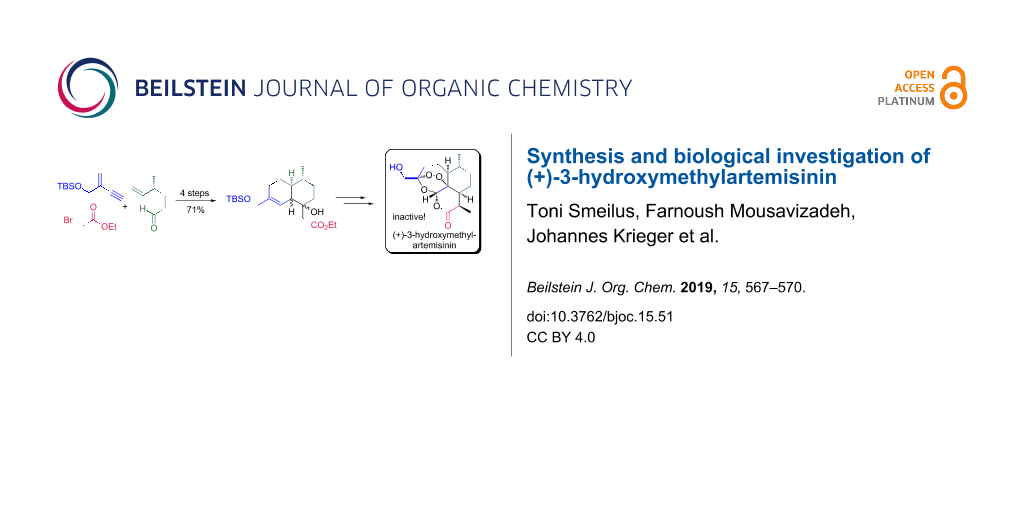
Abstract
Herein, we describe a biomimetic entry to (+)-3-hydroxymethylartemisinin (2) as well as to the artemisinin derivatives (+)-3-hydroxymethyl-9-desmethylartemisinin (16) and (+)-3-hydroxymethyl-9-epi-artemisinin (18), starting from the known and readily available chiral aldehyde 3 and alkyne 4. Subsequently, the synthesized compounds have been evaluated for their antimalarial activity against the drug-sensitive P. falciparum NF54 strain. All of them were inactive. In addition, they did not show any toxicity against L6 cells (a primary cell line derived from rat skeletal myoblasts). These results contribute to a better understanding of artemisinins mechanism of action.

Graphical Abstract
Introduction
The isolation of artemisinin (1; qinghaosu, Figure 1) from Artemisia annua L. and the discovery of its antimalarial properties in 1971 represent one of the greatest medical breakthroughs of the 20th century [1,2]. For these achievements Youyou Tu was awarded the Nobel Prize in Physiology or Medicine in 2015 [3]. As artemisinin has poor solubility and bioavailability, several derivatives of this natural product like artesunate and artemether were developed and are currently used in combination with other drugs for the treatment of malaria [4]. However, the exact mechanism of action of this endoperoxide sesquiterpene lactone is still unknown [5].
![[1860-5397-15-51-1]](/bjoc/content/figures/1860-5397-15-51-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of the natural (+)-artemisinin (1) and the synthesized (+)-3-hydroxymethylartemisinin (2).
Figure 1: Structures of the natural (+)-artemisinin (1) and the synthesized (+)-3-hydroxymethylartemisinin (2...
Whereas artemisinins are almost non-toxic to normal cells, several studies have confirmed their potent antitumor activity [6,7]. In addition, they have been reported to possess immunosuppressive, anti-inflammatory, antiviral, antifungal and antiparasitic activities [8-10]. Recently, it was shown that artemisinin interacts with the mammalian protein gephyrin and by stabilizing it, it enhances GABAA receptor signaling resulting in in vivo conversion of pancreatic α-cells into functional β-like cells [11]. Therefore, this sesquiterpene lactone may also find an application in the treatment of diabetes mellitus. These facts indicate the clinical potential of artemisinins and “it is likely that artemisinin drugs will become a major armamentarium combating a variety of human diseases beyond malaria” [9].
In the past, several semi-synthetic artemisinin derivatives were prepared for experimental therapy of pathologies like the ones mentioned before [12,13]. However, the variety of these derivatives is limited as all of them are produced modifying the artemisinin skeleton at the same positions due to intrinsic synthetic challenges. Recently, we reported a biomimetic artemisinin synthesis that addresses these challenges and pave the way for derivatization of the (+)-artemisinin skeleton at positions not accessible using current methodology [14]. Based on our approach we report here on the synthesis and biological activity of the title compound (+)-3-hydroxymethylartemisinin (2, Figure 1).
Results and Discussion
Our synthesis (Scheme 1) started from the known and readily available aldehyde 3 [14] which was treated with the organolithium species obtained from derivative 4 [15] and n-BuLi to yield derivative 5. The latter afforded allylic alcohol 6 after reduction of the propargylic moiety with Red-Al. BAIB/TEMPO oxidation of this alcohol gave ketone 7. By a Reformatsky reaction of 7 using Zn/ethyl bromoacetate derivative 8 was obtained, which was subjected to a thermal (190 °C, toluene) intramolecular Diels–Alder reaction resulting in the formation of the β-hydroxy esters 9, 10 and 11 (dr = 1.74:0:38:0.24) in a total yield of 84% (Scheme 2). Gratifyingly, the percentage of derivatives 9 and 10 that show the desired stereochemistry at carbon center 5a is around 90%.
![[1860-5397-15-51-i1]](/bjoc/content/inline/1860-5397-15-51-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of the Diels–Alder precursor 8 over four steps in 71% yield, starting from aldehyde 3 and alkyne 4. Reaction conditions: a) alkyne 4, n-BuLi, THF, −78 °C to rt, 16 h, 86%; b) Red-Al, THF, 0 °C, 10 min, 96%; c) BAIB, TEMPO, DCM, rt, 16 h, 92%; d) BrCH2CO2Et, Zn, toluene, reflux, 30 min, 93%. Red-Al = sodium bis(2-methoxyethoxy)aluminum dihydride, BAIB = (diacetoxyiodo)benzene, TEMPO = (2,2,6,6-tetramethylpiperidin-1-yl)oxyl.
Scheme 1: Synthesis of the Diels–Alder precursor 8 over four steps in 71% yield, starting from aldehyde 3 and...
![[1860-5397-15-51-i2]](/bjoc/content/inline/1860-5397-15-51-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of (+)-3-hydroxymethyl-9-desmethylartemisinin (16), starting from Diels–Alder derivatives 9 and 10. Reaction conditions: a) toluene, 190 °C, 24 h, 84% (dr 9:10:11 = 1.74:0.38:0.24); b) Martin sulfurane, DCM, 0 °C, 10 min, 97% [(E)/(Z) = 1:1]; c) Martin sulfurane, DCM, 0 °C, 10 min, 95%; d) NiCl2, NaBH4, −60 °C to −40 °C, 1 h, 96% (dr = 1:0.03); e) Li, EtOH, NH3, −70 °C, 10 min, 61% (dr = 1:0.4); f) i. O2, methylene blue, light, DCM, −30 °C, 30 h; ii. then O2, cat. TFA, DCM, rt, 2 d, 24%; g) i. O2, methylene blue, light, DCM, −30 °C, 30 h; ii. then O2, cat. TFA, DCM, rt, 2 d, 16% [13].
Scheme 2: Synthesis of (+)-3-hydroxymethyl-9-desmethylartemisinin (16), starting from Diels–Alder derivatives ...
The structure of all these isomers was confirmed by NOE experiments (see Supporting Information File 1 for full experimental details). Both derivatives 9 and 10 (Scheme 2) yielded α,β-unsaturated esters 12 and 13 after treatment with Martin sulfurane [16]. Reduction of compound 12 using NiCl2/NaBH4 in methanol as solvent yielded derivative 14 with excellent diastereomeric ratio (dr = 1:0.03). On the other hand, derivative 13 was reduced under Birch conditions (Li/NH3) to afford ester 15 in 61% yield (dr = 1:0.4). Gratifyingly, both artemisinin precursors 14 and 15 possess the desired stereoconfiguration at carbon center 8a.
Subsequently, a solution of 14 and 15 in dichloromethane containing methylene blue as photosensitizer was exposed to sunlight and oxygen. The treatment of the resulting intermediate hydroperoxide with a small amount of trifluoroacetic acid as previously described [17,18], afforded in the frame of a Hock cleavage (+)-3-hydroxymethyl-9-desmethylartemisinin (16) in 24% and 16% yield, respectively. Protection of the free hydroxy group of 16 as a silyl ether and methylation of the obtained lactone in α-position (LDA/MeI/HMPA) afforded derivative 17. After removal of the TES protecting group (+)-3-hydroxymethyl-9-epi-artemisinin (18, Scheme 3) was obtained, whereas the desired (+)-3-hydroxymethylartemisinin (2) was produced from 17 in two steps including epimerization and cleavage of the TES group in excellent yield (Scheme 3).
![[1860-5397-15-51-i3]](/bjoc/content/inline/1860-5397-15-51-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of (+)-3-hydroxymethyl-9-epi-artemisinin (18) and (+)-3-hydroxymethylartemisinin (2). Reaction conditions: a) i. TESCl, NEt3, 0 °C, 16 h; ii. LDA, MeI, HMPA, THF, −78 °C to −50 °C, 1 h, 73% (over two steps); b) TBAF, THF, 0 °C, 10 min, 95%; c) i. DBU, DCM, rt, 1 d; ii. TBAF, THF, 0 °C, 10 min, 87% (over two steps). LDA = lithium diisopropylamide, HMPA = hexamethylphosphoramide, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, TESCl = triethylsilyl chloride.
Scheme 3: Synthesis of (+)-3-hydroxymethyl-9-epi-artemisinin (18) and (+)-3-hydroxymethylartemisinin (2). Rea...
Finally, we evaluated the antimalarial activity of (+)-3-hydroxymethylartemisinin (2) as well as of the derivatives 16 and 18 against the drug-sensitive P. falciparum NF54 strain as described previously [14]. We found them to be inactive. In addition, 2 did not show any toxicity against L6 cells (a primary cell line derived from rat skeletal myoblasts). In both assays the highest concentration used was 100 μg/mL.
Conclusion
In the past it has been postulated that artemisinins kill intraerythrocytic parasites such as P. falciparum in the presence of Fe2+ forming reactive oxygen species (ROS) [4]. As artemisinin and similar derivatives are destroyed by Fe2+ in vitro (Fenton reaction), it is difficult for us to explain their inactivity against Plasmodium. Furthermore, in cellular systems free ferrous iron [19] as well as haem [20,21] have been proposed to be the main iron sources for artemisinin activation. However, the results on that matter remain controversial [4,22]. Our results challenge the radical hypothesis of artemisinins action and also indicate that the methyl group at the C-3 position of artemisinin is possibly involved in lipophilic interactions with putative proteins essential for survival of the Plasmodium parasite. Derivative 2 will be used to attach further substituents at position 14 of artemisinin to prove this hypothesis.
Experimental
Experimental details, NMR spectra and other physical data are shown in Supporting Information File 1.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 6.4 MB | Download |
References
-
White, N. J. Science 2008, 320, 330–334. doi:10.1126/science.1155165
Return to citation in text: [1] -
White, N. J.; Hien, T. T.; Nosten, F. H. Trends Parasitol. 2015, 31, 607–610. doi:10.1016/j.pt.2015.10.010
Return to citation in text: [1] -
Tu, Y. Angew. Chem., Int. Ed. 2016, 55, 10210–10226. doi:10.1002/anie.201601967
Angew. Chem. 2016, 128, 10366–10382. doi:10.1002/ange.201601967
Return to citation in text: [1] -
Phillips, M. A.; Burrows, J. N.; Manyando, C.; van Huijsduijnen, R. H.; Van Voorhis, W. C.; Wells, T. N. C. Nat. Rev. Dis. Primers 2017, 3, 17050. doi:10.1038/nrdp.2017.50
Return to citation in text: [1] [2] [3] -
Wang, J.; Zhang, C.-J.; Chia, W. N.; Loh, C. C. Y.; Li, Z.; Lee, Y. M.; He, Y.; Yuan, L.-X.; Lim, T. K.; Liu, M.; Liew, C. X.; Lee, Y. Q.; Zhang, J.; Lu, N.; Lim, C. T.; Hua, Z.-C.; Liu, B.; Shen, H.-M.; Tan, K. S. W.; Lin, Q. Nat. Commun. 2015, 6, 10111. doi:10.1038/ncomms10111
Return to citation in text: [1] -
Krishna, S.; Ganapathi, S.; Ster, I. C.; Saeed, M. E. M.; Cowan, M.; Finlayson, C.; Kovacsevics, H.; Jansen, H.; Kremsner, P. G.; Efferth, T.; Kumar, D. EBioMedicine 2015, 2, 82–90. doi:10.1016/j.ebiom.2014.11.010
Return to citation in text: [1] -
Ericsson, T.; Blank, A.; von Hagens, C.; Ashton, M.; Äbelö, A. Eur. J. Clin. Pharmacol. 2014, 70, 1453–1463. doi:10.1007/s00228-014-1754-2
Return to citation in text: [1] -
Raffetin, A.; Bruneel, F.; Roussel, C.; Thellier, M.; Buffet, P.; Caumes, E.; Jauréguiberry, S. Med. Mal. Infect. 2018, 48, 238–249. doi:10.1016/j.medmal.2018.01.004
Return to citation in text: [1] -
Krishna, S.; Bustamante, L.; Haynes, R. K.; Staines, H. M. Trends Pharmacol. Sci. 2008, 29, 520–527. doi:10.1016/j.tips.2008.07.004
Return to citation in text: [1] [2] -
Ho, W. E.; Peh, H. Y.; Chan, T. K.; Wong, W. S. F. Pharmacol. Ther. 2014, 142, 126–139. doi:10.1016/j.pharmthera.2013.12.001
Return to citation in text: [1] -
Li, J.; Casteels, T.; Frogne, T.; Ingvorsen, C.; Honoré, C.; Courtney, M.; Huber, K. V. M.; Schmitner, N.; Kimmel, R. A.; Romanov, R. A.; Sturtzel, C.; Lardeau, C.-H.; Klughammer, J.; Farlik, M.; Sdelci, S.; Vieira, A.; Avolio, F.; Briand, F.; Baburin, I.; Májek, P.; Pauler, F. M.; Penz, T.; Stukalov, A.; Gridling, M.; Parapatics, K.; Barbieux, C.; Berishvili, E.; Spittler, A.; Colinge, J.; Bennett, K. L.; Hering, S.; Sulpice, T.; Bock, C.; Distel, M.; Harkany, T.; Meyer, D.; Superti-Furga, G.; Collombat, P.; Hecksher-Sørensen, J.; Kubicek, S. Cell 2017, 168, 86–100. doi:10.1016/j.cell.2016.11.010
Return to citation in text: [1] -
Njuguna, N. M.; Ongarora, D. S. B.; Chibale, K. Expert Opin. Ther. Pat. 2012, 22, 1179–1203. doi:10.1517/13543776.2012.724063
Return to citation in text: [1] -
Mao, W.; Zhang, Y.; Zhang, A. Discovery of Antimalarial Drug Artemisinin and Beyond. In Case Studies in Modern Drug Discovery and Development; Huang, X.; Aslanian, R. G., Eds.; John Wiley & Sons: Hoboken, NJ, U.S.A., 2012; pp 227–256. doi:10.1002/9781118219683.ch10
Return to citation in text: [1] [2] -
Krieger, J.; Smeilus, T.; Kaiser, M.; Seo, E.-J.; Efferth, T.; Giannis, A. Angew. Chem., Int. Ed. 2018, 57, 8293–8296. doi:10.1002/anie.201802015
Angew. Chem. 2018, 130, 8425–8428. doi:10.1002/ange.201802015
Return to citation in text: [1] [2] [3] -
Charpenay, M.; Boudhar, A.; Hulot, C.; Blond, G.; Suffert, J. Tetrahedron 2013, 69, 7568–7591. doi:10.1016/j.tet.2013.05.089
Return to citation in text: [1] -
Martin, J. C.; Arhart, R. J. J. Am. Chem. Soc. 1971, 93, 4327–4329. doi:10.1021/ja00746a059
Return to citation in text: [1] -
Lévesque, F.; Seeberger, P. H. Angew. Chem., Int. Ed. 2012, 51, 1706–1709. doi:10.1002/anie.201107446
Angew. Chem. 2012, 124, 1738–1741. doi:10.1002/ange.201107446
Return to citation in text: [1] -
Acton, N.; Roth, R. J. J. Org. Chem. 1992, 57, 3610–3614. doi:10.1021/jo00039a020
Return to citation in text: [1] -
Stocks, P. A.; Bray, P. G.; Barton, V. E.; Al-Helal, M.; Jones, M.; Araujo, N. C.; Gibbons, P.; Ward, S. A.; Hughes, R. H.; Biagini, G. A.; Davies, J.; Amewu, R.; Mercer, A. E.; Ellis, G.; O'Neill, P. M. Angew. Chem., Int. Ed. 2007, 46, 6278–6283. doi:10.1002/anie.200604697
Return to citation in text: [1] -
Klonis, N.; Crespo-Ortiz, M. P.; Bottova, I.; Abu-Bakar, N.; Kenny, S.; Rosenthal, P. J.; Tilley, L. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 11405–11410. doi:10.1073/pnas.1104063108
Return to citation in text: [1] -
Robert, A.; Benoit-Vical, F.; Claparols, C.; Meunier, B. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 13676–13680. doi:10.1073/pnas.0500972102
Return to citation in text: [1] -
Haynes, R. K.; Krishna, S. Microbes Infect. 2004, 6, 1339–1346. doi:10.1016/j.micinf.2004.09.002
Return to citation in text: [1]
| 20. | Klonis, N.; Crespo-Ortiz, M. P.; Bottova, I.; Abu-Bakar, N.; Kenny, S.; Rosenthal, P. J.; Tilley, L. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 11405–11410. doi:10.1073/pnas.1104063108 |
| 21. | Robert, A.; Benoit-Vical, F.; Claparols, C.; Meunier, B. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 13676–13680. doi:10.1073/pnas.0500972102 |
| 4. | Phillips, M. A.; Burrows, J. N.; Manyando, C.; van Huijsduijnen, R. H.; Van Voorhis, W. C.; Wells, T. N. C. Nat. Rev. Dis. Primers 2017, 3, 17050. doi:10.1038/nrdp.2017.50 |
| 19. | Stocks, P. A.; Bray, P. G.; Barton, V. E.; Al-Helal, M.; Jones, M.; Araujo, N. C.; Gibbons, P.; Ward, S. A.; Hughes, R. H.; Biagini, G. A.; Davies, J.; Amewu, R.; Mercer, A. E.; Ellis, G.; O'Neill, P. M. Angew. Chem., Int. Ed. 2007, 46, 6278–6283. doi:10.1002/anie.200604697 |
| 1. | White, N. J. Science 2008, 320, 330–334. doi:10.1126/science.1155165 |
| 2. | White, N. J.; Hien, T. T.; Nosten, F. H. Trends Parasitol. 2015, 31, 607–610. doi:10.1016/j.pt.2015.10.010 |
| 6. | Krishna, S.; Ganapathi, S.; Ster, I. C.; Saeed, M. E. M.; Cowan, M.; Finlayson, C.; Kovacsevics, H.; Jansen, H.; Kremsner, P. G.; Efferth, T.; Kumar, D. EBioMedicine 2015, 2, 82–90. doi:10.1016/j.ebiom.2014.11.010 |
| 7. | Ericsson, T.; Blank, A.; von Hagens, C.; Ashton, M.; Äbelö, A. Eur. J. Clin. Pharmacol. 2014, 70, 1453–1463. doi:10.1007/s00228-014-1754-2 |
| 17. |
Lévesque, F.; Seeberger, P. H. Angew. Chem., Int. Ed. 2012, 51, 1706–1709. doi:10.1002/anie.201107446
Angew. Chem. 2012, 124, 1738–1741. doi:10.1002/ange.201107446 |
| 18. | Acton, N.; Roth, R. J. J. Org. Chem. 1992, 57, 3610–3614. doi:10.1021/jo00039a020 |
| 5. | Wang, J.; Zhang, C.-J.; Chia, W. N.; Loh, C. C. Y.; Li, Z.; Lee, Y. M.; He, Y.; Yuan, L.-X.; Lim, T. K.; Liu, M.; Liew, C. X.; Lee, Y. Q.; Zhang, J.; Lu, N.; Lim, C. T.; Hua, Z.-C.; Liu, B.; Shen, H.-M.; Tan, K. S. W.; Lin, Q. Nat. Commun. 2015, 6, 10111. doi:10.1038/ncomms10111 |
| 14. |
Krieger, J.; Smeilus, T.; Kaiser, M.; Seo, E.-J.; Efferth, T.; Giannis, A. Angew. Chem., Int. Ed. 2018, 57, 8293–8296. doi:10.1002/anie.201802015
Angew. Chem. 2018, 130, 8425–8428. doi:10.1002/ange.201802015 |
| 4. | Phillips, M. A.; Burrows, J. N.; Manyando, C.; van Huijsduijnen, R. H.; Van Voorhis, W. C.; Wells, T. N. C. Nat. Rev. Dis. Primers 2017, 3, 17050. doi:10.1038/nrdp.2017.50 |
| 13. | Mao, W.; Zhang, Y.; Zhang, A. Discovery of Antimalarial Drug Artemisinin and Beyond. In Case Studies in Modern Drug Discovery and Development; Huang, X.; Aslanian, R. G., Eds.; John Wiley & Sons: Hoboken, NJ, U.S.A., 2012; pp 227–256. doi:10.1002/9781118219683.ch10 |
| 3. |
Tu, Y. Angew. Chem., Int. Ed. 2016, 55, 10210–10226. doi:10.1002/anie.201601967
Angew. Chem. 2016, 128, 10366–10382. doi:10.1002/ange.201601967 |
| 16. | Martin, J. C.; Arhart, R. J. J. Am. Chem. Soc. 1971, 93, 4327–4329. doi:10.1021/ja00746a059 |
| 12. | Njuguna, N. M.; Ongarora, D. S. B.; Chibale, K. Expert Opin. Ther. Pat. 2012, 22, 1179–1203. doi:10.1517/13543776.2012.724063 |
| 13. | Mao, W.; Zhang, Y.; Zhang, A. Discovery of Antimalarial Drug Artemisinin and Beyond. In Case Studies in Modern Drug Discovery and Development; Huang, X.; Aslanian, R. G., Eds.; John Wiley & Sons: Hoboken, NJ, U.S.A., 2012; pp 227–256. doi:10.1002/9781118219683.ch10 |
| 14. |
Krieger, J.; Smeilus, T.; Kaiser, M.; Seo, E.-J.; Efferth, T.; Giannis, A. Angew. Chem., Int. Ed. 2018, 57, 8293–8296. doi:10.1002/anie.201802015
Angew. Chem. 2018, 130, 8425–8428. doi:10.1002/ange.201802015 |
| 9. | Krishna, S.; Bustamante, L.; Haynes, R. K.; Staines, H. M. Trends Pharmacol. Sci. 2008, 29, 520–527. doi:10.1016/j.tips.2008.07.004 |
| 15. | Charpenay, M.; Boudhar, A.; Hulot, C.; Blond, G.; Suffert, J. Tetrahedron 2013, 69, 7568–7591. doi:10.1016/j.tet.2013.05.089 |
| 11. | Li, J.; Casteels, T.; Frogne, T.; Ingvorsen, C.; Honoré, C.; Courtney, M.; Huber, K. V. M.; Schmitner, N.; Kimmel, R. A.; Romanov, R. A.; Sturtzel, C.; Lardeau, C.-H.; Klughammer, J.; Farlik, M.; Sdelci, S.; Vieira, A.; Avolio, F.; Briand, F.; Baburin, I.; Májek, P.; Pauler, F. M.; Penz, T.; Stukalov, A.; Gridling, M.; Parapatics, K.; Barbieux, C.; Berishvili, E.; Spittler, A.; Colinge, J.; Bennett, K. L.; Hering, S.; Sulpice, T.; Bock, C.; Distel, M.; Harkany, T.; Meyer, D.; Superti-Furga, G.; Collombat, P.; Hecksher-Sørensen, J.; Kubicek, S. Cell 2017, 168, 86–100. doi:10.1016/j.cell.2016.11.010 |
| 4. | Phillips, M. A.; Burrows, J. N.; Manyando, C.; van Huijsduijnen, R. H.; Van Voorhis, W. C.; Wells, T. N. C. Nat. Rev. Dis. Primers 2017, 3, 17050. doi:10.1038/nrdp.2017.50 |
| 22. | Haynes, R. K.; Krishna, S. Microbes Infect. 2004, 6, 1339–1346. doi:10.1016/j.micinf.2004.09.002 |
| 8. | Raffetin, A.; Bruneel, F.; Roussel, C.; Thellier, M.; Buffet, P.; Caumes, E.; Jauréguiberry, S. Med. Mal. Infect. 2018, 48, 238–249. doi:10.1016/j.medmal.2018.01.004 |
| 9. | Krishna, S.; Bustamante, L.; Haynes, R. K.; Staines, H. M. Trends Pharmacol. Sci. 2008, 29, 520–527. doi:10.1016/j.tips.2008.07.004 |
| 10. | Ho, W. E.; Peh, H. Y.; Chan, T. K.; Wong, W. S. F. Pharmacol. Ther. 2014, 142, 126–139. doi:10.1016/j.pharmthera.2013.12.001 |
| 14. |
Krieger, J.; Smeilus, T.; Kaiser, M.; Seo, E.-J.; Efferth, T.; Giannis, A. Angew. Chem., Int. Ed. 2018, 57, 8293–8296. doi:10.1002/anie.201802015
Angew. Chem. 2018, 130, 8425–8428. doi:10.1002/ange.201802015 |
© 2019 Smeilus et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)