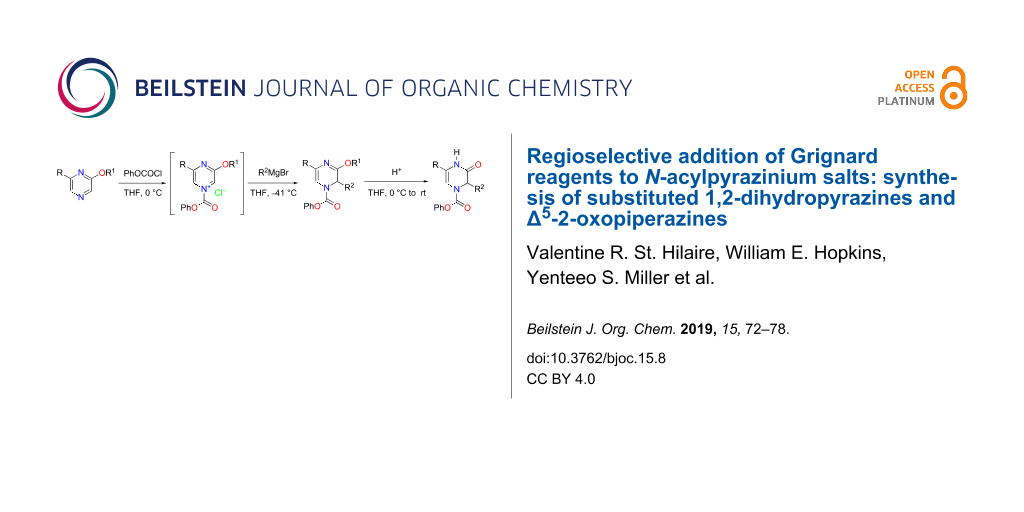
Abstract
The regioselective addition of Grignard reagents to mono- and disubstituted N-acylpyrazinium salts affording substituted 1,2-dihydropyrazines in modest to excellent yields (45–100%) is described. Under acidic conditions, these 1,2-dihydropyrazines can be converted to substituted Δ5-2-oxopiperazines providing a simple and efficient approach towards their preparation.

Graphical Abstract
Introduction
Pyrazine and piperazine ring systems are key structural elements in a large number of biologically active molecules [1-6]. Compounds containing these scaffolds were shown to behave as anticancer agents [2-5], sodium channel blockers [5], and also display antiviral activity [7]. Due to their appearance in an array of biologically active small molecules and natural products, efficient synthetic routes into this class of privileged structures would be very beneficial [7,8]. One approach towards their synthesis involves the addition of nucleophiles to activated pyrazines. We recently showed that 3-alkoxy-substituted N-acylpyrazinium salts can be selectively reduced by tributyltin hydride to afford 1,2-dihydropyrazines in good to excellent yields [9]. There have been other reports involving the addition of TMS-ketene acetals to pyrazinium salts [10-12]. A double nucleophilic addition of bis(trimethylsilyl)ketene acetals to pyrazines activated with methyl chloroformate was found to afford polycyclic γ-lactones in moderate yields [3,10,11]. The work by Garduño-Alva and co-workers demonstrated that these TMS-ketene acetals can be regioselectively added to substituted N-triflate pyrazinium salts to also generate γ-lactones [12].
Grignard reagents have been used as nucleophiles on a variety of N-acyl-activated pyridines in the production of natural products and biologically active small molecules [13-20]. To our surprise, there are no reports on the nucleophilic addition of Grignard reagents to N-acylpyrazinium salts. A literature search showed this organometallic reagent reacting with pyrazine N-oxides towards the one-pot synthesis of N-Boc-protected N-hydroxy-substituted piperazines in good yields [6]. Methylmagnesium iodide was observed adding to 2-cyano-6-morpholinylpyrazine [21]. As a part of our continued exploration into the synthetic utility of N-acylpyrazinium salts, herein we report the regioselective addition of Grignard reagents to mono- and disubstituted N-acylpyrazinium salts towards the synthesis of 1,2-dihydropyrazines and Δ5-2-oxopiperazines.
Results and Discussion
Our journey began by first reacting 2-methoxypyrazine with phenyl chloroformate to generate the N-acylpyrazinium salt 2 using DCM as the solvent. Next, phenylmagnesium bromide in THF was added at −41 °C. After stirring for 60 min, dihydropyrazine 3a was isolated in a yield of 40% (entry 1, Table 1). This initial low yield prompted us to switch the Grignard solvent from THF to DCM. Using modified reaction conditions from Andersson and co-wokers [22], phenylmagnesium bromide in DCM was added to 2 at −41 °C and after 35 min, dihydropyrazine 3a was only isolated in a 32% yield (entry 2, Table 1). The lower yields appear to be caused by the Grignard attacking the carbonyl of the N-acyl salt 2 causing the formation of phenyl benzoate. When toluene was used as the solvent, both 3a and the ester were obtained in yields of 41% and 39%, respectively (entry 3, Table 1). Changing the solvent to diethyl ether showed no improvement in the yield of 3a but when THF was used, an excellent yield of 87% was produced (entries 4 and 5, Table 1).
Table 1: Phenyl Grignard addition to methoxy-substituted N-acylpyrazinium salts.
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-8-i1.svg?max-width=637&scale=1.0)
|
|||
| entrya | solvent | time (min) | yield 3a (%)b |
| 1 | DCM | 60 | 40 |
| 2c | DCM | 35 | 32 |
| 3 | toluene | 40 | 41d |
| 4 | diethyl ether | 60 | 15 |
| 5 | THF | 30 | 87e |
aGrignard reagent in THF. bIsolated yields. cGrignard reagent added as solution in DCM. dPhCO2Ph (39%) was also isolated. eTrace amounts of the ester byproduct PhCO2Ph was isolated.
Based on our previously developed selective tin hydride reduction of monosubstituted pyrazinium salts [9], we expected the Grignard reagent to add regioselectively to give 1,2-dihydropyrazine 3a. DFT calculations support the observations that the isolated regioisomer we obtained was the result of a thermodynamically favored 1,2-addition over a 1,6-addition [9]. It has also been shown that TMS-ketene acetals add selectively to N-triflate pyrazinium salts [12]. 1H NMR analysis confirmed this by showing a rotameric pair of singlets at 3.89 and 3.84 ppm for the methoxy group at C3, a rotameric pair of singlets at 5.89 and 5.58 ppm for the proton at C2 and a rotameric pair of doublets at 6.17, 6.15 ppm and 6.68, 6.63 ppm for the two vinyl protons at C5 and C6, respectively. This result is in agreement with our observation that nucleophiles favored 1,2-addition over a 1,6-addition [9]. A formation of 1,6-dihydropyrazine was not observed.
With THF identified as the optimal solvent to use, we sought to expand the scope of the Grignard addition to various mono- and disubstituted N-acylpyrazinium salts (Figure 1). Phenyl chloroformate was the acylating reagent of choice for this study due to benzyl or methyl chloroformates producing products in very poor yields. A variety of alkyl Grignard reagents were shown to add regioselectively to methoxy-substituted salts to give the dihydropyrazines in yields ranging from 48% to 73% (Figure 1, compounds 3b–g). Aryl Grignard reagents containing an electron-withdrawing and donating group proceeded to give the desired substituted products in moderate yields (Figure 1, compounds 3h–j). The examination of the Grignard addition to benzyloxy- or p-methoxybenzyloxy (PMB)-substituted pyrazinium salts, resulted in obtaining dihydropyrazines in yields that were comparable to the methoxy salts (Figure 1, compounds 4a,b and 5).
![[1860-5397-15-8-1]](/bjoc/content/figures/1860-5397-15-8-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Regioselective addition of Grignard reagents to mono- and disubstituted pyrazinium salts (yields refer to isolated yields).
Figure 1: Regioselective addition of Grignard reagents to mono- and disubstituted pyrazinium salts (yields re...
We next subjected disubstituted N-acylpyrazinium salts to this reaction. Based on our results from the monosubstituted substrates, a 1,2-addition of the Grignard was expected to occur. When an aryl group was present on the ring, trisubstituted dihydropyrazines in good yields ranging from 78–100% were produced (Figure 1, compounds 6, 7a,b and 12). An alkyl substitution with either an ethyl or benzyl group on the pyrazinium salts gave an 81% yield for both 10 and 11 (Figure 1). Reacting a Grignard with pyrazinium salts disubstituted with electron-donating alkoxy groups gave us the desired dihydropyrazine in moderate to good yields of 45–88% (Figure 1, compounds 8, 9a,b) while the presence of an electron-withdrawing ester group generated 13 in a yield of 49%.
With the substituted 1,2-dihydropyrazines in hand, we next wanted to demonstrate their usefulness for synthesizing substituted Δ5-2-oxopiperazines. Processes into this structural motif would be very useful due to their presence in a variety of biologically active small molecules and natural products [23-30]. We previously reported that 3-methoxy-1,2-dihydropyrazines can be easily converted to Δ5-2-oxopiperazines using 1 M HCl(aq) in methanol [9]. When we applied these acidic conditions on the phenyl-substituted 3-methoxy-1,2-dihydropyrazine 3a, only a 5% yield of Δ5-2-oxopiperazine 14a was obtained (Table 2, entry 1). This low yield appears to be due to the presence of a ring-opened side product [31]. The yields of the Δ5-2-oxopiperazines were improved to 61% and 92%, respectively, when the reaction was repeated on phenyl-substituted 1,2-dihydropyrazines containing benzyl and p-methoxybenzyl (PMB) ether groups (Table 2, entries 2 and 3). This enhancement in yield can be attributed to the benzyl and PMB groups’ better sensitivity towards removal under acidic conditions [32]. With the aqueous acidic conditions not being suitable for converting 3-methoxy-1,2-dihydropyrazine 3a, we decided to run this reaction under anhydrous acidic conditions. After stirring dihydropyrazine 3a in the presence of HCl/dioxane at 0 °C for 30 min, we were pleased to see that Δ5-2-oxopiperazine 14a was obtained in an excellent yield of 93% (Table 2, entry 4). Subjecting both benzyl and 2-bromobenzyl-substituted dihydropyrazines 3f and 3g to these acidic conditions gave 95% and 88% yields of 14b and 14c, respectively (Table 2, entries 5 and 6). When we examined the use of anhydrous acidic conditions on 4b and 5, quantitative yields of Δ5-2-oxopiperazine 14a were obtained (Table 2, entries 7 and 8). Finally, under these conditions, the trisubstituted dihydropyrazine 7a was easily converted to a disubstituted Δ5-2-oxopiperazine 15a in a yield of 73% (Table 2, entry 9).
Table 2: Conversion of dihydropyrazine to Δ5-2-oxopiperazines under acidic conditions.
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-8-i2.svg?max-width=637&scale=1.0)
|
|||||||
| entry | R | R1 | R2 | acid | time (min) | product | yield (%)a |
| 1 | H | Me | Ph | HCl(aq)/MeOH | 60 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-15-8-i3.svg?max-width=637&scale=1.0)
14a |
5 |
| 2 | H | Bn | Ph | HCl(aq)/MeOH | 45 | 14a | 61 |
| 3 | H | PMB | Ph | HCl(aq)/MeOH | 60 | 14a | 92 |
| 4b | H | Me | Ph | HCl/dioxane | 30 | 14a | 93 |
| 5b | H | Me | Bn | HCl/dioxane | 30 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-15-8-i4.svg?max-width=637&scale=1.0)
14b |
95 |
| 6b | H | Me | 2-BrBn | HCl/dioxane | 45 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-15-8-i5.svg?max-width=637&scale=1.0)
14c |
85 |
| 7b | H | Bn | Ph | HCl/dioxane | 20 | 14a | 100 |
| 8b | H | PMB | Ph | HCl/dioxane | 10 | 14a | 100 |
| 9b | p-MeOPh | Me | Ph | HCl/dioxane | 45 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-15-8-i6.svg?max-width=637&scale=1.0)
15a |
73 |
aIsolated yields. b4 M HCl/dioxane was used.
Following the step-wise conversion of 1,2-dihydropyrazines to Δ5-2-oxopiperazines, we decided to develop a one-pot approach towards their synthesis (Table 3). As described above, we first synthesized the phenyl-substituted 1,2-dihydropyrazine 4b by adding a phenyl Grignard reagent to benzyloxy-substituted N-acylpyrazinium salt in THF at −41 °C. Next, 1 M HCl(aq)/MeOH was added and the reaction was monitored by TLC. After 1 h, the hydrolysis of 4b was completed to give 14a in a good yield of 80% (Table 3, entry 1). Disubstituted Δ5-2-oxopiperazines 15b–d can be made similarly with yields ranging from 71–80% (Table 3, entries 2–4). This simple approach towards Δ5-2-oxopiperazines provides access into compounds that can be reduced into mono- and disubstituted 2-oxopiperazines [33,34]. This structure is a common scaffold found in natural products and biologically active small molecules [23].
![[Graphic 7]](/bjoc/content/inline/1860-5397-15-8-i7.svg?max-width=637&scale=1.0)
![[Graphic 8]](/bjoc/content/inline/1860-5397-15-8-i8.svg?max-width=637&scale=1.0)
![[Graphic 9]](/bjoc/content/inline/1860-5397-15-8-i9.svg?max-width=637&scale=1.0)
![[Graphic 10]](/bjoc/content/inline/1860-5397-15-8-i10.svg?max-width=637&scale=1.0)
![[Graphic 11]](/bjoc/content/inline/1860-5397-15-8-i11.svg?max-width=637&scale=1.0)
Conclusion
In conclusion, we have demonstrated that various Grignard reagents can be regioselectively added to mono- and disubstituted N-acylpyrazinium salts to give di- and trisubstituted 1,2-dihydropyrazines in moderate to excellent yields. Under acidic conditions, the dihydropyrazines can be easily converted to substituted Δ5-2-oxopiperazines. These compounds can potentially serve as templates for making substituted 2-oxopiperazines. The investigation into the synthetic utility of this reaction is currently underway and will be reported in due course.
Supporting Information
| Supporting Information File 1: Experimental section and NMR spectra. | ||
| Format: PDF | Size: 4.1 MB | Download |
Acknowledgements
We gratefully acknowledge the financial support from the National Institute of General Medical Sciences (Grant number: SC3GM105572) and the Golden LEAF Foundation, Rocky Mount, NC, for their financial support in setting up the BRITE Institute. HRMS data were obtained at the North Carolina State University Department of Chemistry Mass Spectrometry Facility.
References
-
Lee, Y. B.; Gong, Y.-D.; Kim, D. J.; Ahn, C.-H.; Kong, J.-Y.; Kang, N.-S. Bioorg. Med. Chem. 2012, 20, 1303–1309. doi:10.1016/j.bmc.2011.12.026
Return to citation in text: [1] -
Chetan, B.; Bunha, M.; Jagrat, M.; Sinha, B. N.; Saiko, P.; Graser, G.; Szekeres, T.; Raman, G.; Rajendran, P.; Moorthy, D.; Basu, A.; Jayaprakash, V. Bioorg. Med. Chem. Lett. 2010, 20, 3906–3910. doi:10.1016/j.bmcl.2010.05.020
Return to citation in text: [1] [2] -
Garg, N. K.; Stoltz, B. M. Tetrahedron Lett. 2005, 46, 2423–2426. doi:10.1016/j.tetlet.2005.02.054
Return to citation in text: [1] [2] [3] -
Guo, C.; Bhandaru, S.; Fuchs, P. L.; Boyd, M. R. J. Am. Chem. Soc. 1996, 118, 10672–10673. doi:10.1021/ja962646q
Return to citation in text: [1] [2] -
Hirsh, A. J.; Molino, B. F.; Zhang, J.; Astakhova, N.; Geiss, W. B.; Sargent, B. J.; Swenson, B. D.; Usyatinsky, A.; Wyle, M. J.; Boucher, R. C.; Smith, R. T.; Zamurs, A.; Johnson, M. R. J. Med. Chem. 2006, 49, 4098–4115. doi:10.1021/jm051134w
Return to citation in text: [1] [2] [3] -
Walker, J. A.; Liu, W.; Wise, D. S.; Drach, J. C.; Townsend, L. B. J. Med. Chem. 1998, 41, 1236–1241. doi:10.1021/jm970532z
Return to citation in text: [1] [2] -
Horton, D. A.; Bourne, G. T.; Smythe, M. L. Chem. Rev. 2003, 103, 893–930. doi:10.1021/cr020033s
Return to citation in text: [1] [2] -
Evans, B. E.; Rittle, K. E.; Bock, M. G.; DiPardo, R. M.; Freidinger, R. M.; Whitter, W. L.; Lundell, G. F.; Veber, D. F.; Anderson, P. S.; Chang, R. S. L.; Lotti, V. J.; Cerino, D. J.; Chen, T. B.; Kling, P. J.; Kunkel, K. A.; Springer, J. P.; Hirshfield, J. J. Med. Chem. 1988, 31, 2235–2246. doi:10.1021/jm00120a002
Return to citation in text: [1] -
Williams, A. L.; St. Hilaire, V. R.; Lee, T. J. Org. Chem. 2012, 77, 4097–4102. doi:10.1021/jo202498r
Return to citation in text: [1] [2] [3] [4] [5] -
Rudler, H.; Denise, B.; Xu, Y.; Parlier, A.; Vaissermann, J. Eur. J. Org. Chem. 2005, 3724–3744. doi:10.1002/ejoc.200500162
Return to citation in text: [1] [2] -
Rudler, H.; Denise, B.; Xu, Y.; Vaissermann, J. Tetrahedron Lett. 2005, 46, 3449–3451. doi:10.1016/j.tetlet.2005.03.131
Return to citation in text: [1] [2] -
Garduño-Alva, A.; Ortega-Alfaro, M. C.; López-Cortés, J. G.; Chávez, I.; Barroso-Flores, J.; Toscano, R. A.; Rudler, H.; Álvarez-Toledano, C. Can. J. Chem. 2012, 90, 469–482. doi:10.1139/v2012-016
Return to citation in text: [1] [2] [3] -
Hiebel, A.-C.; Comins, D. L. Tetrahedron 2015, 71, 7354–7360. doi:10.1016/j.tet.2015.04.086
Return to citation in text: [1] -
Tsukanov, S. V.; Comins, D. L. J. Org. Chem. 2014, 79, 9074–9085. doi:10.1021/jo501415r
Return to citation in text: [1] -
Cash, B. M.; Prevost, N.; Wagner, F. F.; Comins, D. L. J. Org. Chem. 2014, 79, 5740–5745. doi:10.1021/jo500878v
Return to citation in text: [1] -
Wu, Y.; Li, L.; Li, H.; Gao, L.; Xie, H.; Zhang, Z.; Su, Z.; Hu, C.; Song, Z. Org. Lett. 2014, 16, 1880–1883. doi:10.1021/ol500302r
Return to citation in text: [1] -
Sahn, J. J.; Bharathi, P.; Comins, D. L. Tetrahedron Lett. 2012, 53, 1347–1350. doi:10.1016/j.tetlet.2011.12.127
Return to citation in text: [1] -
Gotchev, D. B.; Comins, D. L. J. Org. Chem. 2006, 71, 9393–9402. doi:10.1021/jo061677t
Return to citation in text: [1] -
Kuethe, J. T.; Comins, D. L. J. Org. Chem. 2004, 69, 2863–2866. doi:10.1021/jo049943v
Return to citation in text: [1] -
Comins, D. L.; Williams, A. L. Tetrahedron Lett. 2000, 41, 2839–2842. doi:10.1016/s0040-4039(00)00298-7
Return to citation in text: [1] -
Foks, H. Acta Pol. Pharm. 1978, 35, 525–528.
Return to citation in text: [1] -
Andersson, H.; Banchelin, T. S.-L.; Das, S.; Gustafsson, M.; Olsson, R.; Almqvist, F. Org. Lett. 2010, 12, 284–286. doi:10.1021/ol902619h
Return to citation in text: [1] -
De Risi, C.; Pelà, M.; Pollini, G. P.; Trapella, C.; Zanirato, V. Tetrahedron: Asymmetry 2010, 21, 255–274. doi:10.1016/j.tetasy.2010.02.008
Return to citation in text: [1] [2] -
Vieth, M.; Siegel, M. G.; Higgs, R. E.; Watson, I. A.; Robertson, D. H.; Savin, K. A.; Durst, G. L.; Hipskind, P. A. J. Med. Chem. 2004, 47, 224–232. doi:10.1021/jm030267j
Return to citation in text: [1] -
Kitamura, S.; Fukushi, H.; Miyawaki, T.; Kawamura, M.; Konishi, N.; Terashita, Z.-i.; Naka, T. J. Med. Chem. 2001, 44, 2438–2450. doi:10.1021/jm0004345
Return to citation in text: [1] -
Endo, A.; Yanagisawa, A.; Abe, M.; Tohma, S.; Kan, T.; Fukuyama, T. J. Am. Chem. Soc. 2002, 124, 6552–6554. doi:10.1021/ja026216d
Return to citation in text: [1] -
Mori, K.; Rikimaru, K.; Kan, T.; Fukuyama, T. Org. Lett. 2004, 6, 3095–3097. doi:10.1021/ol048857e
Return to citation in text: [1] -
Lee, S.-C.; Park, S. B. J. Comb. Chem. 2007, 9, 828–835. doi:10.1021/cc0700492
and references within.
Return to citation in text: [1] -
Candelon, N.; Shinkaruk, S.; Bennetau, B.; Bennetau-Pelissero, C.; Dumartin, M.-L.; Degueil, M.; Babin, P. Tetrahedron 2010, 66, 2463–2469. doi:10.1016/j.tet.2010.01.088
Return to citation in text: [1] -
El Kaïm, L.; Grimaud, L.; Reddy Purumandla, S. J. Org. Chem. 2011, 76, 4728–4733. doi:10.1021/jo200397m
Return to citation in text: [1] -
Major product present was the ring-opened compound 16; see Supporting Information File 1.
Return to citation in text: [1] -
Wuts, P. G. M.; Greene, T. W. Greene's protective groups in organic synthesis, 4th ed.; Wiley-Interscience: Hoboken, NJ, 2007.
Return to citation in text: [1] -
Peng, H.; Carrico, D.; Thai, V.; Blaskovich, M.; Bucher, C.; Pusateri, E. E.; Sebti, S. M.; Hamilton, A. D. Org. Biomol. Chem. 2006, 4, 1768–1784. doi:10.1039/b517572k
Return to citation in text: [1] -
Horwell, D. C.; Lewthwaite, R. A.; Pritchard, M. C.; Ratcliffe, G. S.; Ronald Rubin, J. Tetrahedron 1998, 54, 4591–4606. doi:10.1016/s0040-4020(98)00092-1
Return to citation in text: [1]
| 9. | Williams, A. L.; St. Hilaire, V. R.; Lee, T. J. Org. Chem. 2012, 77, 4097–4102. doi:10.1021/jo202498r |
| 9. | Williams, A. L.; St. Hilaire, V. R.; Lee, T. J. Org. Chem. 2012, 77, 4097–4102. doi:10.1021/jo202498r |
| 23. | De Risi, C.; Pelà, M.; Pollini, G. P.; Trapella, C.; Zanirato, V. Tetrahedron: Asymmetry 2010, 21, 255–274. doi:10.1016/j.tetasy.2010.02.008 |
| 24. | Vieth, M.; Siegel, M. G.; Higgs, R. E.; Watson, I. A.; Robertson, D. H.; Savin, K. A.; Durst, G. L.; Hipskind, P. A. J. Med. Chem. 2004, 47, 224–232. doi:10.1021/jm030267j |
| 25. | Kitamura, S.; Fukushi, H.; Miyawaki, T.; Kawamura, M.; Konishi, N.; Terashita, Z.-i.; Naka, T. J. Med. Chem. 2001, 44, 2438–2450. doi:10.1021/jm0004345 |
| 26. | Endo, A.; Yanagisawa, A.; Abe, M.; Tohma, S.; Kan, T.; Fukuyama, T. J. Am. Chem. Soc. 2002, 124, 6552–6554. doi:10.1021/ja026216d |
| 27. | Mori, K.; Rikimaru, K.; Kan, T.; Fukuyama, T. Org. Lett. 2004, 6, 3095–3097. doi:10.1021/ol048857e |
| 28. |
Lee, S.-C.; Park, S. B. J. Comb. Chem. 2007, 9, 828–835. doi:10.1021/cc0700492
and references within. |
| 29. | Candelon, N.; Shinkaruk, S.; Bennetau, B.; Bennetau-Pelissero, C.; Dumartin, M.-L.; Degueil, M.; Babin, P. Tetrahedron 2010, 66, 2463–2469. doi:10.1016/j.tet.2010.01.088 |
| 30. | El Kaïm, L.; Grimaud, L.; Reddy Purumandla, S. J. Org. Chem. 2011, 76, 4728–4733. doi:10.1021/jo200397m |
| 1. | Lee, Y. B.; Gong, Y.-D.; Kim, D. J.; Ahn, C.-H.; Kong, J.-Y.; Kang, N.-S. Bioorg. Med. Chem. 2012, 20, 1303–1309. doi:10.1016/j.bmc.2011.12.026 |
| 2. | Chetan, B.; Bunha, M.; Jagrat, M.; Sinha, B. N.; Saiko, P.; Graser, G.; Szekeres, T.; Raman, G.; Rajendran, P.; Moorthy, D.; Basu, A.; Jayaprakash, V. Bioorg. Med. Chem. Lett. 2010, 20, 3906–3910. doi:10.1016/j.bmcl.2010.05.020 |
| 3. | Garg, N. K.; Stoltz, B. M. Tetrahedron Lett. 2005, 46, 2423–2426. doi:10.1016/j.tetlet.2005.02.054 |
| 4. | Guo, C.; Bhandaru, S.; Fuchs, P. L.; Boyd, M. R. J. Am. Chem. Soc. 1996, 118, 10672–10673. doi:10.1021/ja962646q |
| 5. | Hirsh, A. J.; Molino, B. F.; Zhang, J.; Astakhova, N.; Geiss, W. B.; Sargent, B. J.; Swenson, B. D.; Usyatinsky, A.; Wyle, M. J.; Boucher, R. C.; Smith, R. T.; Zamurs, A.; Johnson, M. R. J. Med. Chem. 2006, 49, 4098–4115. doi:10.1021/jm051134w |
| 6. | Walker, J. A.; Liu, W.; Wise, D. S.; Drach, J. C.; Townsend, L. B. J. Med. Chem. 1998, 41, 1236–1241. doi:10.1021/jm970532z |
| 7. | Horton, D. A.; Bourne, G. T.; Smythe, M. L. Chem. Rev. 2003, 103, 893–930. doi:10.1021/cr020033s |
| 8. | Evans, B. E.; Rittle, K. E.; Bock, M. G.; DiPardo, R. M.; Freidinger, R. M.; Whitter, W. L.; Lundell, G. F.; Veber, D. F.; Anderson, P. S.; Chang, R. S. L.; Lotti, V. J.; Cerino, D. J.; Chen, T. B.; Kling, P. J.; Kunkel, K. A.; Springer, J. P.; Hirshfield, J. J. Med. Chem. 1988, 31, 2235–2246. doi:10.1021/jm00120a002 |
| 9. | Williams, A. L.; St. Hilaire, V. R.; Lee, T. J. Org. Chem. 2012, 77, 4097–4102. doi:10.1021/jo202498r |
| 7. | Horton, D. A.; Bourne, G. T.; Smythe, M. L. Chem. Rev. 2003, 103, 893–930. doi:10.1021/cr020033s |
| 12. | Garduño-Alva, A.; Ortega-Alfaro, M. C.; López-Cortés, J. G.; Chávez, I.; Barroso-Flores, J.; Toscano, R. A.; Rudler, H.; Álvarez-Toledano, C. Can. J. Chem. 2012, 90, 469–482. doi:10.1139/v2012-016 |
| 5. | Hirsh, A. J.; Molino, B. F.; Zhang, J.; Astakhova, N.; Geiss, W. B.; Sargent, B. J.; Swenson, B. D.; Usyatinsky, A.; Wyle, M. J.; Boucher, R. C.; Smith, R. T.; Zamurs, A.; Johnson, M. R. J. Med. Chem. 2006, 49, 4098–4115. doi:10.1021/jm051134w |
| 22. | Andersson, H.; Banchelin, T. S.-L.; Das, S.; Gustafsson, M.; Olsson, R.; Almqvist, F. Org. Lett. 2010, 12, 284–286. doi:10.1021/ol902619h |
| 2. | Chetan, B.; Bunha, M.; Jagrat, M.; Sinha, B. N.; Saiko, P.; Graser, G.; Szekeres, T.; Raman, G.; Rajendran, P.; Moorthy, D.; Basu, A.; Jayaprakash, V. Bioorg. Med. Chem. Lett. 2010, 20, 3906–3910. doi:10.1016/j.bmcl.2010.05.020 |
| 3. | Garg, N. K.; Stoltz, B. M. Tetrahedron Lett. 2005, 46, 2423–2426. doi:10.1016/j.tetlet.2005.02.054 |
| 4. | Guo, C.; Bhandaru, S.; Fuchs, P. L.; Boyd, M. R. J. Am. Chem. Soc. 1996, 118, 10672–10673. doi:10.1021/ja962646q |
| 5. | Hirsh, A. J.; Molino, B. F.; Zhang, J.; Astakhova, N.; Geiss, W. B.; Sargent, B. J.; Swenson, B. D.; Usyatinsky, A.; Wyle, M. J.; Boucher, R. C.; Smith, R. T.; Zamurs, A.; Johnson, M. R. J. Med. Chem. 2006, 49, 4098–4115. doi:10.1021/jm051134w |
| 9. | Williams, A. L.; St. Hilaire, V. R.; Lee, T. J. Org. Chem. 2012, 77, 4097–4102. doi:10.1021/jo202498r |
| 12. | Garduño-Alva, A.; Ortega-Alfaro, M. C.; López-Cortés, J. G.; Chávez, I.; Barroso-Flores, J.; Toscano, R. A.; Rudler, H.; Álvarez-Toledano, C. Can. J. Chem. 2012, 90, 469–482. doi:10.1139/v2012-016 |
| 6. | Walker, J. A.; Liu, W.; Wise, D. S.; Drach, J. C.; Townsend, L. B. J. Med. Chem. 1998, 41, 1236–1241. doi:10.1021/jm970532z |
| 33. | Peng, H.; Carrico, D.; Thai, V.; Blaskovich, M.; Bucher, C.; Pusateri, E. E.; Sebti, S. M.; Hamilton, A. D. Org. Biomol. Chem. 2006, 4, 1768–1784. doi:10.1039/b517572k |
| 34. | Horwell, D. C.; Lewthwaite, R. A.; Pritchard, M. C.; Ratcliffe, G. S.; Ronald Rubin, J. Tetrahedron 1998, 54, 4591–4606. doi:10.1016/s0040-4020(98)00092-1 |
| 3. | Garg, N. K.; Stoltz, B. M. Tetrahedron Lett. 2005, 46, 2423–2426. doi:10.1016/j.tetlet.2005.02.054 |
| 10. | Rudler, H.; Denise, B.; Xu, Y.; Parlier, A.; Vaissermann, J. Eur. J. Org. Chem. 2005, 3724–3744. doi:10.1002/ejoc.200500162 |
| 11. | Rudler, H.; Denise, B.; Xu, Y.; Vaissermann, J. Tetrahedron Lett. 2005, 46, 3449–3451. doi:10.1016/j.tetlet.2005.03.131 |
| 23. | De Risi, C.; Pelà, M.; Pollini, G. P.; Trapella, C.; Zanirato, V. Tetrahedron: Asymmetry 2010, 21, 255–274. doi:10.1016/j.tetasy.2010.02.008 |
| 10. | Rudler, H.; Denise, B.; Xu, Y.; Parlier, A.; Vaissermann, J. Eur. J. Org. Chem. 2005, 3724–3744. doi:10.1002/ejoc.200500162 |
| 11. | Rudler, H.; Denise, B.; Xu, Y.; Vaissermann, J. Tetrahedron Lett. 2005, 46, 3449–3451. doi:10.1016/j.tetlet.2005.03.131 |
| 12. | Garduño-Alva, A.; Ortega-Alfaro, M. C.; López-Cortés, J. G.; Chávez, I.; Barroso-Flores, J.; Toscano, R. A.; Rudler, H.; Álvarez-Toledano, C. Can. J. Chem. 2012, 90, 469–482. doi:10.1139/v2012-016 |
| 31. | Major product present was the ring-opened compound 16; see Supporting Information File 1. |
| 9. | Williams, A. L.; St. Hilaire, V. R.; Lee, T. J. Org. Chem. 2012, 77, 4097–4102. doi:10.1021/jo202498r |
| 13. | Hiebel, A.-C.; Comins, D. L. Tetrahedron 2015, 71, 7354–7360. doi:10.1016/j.tet.2015.04.086 |
| 14. | Tsukanov, S. V.; Comins, D. L. J. Org. Chem. 2014, 79, 9074–9085. doi:10.1021/jo501415r |
| 15. | Cash, B. M.; Prevost, N.; Wagner, F. F.; Comins, D. L. J. Org. Chem. 2014, 79, 5740–5745. doi:10.1021/jo500878v |
| 16. | Wu, Y.; Li, L.; Li, H.; Gao, L.; Xie, H.; Zhang, Z.; Su, Z.; Hu, C.; Song, Z. Org. Lett. 2014, 16, 1880–1883. doi:10.1021/ol500302r |
| 17. | Sahn, J. J.; Bharathi, P.; Comins, D. L. Tetrahedron Lett. 2012, 53, 1347–1350. doi:10.1016/j.tetlet.2011.12.127 |
| 18. | Gotchev, D. B.; Comins, D. L. J. Org. Chem. 2006, 71, 9393–9402. doi:10.1021/jo061677t |
| 19. | Kuethe, J. T.; Comins, D. L. J. Org. Chem. 2004, 69, 2863–2866. doi:10.1021/jo049943v |
| 20. | Comins, D. L.; Williams, A. L. Tetrahedron Lett. 2000, 41, 2839–2842. doi:10.1016/s0040-4039(00)00298-7 |
| 32. | Wuts, P. G. M.; Greene, T. W. Greene's protective groups in organic synthesis, 4th ed.; Wiley-Interscience: Hoboken, NJ, 2007. |
© 2019 St. Hilaire et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)