Abstract
At 0 °C in THF in the presence of Grubbs first generation catalyst, cyclobutene derivatives undergo ROMP readily, whereas norbornene derivatives remain intact. When the substrate contains both cyclobutene and norbornene moieties, the conditions using THF as the solvent at 0 °C offer a useful protocol for the selective ROMP of cyclobutene to give norbornene-appended polycyclobutene. Unsymmetrical ladderphane having polycyclobutene and polynorbornene as two strands is obtained by further ROMP of the norbornene appended polycyclobutene in the presence of Grubbs first generation catalyst in DCM at ambient temperature. Methanolysis of this unsymmetrical ladderphane gives polycyclobutene methyl ester and insoluble polynorbornene-amide-alcohol. The latter is converted into the corresponding soluble acetate. Both polymers are well characterized by spectroscopic means. No norbornene moiety is found to be incorporated into polycyclobutene strand at all. The double bonds in the polycyclobutene strand are mainly in cis configuration (ca 70%), whereas the E/Z ratio for polynorbornene strand is 8:1.

Graphical Abstract
Introduction
Ring-opening metathesis polymerizations (ROMP) of strained cycloalkenes offer a powerful arsenal for the synthesis of polymers having a variety of fascinating properties [1-3]. To illustrate this, polynorbornenes and polycyclobutenes are readily obtained from the corresponding monomeric norbornene and cyclobutene derivatives under various conditions. Symmetrical DNA-like double stranded ladderphanes are conveniently synthesized from bisnorbornene [4-15] or from biscyclobutene [16] linked with a range of different rigid linkers. When a flexible linker is used, bisnorbornene derivatives undergo cascade metathetical cyclopolymerization giving the corresponding polynorbornenes with hammock-like pendants [17,18]. Unsymmetrical polynorbornene-based ladderphane is obtained by a replication protocol from a single stranded polynorbornene [19,20]. Alternatively, sequential polymerization of a monomer containing a norbornene moiety and other polymerizable group furnishes an unsymmetrical ladderphane having two structurally different polymeric backbones [21,22]. It seems to be not easy if both strands are arisen from different strained rings by ROMP. It is known that norbornenes having different substituents would have different reaction rates in ROMP [23]. These discrepancies in reactivity have been used for sequence control in polymer synthesis [24]. Since the first living ROMP methods for cyclobutenes were reported in 1992 [25], cyclobutene-containing block copolymers are well documented [26-34]. Alternating cyclobutene–cyclohexene copolymers have been synthesized by ROMP of the corresponding monomers [31-33]. However, to the best of our knowledge, selective ROMPs between cyclobutene and norbornene have not been reported.
The strain energies for norbornene and cyclobutene are 25 and 31 kcal/mol, respectively [35]. It is therefore envisaged that cyclobutene would react faster than norbornene under certain ROMP conditions. As such, when monomer 1 containing a cyclobutene moiety and a norbornene moiety connected by a bridge are subjected to ROMP, it would be feasible that the cyclobutene moiety would react preferentially giving the corresponding norbornene-appended polycyclobutene 2. After all cyclobutene moieties have been consumed and quenched, further ROMP of 2 under different conditions would afford unsymmetrical double-stranded ladderphane 3 having both polycyclobutene and polynorbornene as two polymeric frameworks (Scheme 1). We have tested this viewpoint and now wish to report sequential ROMP of monomers containing both cyclobutene and norbornene moieties tethered by a linker.
![[1860-5397-15-4-i1]](/bjoc/content/inline/1860-5397-15-4-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Strategy for sequential ROMP of 1 to yield 3.
Scheme 1: Strategy for sequential ROMP of 1 to yield 3.
Results and Discussion
A comparison of the reactivity of cyclobutene versus norbornene derivatives 4 and 5 in ROMP catalyzed by Grubbs I catalyst (6)
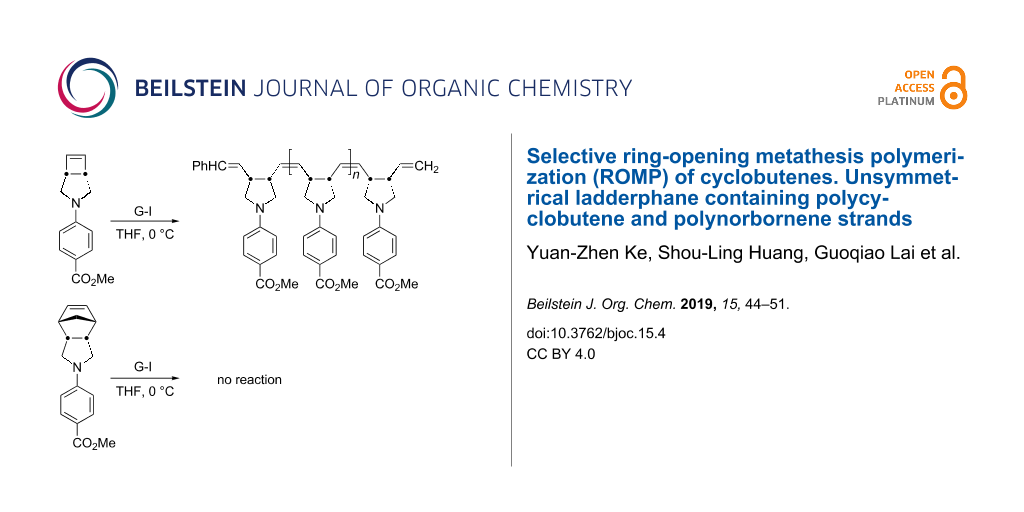
In the beginning of this study, we have examined the first order reaction kinetics of ROMPs of 4 and of 5 in the presence of 10 mol per cent of Grubbs first generation catalyst (6) [36] in DCM at 10 °C [37]. The rate constants for the reactions of 4 and 5 were 1.3 × 10−3 and 5.1 × 10−4 s−1, respectively. On the other hand, when the reaction was carried out in THF-d8 at 273 K, the second order rate constant for 4 was 2.1 × 10−3 M−1s−1, whereas norbornene derivative 5 was inert under these conditions. The details are described in the Experimental section and Supporting Information File 1 (Figures S1, S2 and S8–S10).
It has been suggested that the metathesis reaction may involve a fourteen-electron ruthenium species as the active catalyst [38-40]. This active species might be stabilized when the reaction is carried out in polar solvent having weak coordination ability such as THF [41-43]. As mentioned above, the difference in reactivity between the ROMP of 4 and 5 in THF at 0 °C would offer useful conditions to selectively react with 4 in the presence of 5. Thus, a mixture of an equal molar of 4 and 5 was treated with 10 mol % of 6 in THF-d8 at 0 °C. Only 4 was consumed to give the corresponding polymer 7, whereas 5 remained intact (Scheme 2). This promising observation prompted us to pursue the synthesis of unsymmetrical double-stranded ladderphane 8 by sequential ROMPs of 9 (Scheme 3).
![[1860-5397-15-4-i2]](/bjoc/content/inline/1860-5397-15-4-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: ROMP of 4 and 5 in THF at 0 °C in the presence of 10 mol % of 6.
Scheme 2: ROMP of 4 and 5 in THF at 0 °C in the presence of 10 mol % of 6.
![[1860-5397-15-4-i3]](/bjoc/content/inline/1860-5397-15-4-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Synthesis of monomer 9
4-Aminobutanol (11) was used to link norbornene and cyclobutene moieties via amide and ester groups. The use of such a linker is because the ester group could be selectively hydrolyzed in the presence of amides. This selectivity will be helpful for the structural elucidation of polymer 8. Thus, 10b was allowed to react with 11 to afford amide-alcohol 12 in 79% yield. Esterification of 12 with 13b furnished 70% yield of monomer 9 (Scheme 4).
![[1860-5397-15-4-i4]](/bjoc/content/inline/1860-5397-15-4-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Synthesis of unsymmetrical ladderphane 8 by sequential ROMPs catalyzed by 6
Polymerization of monomer 9 in the presence of 10 mol % of 6 was performed in THF at 0 °C for 4 h, followed by quenching with ethyl vinyl ether to give polymer 14 in 86% yield (Scheme 5). It is worth noting that no incorporation of the norbornene moiety into the polymeric backbone under these conditions was observed. The 1H NMR spectrum of 14 shows the olefinic proton signals at δ 5.49 and 6.12 ppm in 1:1 ratio. These signals were assigned to the absorptions of olefinic protons on the polymeric backbone and the olefinic proton of unreacted norbornene pendants, respectively. In the 13C NMR spectrum, the peak at δ 139 ppm owing to the olefinic carbon of cyclobutene shifts to δ 130 ppm due to ring opening, whereas the olefin carbon of the unreacted norbornene moiety at δ 136 ppm remained unchanged after first polymerization. These observations are consistent with the results of our preliminary studies that only the cyclobutene moiety, but not norbornene in 9, proceeds 6-catalyzed ROMP under these conditions. The degree of polymerization of 14 was estimated to be 10 based on the 1H NMR integration of relevant peaks.
![[1860-5397-15-4-i5]](/bjoc/content/inline/1860-5397-15-4-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of 14 and 8 by selective olefin metathesis.
Scheme 5: Synthesis of 14 and 8 by selective olefin metathesis.
We have previously found that two norbornene derivatives connected by a flexible linker 15 may undergo cascade ring-opening–ring-closing metathesis polymerization to give single-stranded hammock-like appended polynorbornenes 17 (Scheme 6) [17,18]. The linker in 8 is flexible, and, therefore, the possibility for similar intramolecular metathesis cyclopolymerization might take place to form intermediate 16 for further transformations. However, no such reaction was observed in this study. Presumably, the 6-catalyzed metathesis reactivity of cyclobutenes would be much higher than that of norbornene derivatives. Accordingly, intermolecular metathesis reaction between two cyclobutene moieties would be favored over intramolecular ring-closing metathesis between a ruthenium carbene and the norbornene moiety.
![[1860-5397-15-4-i6]](/bjoc/content/inline/1860-5397-15-4-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Cyclopolymerization of 15 with a flexible linker.
Scheme 6: Cyclopolymerization of 15 with a flexible linker.
Polymer 14 was treated with 10 mol % 6 in DCM at rt to give 8 in 95% yield. The 1H NMR spectrum of 8 shows that the relative intensity of the signals around δ 5.4 ppm was doubled, all signals due to olefinic protons in 9 and 14 being diminished.
Methanolysis of unsymmetrical ladderphane 8
In order to confirm the uniformity of the polymerization leading to the formation of unsymmetrical ladderphane 8, methanolysis of 8 with NaOMe in methanol at rt gave 7 and 18. Chloroform was then added and 18 was collected as a grayish precipitate in 56% yield. After filtration, the filtrate was worked up to afford 7 in 64% yield with a degree of polymerization of 10 (Mn = 2500, PDI = 1.11), in good agreement with those of 14 and 8. The 13C NMR spectrum of 7 shows two peaks at δ 40.6 and 45.4 ppm, attributed to the allylic carbons attached to a cis and a trans double bond [13], respectively, and the relative ratio of these two peaks is roughly 7:3. This result suggests that about 70% of the double bonds in 7 might adopt cis configuration. Moreover, no norbornene moiety was detected by NMR on the polymeric backbones in 7 (Scheme 7).
![[1860-5397-15-4-i7]](/bjoc/content/inline/1860-5397-15-4-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Methanolysis of unsymmetrical ladderphane 8.
Scheme 7: Methanolysis of unsymmetrical ladderphane 8.
Since 18 was insoluble in most organic solvents, acetylation of 18 with excess acetic anhydride and pyridine at 70 °C for 10 h gave the corresponding acetate 19, which had good solubility in DCM or chloroform. GPC analysis showed that the degree of polymerization of 19 (DP = 10, PDI = 1.24) was again comparable with that of the corresponding ladderphane 8, polycyclobutene 7 and 14.
The 1H NMR spectrum of 19 shows peaks at δ 5.6 and 5.3 ppm attributed to trans and cis olefinic protons, respectively, in a ratio of 8 to 1. It is well documented that 6-catalyzed ROMP of N-arylpyrrolidene appended norbornene gives polynorbornene with all double bonds in trans configuration [44-46]. The existence of both Z- and E-double bonds in the parent polycyclobutene backbone in 14 may influence the stereoselectivity of the polynorbornene strand in 7 during the course of ROMP.
Conclusion
In summary, we have demonstrated useful ROMP conditions to selectively transform cyclobutene derivatives into the corresponding polycyclobutenes in THF at 0 °C, whereas the corresponding norbornene skeleton appears to be unreactive under these conditions. This protocol has been used for the selective synthesis of unsymmetrical ladderphane having polycyclobutene in one strand and polynorbornene in the other. Further applications of this selectivity to other systems are in progress in our laboratory.
Experimental
General
Unless otherwise specified, all commercially available starting materials were used without further purification. All air and moisture-sensitive reactions were carried out under an atmosphere of dry nitrogen in a glove box. All 1H and 13C NMR spectra were recorded on a Varian 400 Unity Plus NMR spectrometer using CDCl3 as solvent at ambient temperature. Chemical shifts were expressed in parts per million using residual solvent protons as internal standards (1H: chloroform: 7.26 ppm). Gel permeation chromatography (GPC) was performed on a Waters GPC instrument equipped with Waters 1515 HPLC pump using Waters 2487 absorbance detector. Polymer (approximately 0.5 mg) in THF (0.1 mL) was filtered through a 0.5-micron filter and 20 μL of the sample was injected into Shodex KF-G, Styragel HR2, Styragel HR3 and Styragel HR4 column (7.8 × 300 mm) with oven temperature at 40 °C using standard polystyrene samples (1.84 × 105 to 996 Da) for calibration. THF was used as eluent (flow rate 1.0 mL/min).
Synthesis of 12. Under N2 atomosphere, to 10a (560 mg, 2.2 mmol) in DCM (20 mL) was added oxalyl chloride (0.4 mL, 4.3 mmol) at 0 °C. The mixture was gradually warmed to rt and then stirred for 1 h. The solvent was removed in vacuo to give the crude acyl chloride 10b, to which was added DCM (15 mL), DMAP (60 mg, 0.5 mmol) and Et3N (2.0 mL, 15 mmol). 4-Amino-1-butanol (11, 178 mg, 2.0 mmol) was then added slowly at 0 °C. After stirring for 8 h at rt, the mixture was poured into H2O (50 mL) and DCM (50 mL). The organic layer was separated, washed with brine (100 mL) and dried (MgSO4). The solvent was removed in vacuo and the residue was chromatographed on silica gel (DCM/MeOH 20:1) to afford 12 (515 mg, 79%). mp 207–209 °C; IR (KBr): ν 3455, 3306, 3056, 2940, 2867, 1606, 1554, 1514, 1473, 1379, 1309, 1199, 1130, 1047, 969, 826, 768, 733, 683 cm−1; 1H NMR (400 MHz) δ 1.52 (d, J = 8.4 Hz, 1H), 1.61–1.70 (m, 6H), 2.92–2.99 (m, 4H), 2.98–2.99 (m, 2H), 3.09 (m, 2H), 3.25–3.30 (m, 2H), 3.47–3.48 (m, 2H), 3.71 (t, J = 5.6 Hz, 2H), 6.15–6.16 (m, 3H), 6.39 (d, J = 9.0 Hz, 2H), 7.61 (d, J =9.0 Hz, 2H); 13C NMR (100 MHz): δ 26.8, 30.1, 39.8, 45.6, 46.8, 50.6, 52.2, 62.3, 110.7, 120.1, 127.9, 135.3, 148.9, 167.1; HRMS (FAB, m/z): calcd for C20H26N2O2, 326.1994; found, 326.1997.
Synthesis of 9. Under N2 atomosphere, to 13a (321 mg, 1.4 mmol) in DCM (20 mL) was added oxalyl chloride (0.4 mL, 4.3 mmol) at 0 °C. The mixture was gradually warmed to rt and then stirred for 1 h. The solvent was removed in vacuo to give the crude acyl chloride 13b, to which was added DCM (15 mL), DMAP (60 mg, 0.5 mmol) and Et3N (2.0 mL, 15 mmol). Compound 12 (522 mg, 1.6 mmol) was then added slowly at 0 °C. After stirring for 8 h at rt, the mixture was poured into H2O (50 mL) and DCM (50 mL). The organic layer was separated, washed with saturated brine (100 mL) and dried (MgSO4). The solvent was removed in vacuo and the residue was chromatographed on silica gel (DCM/MeOH 20:1) to afford 9 (512 mg, 70%). mp 238–240 °C; IR (KBr): ν 3333, 3051, 2949, 2843, 1699, 1606, 1547, 1511, 1473, 1376, 1274, 1216, 1180, 1106, 1050, 963, 828, 769, 740 cm−1; 1H NMR (400 MHz) δ 1.51 (d, J = 8.2 Hz, 1H), 1.61 (d, J = 8.2 Hz, 1H), 1.72–1.85 (m, 4H), 2.92–2.98 (m, 6H), 3.07–3.09 (m, 2H), 3.25–3.29 (m, 2H), 3.49–3.56 (m, 4H), 3.65 (d, J = 10.0 Hz, 2H), 4.30 (t, J = 6.4 Hz, 2H), 6.03 (m, 1H), 6.13–6.15 (m, 4H), 6.38 (d, J = 8.6 Hz, 2H), 6.62 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 8.6 Hz, 2H), 7.87 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz) δ 26.4, 26.6, 39.4, 45.3, 46.4, 46.5, 48.8, 50.4, 52.0, 63.7, 110.8, 111.8, 117.5, 120.4, 128.0, 130.9, 135.5, 139.1, 149.1, 152.9, 166.6, 167.1; HRMS (FAB, m/z): calcd for C33H37N3O3, 523.2835; found, 523.2839.
Synthesis of 14. Under N2 atomosphere, to a solution of 9 (84.0 mg, 0.16 mmol) in THF (10 mL) was added 6 (12.8 mg, 0.016 mmol) in THF (1 mL) at 0 °C. After stirring at 0 °C for 4 h, ethyl vinyl ether (1.0 mL) was then added and stirring was continued at 0 °C for 2 h. The mixture was concentrated and the residual solution was added to methanol. The precipitate was collected and redissolved in DCM. Reprecipitation by adding the DCM solution to methanol afforded 14 as a grayish powder (74.8 mg, 89%). IR (KBr): ν 3350, 3054, 2954, 2847, 1695, 1605, 1512, 1476, 1381, 1275, 1179, 1107, 967, 827, 768, 733, 698 cm−1; 1H NMR (400 MHz) δ 1.51–1.72 (m, 6H), 2.92–3.48 (m, 16H), 4.26 (br, 2H), 5.49 (m, 2H), 6.12 (br, 2H), 6.36 (m, 5H), 7.63 (br, 2H), 7.86 (br, 2H); degree of polymerization (DP) analysis: δ 7.86/δ 5.07 = 10, indicating a DP of 10; 13C NMR (100 MHz) δ 26.6, 39.6, 40.9, 45.5, 46.6, 50.5, 52.1, 52.9, 64.0, 110.5, 110.9, 117.0, 120.5, 128.2, 129.8, 131.3, 135.6, 149.2, 150.2, 166.8, 167.4.
Synthesis of 8. Under N2 atomosphere, to a solution of 14 (62.8 mg, 0.12 mmol) in DCM (40 mL) was added 6 (9.6 mg, 0.012 mmol) in DCM (5 mL). After stirring at rt for 4 h, ethyl vinyl ether (0.5 mL) was then added and stirring was continued for 30 min. The mixture was concentrated and the residual solution was added to methanol. The precipitate was collected and redissolved in DCM. Reprecipitation by adding the DCM solution to methanol afforded 8 as a grayish powder (59.7 mg, 95%). IR (KBr): ν 3373, 3054, 2929, 2849, 1694, 1605, 1512, 1478, 1381, 1274, 1179, 1106, 966, 827, 767, 733, 697 cm−1; 1H NMR (400 MHz) δ 1.47 (br, 1H), 1.82 (m, 5H), 2.88–3.49 (m, 16H), 4.27 (br, 2H), 5.47 (m, 4H), 6.49 (m, 5H), 7.67–7.89 (m, 4H); DP analysis: δ 4.27/δ 5.05 = 11, indicating a DP of 11. 13C NMR (100MHz) δ 26.6, 40.0, 46.1, 49.7, 53.2, 63.7, 110.6, 111.8, 116.9, 121.8, 126.0, 128.5, 131.3, 136.5, 138.7, 150.1, 166.7, 167.5.
Synthesis of 7 and 18. To a solution of 8 (52 mg, 0.1 mmol [calculated based on the molecular weight of the monomeric unit]) in DCM (20 mL) was added 30% NaOMe in methanol (6 mL). The mixture was stirred at 50 °C for 20 h and cooled to rt. The insoluble solid residue was collected and dried to give crude 18 as a grayish solid (18 mg, 56%). After filtration, the filtrate was washed with water and dried (MgSO4). The mixture was concentrated and the residual solution was added to methanol. The precipitate was collected and redissolved in DCM. Reprecipitation by adding the DCM solution to methanol afforded 7 as a grayish powder (21 mg, 64%). IR (KBr): ν 3066, 2951, 2862, 1702, 1605, 1524, 1478, 1434, 1383, 1281, 1180, 1108, 970, 828, 769, 698, 507 cm−1; 1H NMR (400 MHz) δ 3.02–3.49 (m, 6H), 3.86 (br, 3H), 5.49 (m, 2H), 6.43 (br, 2H), 7.87 (br, 2H), DP analysis by integration of peaks at δ 6.43/δ 5.06 = 10, indicating a DP of 10. 13C NMR (100 MHz) δ 40.8, 45.8, 51.6, 52.7, 110.5, 117.1, 128.4, 129.7, 131.3, 150.2, 167.2. GPC: Mn = 2500, Mw = 2800 , PDI = 1.11.
Synthesis of 19. A mixture of crude 18 (16 mg, 0.05 mmol), obtained from the above experiment, in Ac2O (0.5 mL) and pyridine (5 mL) was stirred at 70 °C for 10 h. The solvent was concentrated and the residue was dissolved in CHCl3 (15 mL) and washed first with diluted HCl (pH 3) and then with water. The organic solvent was concentrated and the residual solution was added to methanol. The precipitate was collected and redissolved in CHCl3. Reprecipitation by adding the CHCl3 solution to methanol afforded 19 as a grayish powder (12 mg, 63%). 1H NMR (400 MHz) δ 1.73 (br, 6H) 2.05 (s, 3H), 2.73–3.62 (m, 10H), 4.07 (br, 2H), 5.50 (m, 2H), 6.48 (br, 2H), 7.73 (br, 2H), DP δ 5.50/δ 5.05 = 10, indicating a DP of 10. 13C NMR (100 MHz) δ 21.1, 28.0, 39.7, 45.0, 46.5, 50.8, 64.3, 112.2, 121.9, 128.5, 131.8, 132.0, 150.5, 168.1, 171.6.
General procedure for kinetic measurements
Monomer 4 or 5 (0.03 mmol) was dissolved in DCM-d2 or THF-d8 (0.5 mL) and was syringed into an NMR tube inside a glove-box under nitrogen atmosphere. The NMR tube was then covered with a standard tube cap and placed in the NMR spectrometer. The tube was left to equilibrate at the desired temperature and all parameters were adjusted. A solution of 6 (24 mg in 1.0 mL of the same solvent) was prepared under nitrogen atmosphere prior to the reaction. Catalyst 6 (10 mol %) was syringed into the NMR tube which was immediately put in the NMR probe again. The reaction was monitored by the decrease of the peak intensity for H-2 using the peaks for H-1 and H-1’ as the internal reference (Supporting Information File 1, Figures S8–S10). The spectra were recorded every ten to twenty minutes interval depending on the reaction (Figures S8–S10). The rate constants were thus obtained (Figures S1 and S2).
Supporting Information
| Supporting Information File 1: 1H and 13C NMR spectra of both monomers and polymers, as well as GPC and kinetic investigation results. | ||
| Format: PDF | Size: 1.0 MB | Download |
References
-
Buchmeiser, M. R. Chapter 19. In Synthesis of Polymers; Schlüter, A. D.; Hawker, C. J.; Sakamoto, J., Eds.; Wiley-VCH, 2012.
Return to citation in text: [1] -
Buchmeiser, M. R. Chem. Rev. 2000, 100, 1565–1604. doi:10.1021/cr990248a
Return to citation in text: [1] -
Grubbs., R. H.; Khosravi, E., Eds. Handbook of Metathesis. Polymer Synthesis, 2nd ed.; Wiley-VCH, 2015; Vol. 3.
Return to citation in text: [1] -
Luh, T.-Y. Acc. Chem. Res. 2013, 46, 378–389. doi:10.1021/ar300170b
Return to citation in text: [1] -
Luh, T.-Y.; Ding, L. Tetrahedron 2017, 73, 6487–6513. doi:10.1016/j.tet.2017.09.029
Return to citation in text: [1] -
Yang, H.-C.; Lin, S.-Y.; Yang, H.-C.; Lin, C.-L.; Tsai, L.; Huang, S.-L.; Chen, I. W.-P.; Chen, C.-h.; Jin, B.-Y.; Luh, T.-Y. Angew. Chem., Int. Ed. 2006, 45, 726–730. doi:10.1002/anie.200503406
Return to citation in text: [1] -
Yang, H.-C.; Lee, S.-L.; Chen, C.-h.; Lin, N.-T.; Yang, H.-C.; Jin, B.-Y.; Luh, T.-Y. Chem. Commun. 2008, 6158–6160. doi:10.1039/b814672a
Return to citation in text: [1] -
Chou, C.-M.; Lee, S.-L.; Chen, C.-H.; Biju, A. T.; Wang, H.-W.; Wu, Y.-L.; Zhang, G.-F.; Yang, K.-W.; Lim, T.-S.; Huang, M.-J.; Tsai, P.-Y.; Lin, K.-C.; Huang, S.-L.; Chen, C.-h.; Luh, T.-Y. J. Am. Chem. Soc. 2009, 131, 12579–12585. doi:10.1021/ja9035362
Return to citation in text: [1] -
Yang, K.-W.; Xu, J.; Chen, C.-H.; Huang, H.-H.; Yu, T. J.-Y.; Lim, T.-S.; Chen, C.-h.; Luh, T.-Y. Macromolecules 2010, 43, 5188–5194. doi:10.1021/ma100550q
Return to citation in text: [1] -
Chen, C.-W.; Chang, H.-Y.; Lee, S.-L.; Hsu, I.-J.; Lee, J.-J.; Chen, C.-h.; Luh, T.-Y. Macromolecules 2010, 43, 8741–8746. doi:10.1021/ma101956n
Return to citation in text: [1] -
Wang, H.-W.; Chen, C.-H.; Lim, T.-S.; Huang, S.-L.; Luh, T.-Y. Chem. – Asian J. 2011, 6, 524–533. doi:10.1002/asia.201000492
Return to citation in text: [1] -
Huang, H.-H.; Chao, C.-G.; Lee, S.-L.; Wu, H.-J.; Chen, C.-h.; Luh, T.-Y. Org. Biomol. Chem. 2012, 10, 5948–5953. doi:10.1039/c2ob25114k
Return to citation in text: [1] -
Yeh, N.-H.; Chen, C.-W.; Lee, S.-L.; Wu, H.-J.; Chen, C.-h.; Luh, T.-Y. Macromolecules 2012, 45, 2662–2667. doi:10.1021/ma300027k
Return to citation in text: [1] [2] -
Xu, J.; Zhang, Z.; Liu, Y.-H.; Guo, Q.; Wang, G.-W.; Lai, G.; Luh, T.-Y. J. Polym. Sci., Part A: Polym. Chem. 2017, 55, 2999–3010. doi:10.1002/pola.28572
Return to citation in text: [1] -
Zhu, L.; Flook, M. M.; Lee, S.-L.; Chan, L.-W.; Huang, S.-L.; Chiu, C.-W.; Chen, C.-H.; Schrock, R. R.; Luh, T.-Y. Macromolecules 2012, 45, 8166–8171. doi:10.1021/ma301686f
Return to citation in text: [1] -
Chen, C.-H.; Satyanarayana, K.; Liu, Y.-H.; Huang, S.-L.; Lim, T.-S.; Luh, T.-Y. Chem. – Eur. J. 2015, 21, 800–807. doi:10.1002/chem.201403806
Return to citation in text: [1] -
Zhu, L.; Lin, N.-T.; Xie, Z.-Y.; Lee, S.-L.; Huang, S.-L.; Yang, J.-H.; Lee, Y.-D.; Chen, C.-h.; Chen, C.-H.; Luh, T.-Y. Macromolecules 2013, 46, 656–663. doi:10.1021/ma302293q
Return to citation in text: [1] [2] -
Lin, N.-T.; Xie, C.-Y.; Huang, S.-L.; Chen, C.-H.; Luh, T.-Y. Chem. – Asian J. 2013, 8, 1436–1440. doi:10.1002/asia.201300222
Return to citation in text: [1] [2] -
Lin, N.-T.; Lin, S.-Y.; Lee, S.-L.; Chen, C.-h.; Hsu, C.-H.; Hwang, L. P.; Xie, Z.-Y.; Chen, C.-H.; Huang, S.-L.; Luh, T.-Y. Angew. Chem., Int. Ed. 2007, 46, 4481–4485. doi:10.1002/anie.200700472
Return to citation in text: [1] -
Lai, G.; Luh, T.-Y. Bull. Chem. Soc. Jpn. 2018, 91, 262–273. doi:10.1246/bcsj.20170354
Return to citation in text: [1] -
Ke, Y.-Z.; Lee, S.-L.; Chen, C.-h.; Luh, T.-Y. Chem. – Asian J. 2011, 6, 1748–1751. doi:10.1002/asia.201000877
Return to citation in text: [1] -
Ke, Y.-Z.; Ji, R.-J.; Wei, T.-C.; Lee, S.-L.; Huang, S.-L.; Huang, M.-J.; Chen, C.-h.; Luh, T.-Y. Macromolecules 2013, 46, 6712–6722. doi:10.1021/ma4012363
Return to citation in text: [1] -
Moatsou, D.; Hansell, C. F.; O'Reilly, R. K. Chem. Sci. 2014, 5, 2246–2250. doi:10.1039/c4sc00752b
Return to citation in text: [1] -
Lutz, J.-F., Ed. Sequence-Controlled Polymers; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2018. doi:10.1002/9783527806096
Return to citation in text: [1] -
Wu, Z.; Wheeler, D. R.; Grubbs, R. H. J. Am. Chem. Soc. 1992, 114, 146–151. doi:10.1021/ja00027a021
Return to citation in text: [1] -
Wu, Z.; Grubbs, R. H. Macromolecules 1994, 27, 6700–6703. doi:10.1021/ma00101a002
Return to citation in text: [1] -
Perrott, M. G.; Novak, B. M. Macromolecules 1995, 28, 3492–3494. doi:10.1021/ma00113a062
Return to citation in text: [1] -
Perrott, M. G.; Novak, B. M. Macromolecules 1996, 29, 1817–1823. doi:10.1021/ma951516j
Return to citation in text: [1] -
Maughon, B. R.; Grubbs, R. H. Macromolecules 1997, 30, 3459–3469. doi:10.1021/ma961780s
Return to citation in text: [1] -
Charvet, R.; Novak, B. M. Macromolecules 2001, 34, 7680–7685. doi:10.1021/ma0109875
Return to citation in text: [1] -
Lee, J. C.; Parker, K. A.; Sampson, N. S. J. Am. Chem. Soc. 2006, 128, 4578–4579. doi:10.1021/ja058801v
Return to citation in text: [1] [2] -
Song, A.; Parker, K. A.; Sampson, N. S. J. Am. Chem. Soc. 2009, 131, 3444–3445. doi:10.1021/ja809661k
Return to citation in text: [1] [2] -
Parker, K. A.; Sampson, N. S. Acc. Chem. Res. 2016, 49, 408–417. doi:10.1021/acs.accounts.5b00490
Return to citation in text: [1] [2] -
Lin, N.-T.; Ke, Y.-Z.; Satyanarayana, K.; Huang, S.-L.; Lan, Y.-K.; Yang, H.-C.; Luh, T.-Y. Macromolecules 2013, 46, 7173–7179. doi:10.1021/ma401007b
Return to citation in text: [1] -
Greenberg, A.; Liebman, J. F. Strained organic molecules; Academic Press: New York, 1978.
Return to citation in text: [1] -
Schwab, P.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1996, 118, 100–110. doi:10.1021/ja952676d
Return to citation in text: [1] -
Rule, J. D.; Moore, J. S. Macromolecules 2002, 35, 7878–7882. doi:10.1021/ma0209489
Return to citation in text: [1] -
Wenzel, A. G.; Grubbs, R. H. J. Am. Chem. Soc. 2006, 128, 16048–16049. doi:10.1021/ja0666598
Return to citation in text: [1] -
Beligny, S.; Blechert, S. In N-Heterocyclic Carbenes in Synthesis; Nolan, S. P., Ed.; Wiley-VCH: Weinheim, 2006.
Return to citation in text: [1] -
van der Eide, E. F.; Piers, W. E. Nat. Chem. 2010, 2, 571–576. doi:10.1038/nchem.653
Return to citation in text: [1] -
Cazalis, C.; Héroguez, V.; Fontanille, M. Macromol. Chem. Phys. 2000, 201, 869–876. doi:10.1002/(sici)1521-3935(20000501)201:8<869::aid-macp869>3.3.co;2-q
Return to citation in text: [1] -
Al Samak, B.; Amir-Ebrahimi, V.; Corry, D. G.; Hamilton, J. G.; Rigby, S.; Rooney, J. J.; Thompson, J. M. J. Mol. Catal. A: Chem. 2000, 160, 13–21. doi:10.1016/s1381-1169(00)00228-4
Return to citation in text: [1] -
Matos, J. M. E.; Lima-Neto, B. S. J. Mol. Catal. A: Chem. 2005, 240, 233–238. doi:10.1016/j.molcata.2005.07.003
Return to citation in text: [1] -
Sattigeri, J. A.; Shiau, C.-W.; Hsu, C. C.; Yeh, F.-F.; Liou, S.; Jin, B.-Y.; Luh, T.-Y. J. Am. Chem. Soc. 1999, 121, 1607–1608. doi:10.1021/ja983433z
Return to citation in text: [1] -
Lin, W.-Y.; Murugesh, M. G.; Sudhakar, S.; Yang, H.-C.; Tai, H.-C.; Chang, C.-S.; Liu, Y.-H.; Wang, Y.; Chen, I.-W. P.; Chen, C.-h.; Luh, T.-Y. Chem. – Eur. J. 2006, 12, 324–330. doi:10.1002/chem.200500770
Return to citation in text: [1] -
Lin, W.-Y.; Wang, H.-W.; Liu, Z.-C.; Xu, J.; Chen, C.-W.; Yang, Y.-C.; Huang, S.-L.; Yang, H.-C.; Luh, T.-Y. Chem. – Asian J. 2007, 2, 764–774. doi:10.1002/asia.200700011
Return to citation in text: [1]
| 44. | Sattigeri, J. A.; Shiau, C.-W.; Hsu, C. C.; Yeh, F.-F.; Liou, S.; Jin, B.-Y.; Luh, T.-Y. J. Am. Chem. Soc. 1999, 121, 1607–1608. doi:10.1021/ja983433z |
| 45. | Lin, W.-Y.; Murugesh, M. G.; Sudhakar, S.; Yang, H.-C.; Tai, H.-C.; Chang, C.-S.; Liu, Y.-H.; Wang, Y.; Chen, I.-W. P.; Chen, C.-h.; Luh, T.-Y. Chem. – Eur. J. 2006, 12, 324–330. doi:10.1002/chem.200500770 |
| 46. | Lin, W.-Y.; Wang, H.-W.; Liu, Z.-C.; Xu, J.; Chen, C.-W.; Yang, Y.-C.; Huang, S.-L.; Yang, H.-C.; Luh, T.-Y. Chem. – Asian J. 2007, 2, 764–774. doi:10.1002/asia.200700011 |
| 17. | Zhu, L.; Lin, N.-T.; Xie, Z.-Y.; Lee, S.-L.; Huang, S.-L.; Yang, J.-H.; Lee, Y.-D.; Chen, C.-h.; Chen, C.-H.; Luh, T.-Y. Macromolecules 2013, 46, 656–663. doi:10.1021/ma302293q |
| 18. | Lin, N.-T.; Xie, C.-Y.; Huang, S.-L.; Chen, C.-H.; Luh, T.-Y. Chem. – Asian J. 2013, 8, 1436–1440. doi:10.1002/asia.201300222 |
| 13. | Yeh, N.-H.; Chen, C.-W.; Lee, S.-L.; Wu, H.-J.; Chen, C.-h.; Luh, T.-Y. Macromolecules 2012, 45, 2662–2667. doi:10.1021/ma300027k |
| 1. | Buchmeiser, M. R. Chapter 19. In Synthesis of Polymers; Schlüter, A. D.; Hawker, C. J.; Sakamoto, J., Eds.; Wiley-VCH, 2012. |
| 2. | Buchmeiser, M. R. Chem. Rev. 2000, 100, 1565–1604. doi:10.1021/cr990248a |
| 3. | Grubbs., R. H.; Khosravi, E., Eds. Handbook of Metathesis. Polymer Synthesis, 2nd ed.; Wiley-VCH, 2015; Vol. 3. |
| 19. | Lin, N.-T.; Lin, S.-Y.; Lee, S.-L.; Chen, C.-h.; Hsu, C.-H.; Hwang, L. P.; Xie, Z.-Y.; Chen, C.-H.; Huang, S.-L.; Luh, T.-Y. Angew. Chem., Int. Ed. 2007, 46, 4481–4485. doi:10.1002/anie.200700472 |
| 20. | Lai, G.; Luh, T.-Y. Bull. Chem. Soc. Jpn. 2018, 91, 262–273. doi:10.1246/bcsj.20170354 |
| 38. | Wenzel, A. G.; Grubbs, R. H. J. Am. Chem. Soc. 2006, 128, 16048–16049. doi:10.1021/ja0666598 |
| 39. | Beligny, S.; Blechert, S. In N-Heterocyclic Carbenes in Synthesis; Nolan, S. P., Ed.; Wiley-VCH: Weinheim, 2006. |
| 40. | van der Eide, E. F.; Piers, W. E. Nat. Chem. 2010, 2, 571–576. doi:10.1038/nchem.653 |
| 17. | Zhu, L.; Lin, N.-T.; Xie, Z.-Y.; Lee, S.-L.; Huang, S.-L.; Yang, J.-H.; Lee, Y.-D.; Chen, C.-h.; Chen, C.-H.; Luh, T.-Y. Macromolecules 2013, 46, 656–663. doi:10.1021/ma302293q |
| 18. | Lin, N.-T.; Xie, C.-Y.; Huang, S.-L.; Chen, C.-H.; Luh, T.-Y. Chem. – Asian J. 2013, 8, 1436–1440. doi:10.1002/asia.201300222 |
| 41. | Cazalis, C.; Héroguez, V.; Fontanille, M. Macromol. Chem. Phys. 2000, 201, 869–876. doi:10.1002/(sici)1521-3935(20000501)201:8<869::aid-macp869>3.3.co;2-q |
| 42. | Al Samak, B.; Amir-Ebrahimi, V.; Corry, D. G.; Hamilton, J. G.; Rigby, S.; Rooney, J. J.; Thompson, J. M. J. Mol. Catal. A: Chem. 2000, 160, 13–21. doi:10.1016/s1381-1169(00)00228-4 |
| 43. | Matos, J. M. E.; Lima-Neto, B. S. J. Mol. Catal. A: Chem. 2005, 240, 233–238. doi:10.1016/j.molcata.2005.07.003 |
| 16. | Chen, C.-H.; Satyanarayana, K.; Liu, Y.-H.; Huang, S.-L.; Lim, T.-S.; Luh, T.-Y. Chem. – Eur. J. 2015, 21, 800–807. doi:10.1002/chem.201403806 |
| 36. | Schwab, P.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1996, 118, 100–110. doi:10.1021/ja952676d |
| 4. | Luh, T.-Y. Acc. Chem. Res. 2013, 46, 378–389. doi:10.1021/ar300170b |
| 5. | Luh, T.-Y.; Ding, L. Tetrahedron 2017, 73, 6487–6513. doi:10.1016/j.tet.2017.09.029 |
| 6. | Yang, H.-C.; Lin, S.-Y.; Yang, H.-C.; Lin, C.-L.; Tsai, L.; Huang, S.-L.; Chen, I. W.-P.; Chen, C.-h.; Jin, B.-Y.; Luh, T.-Y. Angew. Chem., Int. Ed. 2006, 45, 726–730. doi:10.1002/anie.200503406 |
| 7. | Yang, H.-C.; Lee, S.-L.; Chen, C.-h.; Lin, N.-T.; Yang, H.-C.; Jin, B.-Y.; Luh, T.-Y. Chem. Commun. 2008, 6158–6160. doi:10.1039/b814672a |
| 8. | Chou, C.-M.; Lee, S.-L.; Chen, C.-H.; Biju, A. T.; Wang, H.-W.; Wu, Y.-L.; Zhang, G.-F.; Yang, K.-W.; Lim, T.-S.; Huang, M.-J.; Tsai, P.-Y.; Lin, K.-C.; Huang, S.-L.; Chen, C.-h.; Luh, T.-Y. J. Am. Chem. Soc. 2009, 131, 12579–12585. doi:10.1021/ja9035362 |
| 9. | Yang, K.-W.; Xu, J.; Chen, C.-H.; Huang, H.-H.; Yu, T. J.-Y.; Lim, T.-S.; Chen, C.-h.; Luh, T.-Y. Macromolecules 2010, 43, 5188–5194. doi:10.1021/ma100550q |
| 10. | Chen, C.-W.; Chang, H.-Y.; Lee, S.-L.; Hsu, I.-J.; Lee, J.-J.; Chen, C.-h.; Luh, T.-Y. Macromolecules 2010, 43, 8741–8746. doi:10.1021/ma101956n |
| 11. | Wang, H.-W.; Chen, C.-H.; Lim, T.-S.; Huang, S.-L.; Luh, T.-Y. Chem. – Asian J. 2011, 6, 524–533. doi:10.1002/asia.201000492 |
| 12. | Huang, H.-H.; Chao, C.-G.; Lee, S.-L.; Wu, H.-J.; Chen, C.-h.; Luh, T.-Y. Org. Biomol. Chem. 2012, 10, 5948–5953. doi:10.1039/c2ob25114k |
| 13. | Yeh, N.-H.; Chen, C.-W.; Lee, S.-L.; Wu, H.-J.; Chen, C.-h.; Luh, T.-Y. Macromolecules 2012, 45, 2662–2667. doi:10.1021/ma300027k |
| 14. | Xu, J.; Zhang, Z.; Liu, Y.-H.; Guo, Q.; Wang, G.-W.; Lai, G.; Luh, T.-Y. J. Polym. Sci., Part A: Polym. Chem. 2017, 55, 2999–3010. doi:10.1002/pola.28572 |
| 15. | Zhu, L.; Flook, M. M.; Lee, S.-L.; Chan, L.-W.; Huang, S.-L.; Chiu, C.-W.; Chen, C.-H.; Schrock, R. R.; Luh, T.-Y. Macromolecules 2012, 45, 8166–8171. doi:10.1021/ma301686f |
| 37. | Rule, J. D.; Moore, J. S. Macromolecules 2002, 35, 7878–7882. doi:10.1021/ma0209489 |
| 25. | Wu, Z.; Wheeler, D. R.; Grubbs, R. H. J. Am. Chem. Soc. 1992, 114, 146–151. doi:10.1021/ja00027a021 |
| 31. | Lee, J. C.; Parker, K. A.; Sampson, N. S. J. Am. Chem. Soc. 2006, 128, 4578–4579. doi:10.1021/ja058801v |
| 32. | Song, A.; Parker, K. A.; Sampson, N. S. J. Am. Chem. Soc. 2009, 131, 3444–3445. doi:10.1021/ja809661k |
| 33. | Parker, K. A.; Sampson, N. S. Acc. Chem. Res. 2016, 49, 408–417. doi:10.1021/acs.accounts.5b00490 |
| 24. | Lutz, J.-F., Ed. Sequence-Controlled Polymers; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2018. doi:10.1002/9783527806096 |
| 35. | Greenberg, A.; Liebman, J. F. Strained organic molecules; Academic Press: New York, 1978. |
| 23. | Moatsou, D.; Hansell, C. F.; O'Reilly, R. K. Chem. Sci. 2014, 5, 2246–2250. doi:10.1039/c4sc00752b |
| 21. | Ke, Y.-Z.; Lee, S.-L.; Chen, C.-h.; Luh, T.-Y. Chem. – Asian J. 2011, 6, 1748–1751. doi:10.1002/asia.201000877 |
| 22. | Ke, Y.-Z.; Ji, R.-J.; Wei, T.-C.; Lee, S.-L.; Huang, S.-L.; Huang, M.-J.; Chen, C.-h.; Luh, T.-Y. Macromolecules 2013, 46, 6712–6722. doi:10.1021/ma4012363 |
| 26. | Wu, Z.; Grubbs, R. H. Macromolecules 1994, 27, 6700–6703. doi:10.1021/ma00101a002 |
| 27. | Perrott, M. G.; Novak, B. M. Macromolecules 1995, 28, 3492–3494. doi:10.1021/ma00113a062 |
| 28. | Perrott, M. G.; Novak, B. M. Macromolecules 1996, 29, 1817–1823. doi:10.1021/ma951516j |
| 29. | Maughon, B. R.; Grubbs, R. H. Macromolecules 1997, 30, 3459–3469. doi:10.1021/ma961780s |
| 30. | Charvet, R.; Novak, B. M. Macromolecules 2001, 34, 7680–7685. doi:10.1021/ma0109875 |
| 31. | Lee, J. C.; Parker, K. A.; Sampson, N. S. J. Am. Chem. Soc. 2006, 128, 4578–4579. doi:10.1021/ja058801v |
| 32. | Song, A.; Parker, K. A.; Sampson, N. S. J. Am. Chem. Soc. 2009, 131, 3444–3445. doi:10.1021/ja809661k |
| 33. | Parker, K. A.; Sampson, N. S. Acc. Chem. Res. 2016, 49, 408–417. doi:10.1021/acs.accounts.5b00490 |
| 34. | Lin, N.-T.; Ke, Y.-Z.; Satyanarayana, K.; Huang, S.-L.; Lan, Y.-K.; Yang, H.-C.; Luh, T.-Y. Macromolecules 2013, 46, 7173–7179. doi:10.1021/ma401007b |
© 2019 Ke et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)