Cytotoxicity assay: Viability (live/dead) assessment was performed by staining cells with 7-AAD (7-aminoactinomycin D, Biolegend) in a similar manner as described in [62]. In brief, after treatment cells were harvested, washed 1–2 times with phosphate-buffered saline (PBS) and centrifuged at 400g for 5 minutes. Cell pellets were resuspended in 200 µL of flow cytometry staining buffer (PBS without Ca2+and Mg2+, 2.5% FBS) and stained with 5 µL of 7-AAD staining solution for 15 minutes at room temperature in the dark. Samples were acquired on a NovoCyteTM 2000 Flow Cytometry System (ACEA) equipped with a 488 nm argon laser. Detection of 7-AAD emission was collected through a 675/30 nm filter in FL4 channel.
Annexin V-FITC/PI assay: The apoptosis-inducing activity of the compounds was analyzed quantitatively using an Alexa Fluor® 488 Annexin V/Dead Cell Apoptosis Kit in a similar way as described in [60]. Phosphatidylserine externalization on the outer surface of the plasma membrane is an exact and reliable indication of cell apoptosis or necrosis. The difference between these two forms of cell death is that in the early stages of apoptosis, the cell membrane remains relatively undamaged, whereas upon necrosis, the cell membrane loses integrity. In the living, normally functioning cells, phosphatidylserine is present on the cell surface membrane in minor quantity; hence, its interaction with the V Alexa Fluor® 488 annexin is insignificant. Furthermore, undamaged cell membrane is impermeable for propidium iodide. At the early stage of apoptosis, phosphatidylserine molecules appear on the cell surface but the membrane is still impermeable for DNA-binding dyes (such as propidium iodide). The membrane integrity is lost at later stages of the cell death. Thus, while detecting the apoptosis, one can distinguish four types of cells: living cells (annexin V−/PI−), early apoptosis (annexin V+/PI−), late apoptosis (annexin V+/PI+), and necrosis (annexin V−/PI+).
Apoptosis was quantified by the detection of phosphatidylserine surface exposure on apoptotic cells using an Alexa Fluor® 488 Annexin V/Dead Cell Apoptosis Kit. HEK293 cells were incubated with or without the testing compound for 24 h. The adherent and floating cells were collected and washed twice with cold PBS. Then, the cells were resuspended in 90 μL of annexin V binding buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2; pH 7.4). Annexin V (5 μL) and 1 μL of propidium iodide were added to the reaction mixture and incubated for 15 min at room temperature in the dark. After the addition of 300 μL of binding buffer, the stained cells were analyzed with a NovoCyteTM 2000 FlowCytometry System (ACEA).
Supporting Information
| Supporting Information File 1: Experimental details, characterization data and copies of spectra. | ||
| Format: PDF | Size: 3.5 MB | Download |
Acknowledgements
S. K. thanks the Department of Science and Technology for the award of a J. C. Bose fellowship and Praj Industries for the award of chair professor (Green chemistry). G. S. thanks the CSIR-New Delhi for the award of a research fellowship. We also thank DST, New Delhi for the financial support. V. A. D. thanks the Russian Foundation for Basic Research (Grants No. 18-33-20058, 18-29-09068) and the Grant of the RF President (Sci. Sh.-5240.2018.3) for financial support.
References
-
Saini, M. S.; Kumar, A.; Dwivedi, J.; Singh, R. Int. J. Pharma Sci. Res. 2013, 4, 66–77.
Return to citation in text: [1] -
Arora, P.; Arora, V.; Lamba, H. S.; Wadhwa, D. Int. J. Pharm. Sci. Res. 2012, 3, 2947–2954. doi:10.13040/ijpsr.0975-8232.3(9).2947-54
Return to citation in text: [1] -
Visentini, P. Farmaco, Ed. Sci. 1954, 9, 274–277.
Return to citation in text: [1] [2] -
Thompson, L. A.; Ellman, J. A. Chem. Rev. 1996, 96, 555–600. doi:10.1021/cr9402081
Return to citation in text: [1] -
Fujita, T.; Sugiyama, Y.; Taketomi, S.; Sohda, T.; Kawamatsu, Y.; Iwatsuka, H.; Suzuoki, Z. Diabetes 1983, 32, 804–810. doi:10.2337/diab.32.9.804
Return to citation in text: [1] -
Bruno, G.; Costantino, L.; Curinga, C.; Maccari, R.; Monforte, F.; Nicolò, F.; Ottanà, R.; Vigorita, M. G. Bioorg. Med. Chem. 2002, 10, 1077–1084. doi:10.1016/s0968-0896(01)00366-2
Return to citation in text: [1] -
Suzen, S.; Buyukbingol, E. Curr. Med. Chem. 2003, 10, 1329–1352. doi:10.2174/0929867033457377
Return to citation in text: [1] -
Rekha, S.; Shantharam, U.; Chandy, V. Int. Res. J. Pharm. 2011, 2, 81–84.
Return to citation in text: [1] -
Youssef, A. M.; White, M. S.; Villanueva, E. B.; El-Ashmawy, I. M.; Klegeris, A. Bioorg. Med. Chem. 2010, 18, 2019–2028. doi:10.1016/j.bmc.2010.01.021
Return to citation in text: [1] -
da Rocha Junior, L. F.; de Melo Rêgo, M. J. B.; Cavalcanti, M. B.; Pereira, M. C.; Pitta, M. G. d. R.; de Oliveira, P. S. S.; Gonçalves, S. M. C.; Duarte, A. L. B. P.; de Lima, M. d. C. A.; Pitta, I. d. R.; Pitta, M. G. d. R. BioMed Res. Int. 2013, No. 926060. doi:10.1155/2013/926060
Return to citation in text: [1] -
Ip, M. M.; Sylvester, P. W.; Schenkel, L. Cancer Res. 1986, 46, 1735–1740.
Return to citation in text: [1] -
Shimazaki, N.; Togashi, N.; Hanai, M.; Isoyama, T.; Wada, K.; Fujita, T.; Fujiwara, K.; Kurakata, S. Eur. J. Cancer 2008, 44, 1734–1743. doi:10.1016/j.ejca.2008.04.016
Return to citation in text: [1] -
Subtel’na, I.; Atamanyuk, D.; Szymańska, E.; Kieć-Kononowicz, K.; Zimenkovsky, B.; Vasylenko, O.; Gzella, A.; Lesyk, R. Bioorg. Med. Chem. 2010, 18, 5090–5102. doi:10.1016/j.bmc.2010.05.073
Return to citation in text: [1] -
Mulwad, V. V.; Mir, A. A.; Parmar, H. T. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 2009, 48, 137–141. doi:10.1002/chin.200925122
Return to citation in text: [1] -
Sharma, R.; Nagda, D. P.; Talesara, G. L. ARKIVOC 2006, No. i, 1–12. doi:10.3998/ark.5550190.0007.101
Return to citation in text: [1] -
Ceylan, M. Turk. J. Chem. 2006, 30, 355–360.
Return to citation in text: [1] -
Chavan, A. A.; Pai, N. R. ARKIVOC 2007, No. xvi, 148–155. doi:10.3998/ark.5550190.0008.g16
Return to citation in text: [1] -
Albrecht, U.; Gördes, D.; Schmidt, E.; Thurow, K.; Lalk, M.; Langer, P. Bioorg. Med. Chem. 2005, 13, 4402–4407. doi:10.1016/j.bmc.2005.04.046
Return to citation in text: [1] -
Jain, V. S.; Vora, D. K.; Ramaa, C. S. Bioorg. Med. Chem. 2013, 21, 1599–1620. doi:10.1016/j.bmc.2013.01.029
Return to citation in text: [1] -
Singh, S. P.; Parmar, S. S.; Raman, K.; Stenberg, V. I. Chem. Rev. 1981, 81, 175–203. doi:10.1021/cr00042a003
Return to citation in text: [1] -
Napoleon, A. A. Int. J. PharmTech Res. 2016, 9, 429–443.
Return to citation in text: [1] -
Chadha, N.; Bahia, M. S.; Kaur, M.; Silakari, O. Bioorg. Med. Chem. 2015, 23, 2953–2974. doi:10.1016/j.bmc.2015.03.071
Return to citation in text: [1] -
Sohda, T.; Mizuno, K.; Tawada, H.; Sugiyama, Y.; Fujita, T.; Kawamatsu, Y. Chem. Pharm. Bull. 1982, 30, 3563–3573. doi:10.1248/cpb.30.3563
Return to citation in text: [1] [2] -
Lehmann, J. M.; Moore, L. B.; Smith-Oliver, T. A.; Wilkison, W. O.; Willson, T. M.; Kliewer, S. A. J. Biol. Chem. 1995, 270, 12953–12956. doi:10.1074/jbc.270.22.12953
Return to citation in text: [1] [2] -
Brown, F. C. Chem. Rev. 1961, 61, 463–521. doi:10.1021/cr60213a002
Return to citation in text: [1] -
Jones, E. R. H.; Robinson, F. A.; Strachan, M. N. J. Chem. Soc. 1946, 91–92. doi:10.1039/jr9460000091
Return to citation in text: [1] -
Bozdag, O.; Ertan, R. Turk. J. Chem. 1999, 23, 163–169.
Return to citation in text: [1] -
Dhara, K.; Paladhi, S.; Midya, G. C.; Dash, J. Org. Biomol. Chem. 2011, 9, 3801–3807. doi:10.1039/c0ob01248c
Return to citation in text: [1] -
Okamoto, S.; Sugiyama, Y.-k. Synlett 2013, 24, 1044–1060. doi:10.1055/s-0032-1316913
Return to citation in text: [1] -
Shibata, Y.; Tanaka, K. Synthesis 2012, 44, 323–350. doi:10.1055/s-0031-1289665
Return to citation in text: [1] -
Kotha, S.; Brahmachary, E.; Lahiri, K. Eur. J. Org. Chem. 2005, 4741–4767. doi:10.1002/ejoc.200500411
Return to citation in text: [1] -
Galan, B. R.; Rovis, T. Angew. Chem., Int. Ed. 2009, 48, 2830–2834. doi:10.1002/anie.200804651
Return to citation in text: [1] -
Chopade, P. R.; Louie, J. Adv. Synth. Catal. 2006, 348, 2307–2327. doi:10.1002/adsc.200600325
Return to citation in text: [1] -
Domínguez, G.; Pérez-Castells, J. Chem. Soc. Rev. 2011, 40, 3430–3444. doi:10.1039/c1cs15029d
Return to citation in text: [1] -
Gandon, V.; Aubert, C.; Malacria, M. Chem. Commun. 2006, 2209–2217. doi:10.1039/b517696b
Return to citation in text: [1] -
Varela, J. A.; Saá, C. Chem. Rev. 2003, 103, 3787–3802. doi:10.1021/cr030677f
Return to citation in text: [1] -
Yamamoto, Y. Curr. Org. Chem. 2005, 9, 503–519. doi:10.2174/1385272053544399
Return to citation in text: [1] -
Agenet, N.; Buisine, O.; Slowinski, F.; Gandon, V.; Aubert, C.; Malacria, M. In Cotrimerizations of Acetylenic Compounds; Rajan Babu, T. V., Ed.; Organic Reactions, Vol. 68; John Wiley and Sons: Hoboken, New Jersey; pp 1–302. doi:10.1002/0471264180.or068.01
Return to citation in text: [1] -
Heller, B.; Hapke, M. Chem. Soc. Rev. 2007, 36, 1085–1094. doi:10.1039/b607877j
Return to citation in text: [1] -
Inglesby, P. A.; Evans, P. A. Chem. Soc. Rev. 2010, 39, 2791–2805. doi:10.1039/b913110h
Return to citation in text: [1] -
Tanaka, K. Transition Metal-Mediated Aromatic Ring Construction. In Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds; Mortier, J., Ed.; John Wiley and Sons: Hoboken, New Jersey, 2016; pp 587–614. doi:10.1002/9781118754887.ch21
Return to citation in text: [1] -
Weding, N.; Hapke, M. Chem. Soc. Rev. 2011, 40, 4525–4538. doi:10.1039/c0cs00189a
Return to citation in text: [1] -
Vollhardt, K. P. C. Acc. Chem. Res. 1977, 10, 1–8. doi:10.1021/ar50109a001
Return to citation in text: [1] -
Paymode, D. J.; Ramana, C. V. ACS Omega 2017, 2, 5591–5600. doi:10.1021/acsomega.7b00980
Return to citation in text: [1] -
Wakatsuki, Y.; Yamazaki, H. Tetrahedron Lett. 1973, 14, 3383–3384. doi:10.1016/s0040-4039(01)86920-3
Return to citation in text: [1] -
Yamamoto, Y.; Kinpara, K.; Saigoku, T.; Takagishi, H.; Okuda, S.; Nishiyama, H.; Itoh, K. J. Am. Chem. Soc. 2005, 127, 605–613. doi:10.1021/ja045694g
Return to citation in text: [1] -
Domínguez, G.; Pérez-Castells, J. Chem. – Eur. J. 2016, 22, 6720–6739. doi:10.1002/chem.201504987
Return to citation in text: [1] -
Kotha, S.; Sreevani, G. Synthesis 2018, 50, 4883–4888. doi:10.1055/s-0037-1610238
Return to citation in text: [1] -
Kotha, S.; Mohanraja, K.; Durani, S. Chem. Commun. 2000, 1909–1910. doi:10.1039/b005605g
Return to citation in text: [1] -
Kotha, S.; Brahmachary, E. Bioorg. Med. Chem. 2002, 10, 2291–2295. doi:10.1016/s0968-0896(02)00039-1
Return to citation in text: [1] -
Kotha, S.; Sreenivasachary, N. Bioorg. Med. Chem. Lett. 2000, 10, 1413–1415. doi:10.1016/s0960-894x(00)00259-6
Return to citation in text: [1] -
Paladhi, S.; Chauhan, A.; Dhara, K.; Tiwari, A. K.; Dash, J. Green Chem. 2012, 14, 2990–2995. doi:10.1039/c2gc35819k
Return to citation in text: [1] -
Kotha, S.; Brahmachary, E. Tetrahedron Lett. 1997, 38, 3561–3564. doi:10.1016/s0040-4039(97)00663-1
Return to citation in text: [1] -
Bhat, B. A.; Ponnala, S.; Sahu, D. P.; Tiwari, P.; Tripathi, B. K.; Srivastava, A. K. Bioorg. Med. Chem. 2004, 12, 5857–5864. doi:10.1016/j.bmc.2004.08.031
Return to citation in text: [1] -
Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Chem. Rev. 2013, 113, 4905–4979. doi:10.1021/cr200409f
Return to citation in text: [1] -
Wang, Y.-C.; Xie, Y.-Y.; Qu, H.-E.; Wang, H.-S.; Pan, Y.-M.; Huang, F.-P. J. Org. Chem. 2014, 79, 4463–4469. doi:10.1021/jo5004339
Return to citation in text: [1] -
Xie, Y.-Y.; Wang, Y.-C.; He, Y.; Hu, D.-C.; Wang, H.-S.; Pan, Y.-M. Green Chem. 2017, 19, 656–659. doi:10.1039/c6gc01553k
Return to citation in text: [1] -
Kotha, S.; Goyal, D.; Bitra, A.; Thota, N.; Kruger, G.; Anand, R. RSC Adv. 2013, 3, 24447–24454. doi:10.1039/c3ra44318c
Return to citation in text: [1] [2] -
D'yakonov, V. A.; Tuktarova, R. A.; Dzhemileva, L. U.; Ishmukhametova, S. R.; Yunusbaeva, M. M.; Dzhemilev, U. M. Steroids 2018, 138, 6–13. doi:10.1016/j.steroids.2018.06.004
Return to citation in text: [1] -
Dzhemilev, U. M.; D’yakonov, V. A.; Tuktarova, R. A.; Dzhemileva, L. U.; Ishmukhametova, S. R.; Yunusbaeva, M. M.; de Meijere, A. J. Nat. Prod. 2016, 79, 2039–2044. doi:10.1021/acs.jnatprod.6b00335
Return to citation in text: [1] [2] -
Du, K.; Liang, J.; Wang, Y.; Kou, J.; Qian, C.; Ji, L.; Chao, H. Dalton Trans. 2014, 43, 17303–17316. doi:10.1039/c4dt02142h
Return to citation in text: [1] -
D'yakonov, V. A.; Kadikova, G. N.; Dzhemileva, L. U.; Gazizullina, G. F.; Yunusbaeva, M. M.; Dzhemilev, U. M. Tetrahedron 2018, 74, 4071–4077. doi:10.1016/j.tet.2018.06.008
Return to citation in text: [1]
| 59. | D'yakonov, V. A.; Tuktarova, R. A.; Dzhemileva, L. U.; Ishmukhametova, S. R.; Yunusbaeva, M. M.; Dzhemilev, U. M. Steroids 2018, 138, 6–13. doi:10.1016/j.steroids.2018.06.004 |
| 55. | Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Chem. Rev. 2013, 113, 4905–4979. doi:10.1021/cr200409f |
| 56. | Wang, Y.-C.; Xie, Y.-Y.; Qu, H.-E.; Wang, H.-S.; Pan, Y.-M.; Huang, F.-P. J. Org. Chem. 2014, 79, 4463–4469. doi:10.1021/jo5004339 |
| 57. | Xie, Y.-Y.; Wang, Y.-C.; He, Y.; Hu, D.-C.; Wang, H.-S.; Pan, Y.-M. Green Chem. 2017, 19, 656–659. doi:10.1039/c6gc01553k |
| 58. | Kotha, S.; Goyal, D.; Bitra, A.; Thota, N.; Kruger, G.; Anand, R. RSC Adv. 2013, 3, 24447–24454. doi:10.1039/c3ra44318c |
| 58. | Kotha, S.; Goyal, D.; Bitra, A.; Thota, N.; Kruger, G.; Anand, R. RSC Adv. 2013, 3, 24447–24454. doi:10.1039/c3ra44318c |
| 1. | Saini, M. S.; Kumar, A.; Dwivedi, J.; Singh, R. Int. J. Pharma Sci. Res. 2013, 4, 66–77. |
| 2. | Arora, P.; Arora, V.; Lamba, H. S.; Wadhwa, D. Int. J. Pharm. Sci. Res. 2012, 3, 2947–2954. doi:10.13040/ijpsr.0975-8232.3(9).2947-54 |
| 3. | Visentini, P. Farmaco, Ed. Sci. 1954, 9, 274–277. |
| 4. | Thompson, L. A.; Ellman, J. A. Chem. Rev. 1996, 96, 555–600. doi:10.1021/cr9402081 |
| 8. | Rekha, S.; Shantharam, U.; Chandy, V. Int. Res. J. Pharm. 2011, 2, 81–84. |
| 9. | Youssef, A. M.; White, M. S.; Villanueva, E. B.; El-Ashmawy, I. M.; Klegeris, A. Bioorg. Med. Chem. 2010, 18, 2019–2028. doi:10.1016/j.bmc.2010.01.021 |
| 53. | Kotha, S.; Brahmachary, E. Tetrahedron Lett. 1997, 38, 3561–3564. doi:10.1016/s0040-4039(97)00663-1 |
| 6. | Bruno, G.; Costantino, L.; Curinga, C.; Maccari, R.; Monforte, F.; Nicolò, F.; Ottanà, R.; Vigorita, M. G. Bioorg. Med. Chem. 2002, 10, 1077–1084. doi:10.1016/s0968-0896(01)00366-2 |
| 7. | Suzen, S.; Buyukbingol, E. Curr. Med. Chem. 2003, 10, 1329–1352. doi:10.2174/0929867033457377 |
| 54. | Bhat, B. A.; Ponnala, S.; Sahu, D. P.; Tiwari, P.; Tripathi, B. K.; Srivastava, A. K. Bioorg. Med. Chem. 2004, 12, 5857–5864. doi:10.1016/j.bmc.2004.08.031 |
| 5. | Fujita, T.; Sugiyama, Y.; Taketomi, S.; Sohda, T.; Kawamatsu, Y.; Iwatsuka, H.; Suzuoki, Z. Diabetes 1983, 32, 804–810. doi:10.2337/diab.32.9.804 |
| 29. | Okamoto, S.; Sugiyama, Y.-k. Synlett 2013, 24, 1044–1060. doi:10.1055/s-0032-1316913 |
| 30. | Shibata, Y.; Tanaka, K. Synthesis 2012, 44, 323–350. doi:10.1055/s-0031-1289665 |
| 31. | Kotha, S.; Brahmachary, E.; Lahiri, K. Eur. J. Org. Chem. 2005, 4741–4767. doi:10.1002/ejoc.200500411 |
| 32. | Galan, B. R.; Rovis, T. Angew. Chem., Int. Ed. 2009, 48, 2830–2834. doi:10.1002/anie.200804651 |
| 33. | Chopade, P. R.; Louie, J. Adv. Synth. Catal. 2006, 348, 2307–2327. doi:10.1002/adsc.200600325 |
| 34. | Domínguez, G.; Pérez-Castells, J. Chem. Soc. Rev. 2011, 40, 3430–3444. doi:10.1039/c1cs15029d |
| 35. | Gandon, V.; Aubert, C.; Malacria, M. Chem. Commun. 2006, 2209–2217. doi:10.1039/b517696b |
| 36. | Varela, J. A.; Saá, C. Chem. Rev. 2003, 103, 3787–3802. doi:10.1021/cr030677f |
| 37. | Yamamoto, Y. Curr. Org. Chem. 2005, 9, 503–519. doi:10.2174/1385272053544399 |
| 38. | Agenet, N.; Buisine, O.; Slowinski, F.; Gandon, V.; Aubert, C.; Malacria, M. In Cotrimerizations of Acetylenic Compounds; Rajan Babu, T. V., Ed.; Organic Reactions, Vol. 68; John Wiley and Sons: Hoboken, New Jersey; pp 1–302. doi:10.1002/0471264180.or068.01 |
| 39. | Heller, B.; Hapke, M. Chem. Soc. Rev. 2007, 36, 1085–1094. doi:10.1039/b607877j |
| 40. | Inglesby, P. A.; Evans, P. A. Chem. Soc. Rev. 2010, 39, 2791–2805. doi:10.1039/b913110h |
| 41. | Tanaka, K. Transition Metal-Mediated Aromatic Ring Construction. In Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds; Mortier, J., Ed.; John Wiley and Sons: Hoboken, New Jersey, 2016; pp 587–614. doi:10.1002/9781118754887.ch21 |
| 42. | Weding, N.; Hapke, M. Chem. Soc. Rev. 2011, 40, 4525–4538. doi:10.1039/c0cs00189a |
| 43. | Vollhardt, K. P. C. Acc. Chem. Res. 1977, 10, 1–8. doi:10.1021/ar50109a001 |
| 44. | Paymode, D. J.; Ramana, C. V. ACS Omega 2017, 2, 5591–5600. doi:10.1021/acsomega.7b00980 |
| 45. | Wakatsuki, Y.; Yamazaki, H. Tetrahedron Lett. 1973, 14, 3383–3384. doi:10.1016/s0040-4039(01)86920-3 |
| 46. | Yamamoto, Y.; Kinpara, K.; Saigoku, T.; Takagishi, H.; Okuda, S.; Nishiyama, H.; Itoh, K. J. Am. Chem. Soc. 2005, 127, 605–613. doi:10.1021/ja045694g |
| 47. | Domínguez, G.; Pérez-Castells, J. Chem. – Eur. J. 2016, 22, 6720–6739. doi:10.1002/chem.201504987 |
| 48. | Kotha, S.; Sreevani, G. Synthesis 2018, 50, 4883–4888. doi:10.1055/s-0037-1610238 |
| 49. | Kotha, S.; Mohanraja, K.; Durani, S. Chem. Commun. 2000, 1909–1910. doi:10.1039/b005605g |
| 50. | Kotha, S.; Brahmachary, E. Bioorg. Med. Chem. 2002, 10, 2291–2295. doi:10.1016/s0968-0896(02)00039-1 |
| 51. | Kotha, S.; Sreenivasachary, N. Bioorg. Med. Chem. Lett. 2000, 10, 1413–1415. doi:10.1016/s0960-894x(00)00259-6 |
| 52. | Paladhi, S.; Chauhan, A.; Dhara, K.; Tiwari, A. K.; Dash, J. Green Chem. 2012, 14, 2990–2995. doi:10.1039/c2gc35819k |
| 19. | Jain, V. S.; Vora, D. K.; Ramaa, C. S. Bioorg. Med. Chem. 2013, 21, 1599–1620. doi:10.1016/j.bmc.2013.01.029 |
| 20. | Singh, S. P.; Parmar, S. S.; Raman, K.; Stenberg, V. I. Chem. Rev. 1981, 81, 175–203. doi:10.1021/cr00042a003 |
| 21. | Napoleon, A. A. Int. J. PharmTech Res. 2016, 9, 429–443. |
| 22. | Chadha, N.; Bahia, M. S.; Kaur, M.; Silakari, O. Bioorg. Med. Chem. 2015, 23, 2953–2974. doi:10.1016/j.bmc.2015.03.071 |
| 23. | Sohda, T.; Mizuno, K.; Tawada, H.; Sugiyama, Y.; Fujita, T.; Kawamatsu, Y. Chem. Pharm. Bull. 1982, 30, 3563–3573. doi:10.1248/cpb.30.3563 |
| 24. | Lehmann, J. M.; Moore, L. B.; Smith-Oliver, T. A.; Wilkison, W. O.; Willson, T. M.; Kliewer, S. A. J. Biol. Chem. 1995, 270, 12953–12956. doi:10.1074/jbc.270.22.12953 |
| 60. | Dzhemilev, U. M.; D’yakonov, V. A.; Tuktarova, R. A.; Dzhemileva, L. U.; Ishmukhametova, S. R.; Yunusbaeva, M. M.; de Meijere, A. J. Nat. Prod. 2016, 79, 2039–2044. doi:10.1021/acs.jnatprod.6b00335 |
| 14. | Mulwad, V. V.; Mir, A. A.; Parmar, H. T. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 2009, 48, 137–141. doi:10.1002/chin.200925122 |
| 15. | Sharma, R.; Nagda, D. P.; Talesara, G. L. ARKIVOC 2006, No. i, 1–12. doi:10.3998/ark.5550190.0007.101 |
| 16. | Ceylan, M. Turk. J. Chem. 2006, 30, 355–360. |
| 17. | Chavan, A. A.; Pai, N. R. ARKIVOC 2007, No. xvi, 148–155. doi:10.3998/ark.5550190.0008.g16 |
| 18. | Albrecht, U.; Gördes, D.; Schmidt, E.; Thurow, K.; Lalk, M.; Langer, P. Bioorg. Med. Chem. 2005, 13, 4402–4407. doi:10.1016/j.bmc.2005.04.046 |
| 26. | Jones, E. R. H.; Robinson, F. A.; Strachan, M. N. J. Chem. Soc. 1946, 91–92. doi:10.1039/jr9460000091 |
| 27. | Bozdag, O.; Ertan, R. Turk. J. Chem. 1999, 23, 163–169. |
| 28. | Dhara, K.; Paladhi, S.; Midya, G. C.; Dash, J. Org. Biomol. Chem. 2011, 9, 3801–3807. doi:10.1039/c0ob01248c |
| 11. | Ip, M. M.; Sylvester, P. W.; Schenkel, L. Cancer Res. 1986, 46, 1735–1740. |
| 12. | Shimazaki, N.; Togashi, N.; Hanai, M.; Isoyama, T.; Wada, K.; Fujita, T.; Fujiwara, K.; Kurakata, S. Eur. J. Cancer 2008, 44, 1734–1743. doi:10.1016/j.ejca.2008.04.016 |
| 13. | Subtel’na, I.; Atamanyuk, D.; Szymańska, E.; Kieć-Kononowicz, K.; Zimenkovsky, B.; Vasylenko, O.; Gzella, A.; Lesyk, R. Bioorg. Med. Chem. 2010, 18, 5090–5102. doi:10.1016/j.bmc.2010.05.073 |
| 60. | Dzhemilev, U. M.; D’yakonov, V. A.; Tuktarova, R. A.; Dzhemileva, L. U.; Ishmukhametova, S. R.; Yunusbaeva, M. M.; de Meijere, A. J. Nat. Prod. 2016, 79, 2039–2044. doi:10.1021/acs.jnatprod.6b00335 |
| 61. | Du, K.; Liang, J.; Wang, Y.; Kou, J.; Qian, C.; Ji, L.; Chao, H. Dalton Trans. 2014, 43, 17303–17316. doi:10.1039/c4dt02142h |
| 10. | da Rocha Junior, L. F.; de Melo Rêgo, M. J. B.; Cavalcanti, M. B.; Pereira, M. C.; Pitta, M. G. d. R.; de Oliveira, P. S. S.; Gonçalves, S. M. C.; Duarte, A. L. B. P.; de Lima, M. d. C. A.; Pitta, I. d. R.; Pitta, M. G. d. R. BioMed Res. Int. 2013, No. 926060. doi:10.1155/2013/926060 |
| 23. | Sohda, T.; Mizuno, K.; Tawada, H.; Sugiyama, Y.; Fujita, T.; Kawamatsu, Y. Chem. Pharm. Bull. 1982, 30, 3563–3573. doi:10.1248/cpb.30.3563 |
| 24. | Lehmann, J. M.; Moore, L. B.; Smith-Oliver, T. A.; Wilkison, W. O.; Willson, T. M.; Kliewer, S. A. J. Biol. Chem. 1995, 270, 12953–12956. doi:10.1074/jbc.270.22.12953 |
| 62. | D'yakonov, V. A.; Kadikova, G. N.; Dzhemileva, L. U.; Gazizullina, G. F.; Yunusbaeva, M. M.; Dzhemilev, U. M. Tetrahedron 2018, 74, 4071–4077. doi:10.1016/j.tet.2018.06.008 |
© 2019 Kotha et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)