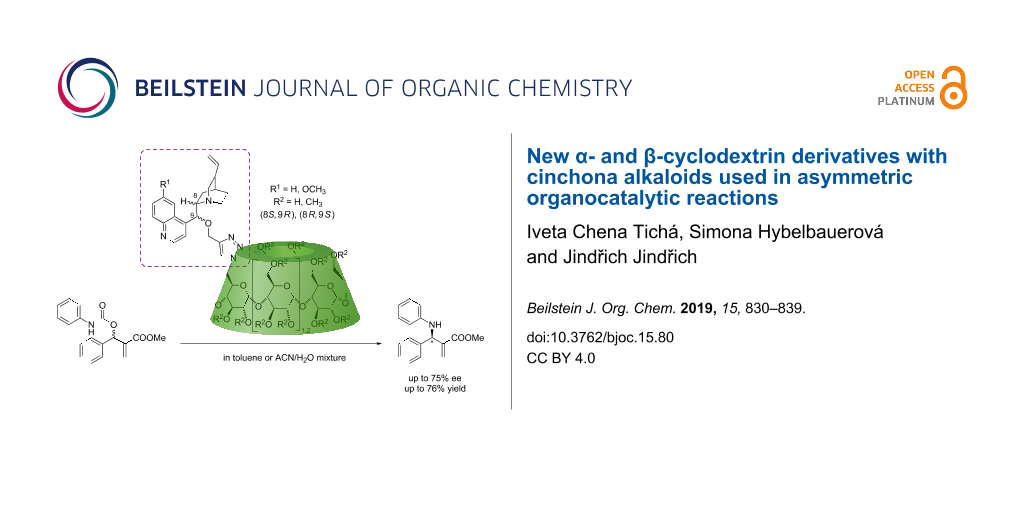
Abstract
The preparation of new organocatalysts for asymmetric syntheses has become a key stage of enantioselective catalysis. In particular, the development of new cyclodextrin (CD)-based organocatalysts allowed to perform enantioselective reactions in water and to recycle catalysts. However, only a limited number of organocatalytic moieties and functional groups have been attached to CD scaffolds so far. Cinchona alkaloids are commonly used to catalyze a wide range of enantioselective reactions. Thus, in this study, we report the preparation of new α- and β-CD derivatives monosubstituted with cinchona alkaloids (cinchonine, cinchonidine, quinine and quinidine) on the primary rim through a CuAAC click reaction. Subsequently, permethylated analogs of these cinchona alkaloid–CD derivatives also were synthesized and the catalytic activity of all derivatives was evaluated in several enantioselective reactions, specifically in the asymmetric allylic amination (AAA), which showed a promising enantiomeric excess of up to 75% ee. Furthermore, a new disubstituted α-CD catalyst was prepared as a pure AD regioisomer and also tested in the AAA. Our results indicate that (i) the cinchona alkaloid moiety can be successfully attached to CD scaffolds through a CuAAC reaction, (ii) the permethylated cinchona alkaloid–CD catalysts showed better results than the non-methylated CDs analogues in the AAA reaction, (iii) promising enantiomeric excesses are achieved, and (iv) the disubstituted CD derivatives performed similarly to monosubstituted CDs. Therefore, these new CD derivatives with cinchona alkaloids effectively catalyze asymmetric allylic aminations and have the potential to be successfully applied in other enantioselective reactions.

Graphical Abstract
Introduction
Cyclodextrins (CDs) [1], cyclic oligosaccharides consisting of α-D-glucopyranoside units, and their derivatives are widely used in many industrial and research areas for their ability to form supramolecular inclusion complexes [2]. CD derivatives have been increasingly applied in catalysis and biomimetic reactions [3,4] thanks to host–guest interactions and to the non-toxic, chiral skeleton of CDs. More specifically, CDs applied in reactions involving metal catalysis [5], organocatalysis [6] and artificial enzymes [7] have been recently studied, thus highlighting their high potential as effective catalysts.
CDs represent an ideal skeleton with a cavity-containing structure for catalysts. Moreover, using native or modified CDs, organic reactions can be performed under green conditions [8-10]. In addition, CDs improve the rate and modulate the regioselectivity and enantioselectivity of reactions [11]. For example, metal-based CD catalytic systems and CD derivatives for organocatalysis have already shown promising results in the studies by Hapiot and Monflier [12], Armspach [13] and others [14,15].
The chemical modification of native CD skeletons with new functional groups enhances the application of CDs and provides access to new organic chemistry transformations and catalytic systems. Among the approaches used for chemical derivatization of CD skeletons, monosubstitution on the primary rim of CD (Figure 1) is a well-explored strategy [2] which can be used to prepare various types of CD derivatives.
![[1860-5397-15-80-1]](/bjoc/content/figures/1860-5397-15-80-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Schematic cone-shaped (a) and structure representations (b) of α-CD (six glucopyranoside units) and β-CD (seven glucopyranoside units).
Figure 1: Schematic cone-shaped (a) and structure representations (b) of α-CD (six glucopyranoside units) and...
Several examples of modified-CD derivatives with a catalytic nucleophilic center have been reported in the area of organocatalytic asymmetric reactions [11]. Initially, Kanagaraj et al. [16] used per-6-amino-β-CD as the promoter (not in a catalytic amount) of a Henry reaction and obtained the product with 99% ee. Subsequently, Doyagüez et al. [17] attached L-proline to β-CD via different linkers (including a triazole linker) and used the resulting organocatalysts in an aldol reaction in water, albeit with a lower enantiomeric excess (54% ee). Conversely, Shen et al. [18] performed an aldol reaction in a buffer using L- and D-proline-derived CDs connected through a pyrrolidine skeleton as catalysts and observed 94% ee. More recently, Liu et al. [19] reported the excellent enantioselectivity of 99% ee in an aldol reaction catalyzed by β-CD with L-proline attached through a urea moiety. Therefore, mainly proline-derived CDs have been previously tested as organocatalysts and mainly in aldol-type reactions.
The limited number of functional groups attached to CD comprising mainly L-proline restricts the potential of asymmetric organocatalytic reactions using CD derivatives. However, a wide range of catalytic groups, especially cinchona alkaloids (Figure 2), have been used in organocatalysis with excellent results. These naturally occurring compounds and their derivatives are commonly applied in various enantioselective reactions (mainly because of the nucleophilic center on the chiral quinuclidine skeleton) [20]. More importantly, they are a privileged class of chiral catalysts, which are well known for their use in Michael additions [21], Morita–Baylis–Hillman reactions [22], and aldol reactions [23], among others [24]. Hence, combining cinchona alkaloids with CDs has the potential to widen the applications of CD derivatives in asymmetric organocatalysis.
![[1860-5397-15-80-2]](/bjoc/content/figures/1860-5397-15-80-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Common cinchona alkaloids (cinchonine, cinchonidine, quinine, quinidine).
Figure 2: Common cinchona alkaloids (cinchonine, cinchonidine, quinine, quinidine).
The combination of cinchona alkaloids with CDs was first reported by Liu et al. [25] who prepared inclusion complexes of cinchona alkaloids and organoselenium-bridged bis-β-CDs. Subsequently, the same research group [26] investigated the performance of inclusion complexes of native and permethylated β-CDs and cinchona alkaloids as pH-responsive binding systems. Nevertheless, to the best of our knowledge, CD derivatives with covalently bonded cinchona alkaloids have never been prepared and tested in asymmetric organocatalysis. Thus, in this study, we investigated methods for attaching cinchona alkaloids to CD skeletons, and we assessed the enantiomeric excess of the resulting CD derivatives as organocatalysts in asymmetric reactions, specifically in the asymmetric allylic amination (AAA).
We successfully prepared a series of monosubstituted α- and β-CDs derivatives with the cinchona alkaloids cinchonine, cinchonidine, quinine, and quinidine with up to 95% isolated yield through CuAAC click reactions. By this simple, high-yielding and quick method we synthesized eight new CD derivatives, four based on the α-CD and four based on β-CD skeleton. In addition, to widen the usability and to improve the solubility of the prepared CD derivatives, the corresponding eight permethylated analogs were also synthesized. Furthermore, to test more advanced types of catalysts, a disubstituted α-CD derivative as a pure AD regioisomer with two identical cinchona alkaloid residues was prepared and tested in the AAA reaction.
Our study shows that the CuAAC reaction is a good and high-yielding method for the functionalization of the CD skeleton when attaching sterically demanding cinchona groups. Additionally, some of these new CD derivatives showed promising results of up to 75% ee in the AAA reactions of Morita–Baylis–Hillman (MBH) carbamates and significant differences depending on the attached cinchona alkaloid (cinchonine, cinchonidine, quinine, quinidine) as well as on the size of the cavity, i.e., β-CD or α-CD derivatives). Thus, this study showed that cinchona-substituted CD catalysts are active in organocatalytic reactions.
Results and Discussion
Synthesis of monosubstituted non-methylated CD derivatives
Initially, the method for attaching cinchona alkaloids to non-methylated CDs was developed. For our purposes of using the derivatives as catalysts for enantioselective reactions, we focused on α- and β-CD skeletons.
Our successful approach consisted of attaching these molecules through copper-catalyzed alkyne–azide cycloaddition (CuAAC). First, the required starting materials 6I-azido-6I-deoxy-α-CD (1) [27] and 6I-azido-6I-deoxy-β-CD (2) [27] and 9-O-propargylated cinchona alkaloid derivatives 3a–d [28] were synthesized followed by optimization of the conditions for the CuAAC click reaction (Scheme 1).
![[1860-5397-15-80-i1]](/bjoc/content/inline/1860-5397-15-80-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: CuAAC click reaction of propargylated cinchona alkaloids 3a–d with 6I-azido-6I-deoxy-α-CD (1) and 6I-azido-6I-deoxy-β-CD (2).
Scheme 1: CuAAC click reaction of propargylated cinchona alkaloids 3a–d with 6I-azido-6I-deoxy-α-CD (1) and 6I...
The CuAAC click reaction conditions were initially optimized using α-CD (Table 1). Initially, the reaction was performed in a THF/H2O mixture with 1.5 equiv of 9-O-propargylated cinchonine (3a) and 50 mol % CuI affording the product in 78% yield (Table 1, entry 1). Reducing both the amount of propargylated cinchona alkaloid 3a to 1.3 equiv and the amount of the Cu salt to 20 mol % resulted in virtually the same yield of the product (Table 1, entry 2). Conversely, the further decreasing the amount of propargylated cinchonine (3a) to 1.05 equiv led to a significantly lower conversion to the product (56%, Table 1, entry 3). Thus, based on the optimal conditions identified for the α-CD skeleton (Table 1, entry 2), the subsequent syntheses were performed using 1.3 equiv of cinchona alkaloids 3b–d in a mixture of THF/H2O with 20 mol % CuI. The corresponding α-CD products (4a–d, 5a–d) were isolated in high yields of up to 86% yield (Table 1, entries 2 and 4–6).
Table 1: Optimized conditions of the CuAAC click reactions of non-methylated azido-CDs with propargylated cinchona alkaloids depicted in Scheme 1.
| Entry | CD | Alkaloid | R1 | x (equiv) | y (mol %) | Solvent | Yielda in % (product) |
| 1 | 1 | 3a (8R,9S) | H | 1.5 | 50 | THF/H2O 1:1 | 78 (4a) |
| 2 | 1 | 3a (8R,9S) | H | 1.3 | 20 | THF/H2O 1:1 | 77 (4a) |
| 3 | 1 | 3a (8R,9S) | H | 1.05 | 20 | THF/H2O 1:1 | 56 (4a) |
| 4 | 1 | 3b (8S,9R) | H | 1.3 | 20 | THF/H2O 1:1 | 86 (4b) |
| 5 | 1 | 3c (8S,9R) | OCH3 | 1.3 | 20 | THF/H2O 1:1 | 72 (4c) |
| 6 | 1 | 3d (8R,9S) | OCH3 | 1.3 | 20 | THF/H2O 1:1 | 74 (4d) |
| 7b | 2 | 3a (8R,9S) | H | 1.3 | 20 | THF/H2O | 20 (5a) |
| 8 | 2 | 3a (8R,9S) | H | 1.3 | 20 | DMF | 89 (5a) |
| 9 | 2 | 3b (8S,9R) | H | 1.3 | 20 | DMF | 70 (5b) |
| 10 | 2 | 3c (8S,9R) | OCH3 | 1.3 | 20 | DMF | 80 (5c) |
| 11 | 2 | 3d (8R,9S) | OCH3 | 1.3 | 20 | DMF | 95 (5d) |
| 12b | 1 | 3a (8R,9S) | H | 1.3 | 20 | DMF | 38 (4a) |
aIsolated yield. bAfter 48 hours.
In the reactions with β-CD (2, Table 1, entries 7–11), no full conversion into the product could be achieved in the solvent mixture THF/H2O even after 48 hours of reaction (Table 1, entry 7). However, when changing the solvent to DMF a full conversion into the product was observed within 2 hours of reaction affording the products with high to excellent yields (95%, 5a–d, Table 1, entries 8–11). Conversely, the product yield was low when using DMF for a CuAAC reaction with α-CD resulting in only 38% of product 4a after 48 hours (Table 1, entry 12).
Synthesis of monosubstituted methylated CD derivatives
After developing the approach for attaching of cinchona alkaloids to non-methylated CD skeletons, we next focused on the functionalization of permethylated CD derivatives. First, we prepared the starting CD compounds, per-Me-N3-α-CD (6) [29] and per-Me-N3-β-CD (7) [30], and subjected them to the previously optimized conditions of the CuAAC click reaction with propargylated cinchona alkaloids (3a–d). The resulting permethylated CD derivatives 8a–d, 9a–d were isolated in high yields of up to 69% (Scheme 2).
![[1860-5397-15-80-i2]](/bjoc/content/inline/1860-5397-15-80-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: CuAAC click reaction of per-Me-N3-α-CD (6) or per-Me-N3-β-CD (7) and propargylated cinchona alkaloids 3a–d.
Scheme 2: CuAAC click reaction of per-Me-N3-α-CD (6) or per-Me-N3-β-CD (7) and propargylated cinchona alkaloi...
As outlined in Table 2, the conditions assessed using the non-methylated CDs were applied to prepare per-Me-α-CD (6) analogs. Thus, reaction 6 with 1.3 molar equivalents of the propargylated cinchona alkaloid 3a in the presence of 20 mol % CuI in DMF afforded product 8a in 59% yield (Table 2, entry 1). Subsequently, we prepared the other permethylated cinchona–α-CD derivatives 8b–d with moderate yields (42–49% yield, Table 2, entries 2–4). In the case of per-Me-β-CD 7, following the same procedure, the products 9a–d were also isolated with good to high yields (up to 69% yield, Table 2, entries 6–9). Concomitantly, the reaction was investigated in the THF/H2O solvent mixture (Table 2, entry 5), however, the reaction in DMF afforded a higher isolated yield (Table 2, entry 3).
Table 2: Yields for optimized conditions of CuAAC click reaction of permethylated azido-CDs with propargylated cinchona alkaloids from Scheme 2.
| Entry | CD | Alkaloid | R1 | Yielda in % (product) |
| 1 | 6 | 3a (8R,9S) | H | 59 (8a) |
| 2 | 6 | 3b (8S,9R) | H | 48 (8b) |
| 3 | 6 | 3c (8S,9R) | OCH3 | 49 (8c) |
| 4 | 6 | 3d (8R,9S) | OCH3 | 42 (8d) |
| 5b | 6 | 3c (8S,9R) | OCH3 | 34 (8c) |
| 6 | 7 | 3a (8R,9S) | H | 64 (9a) |
| 7 | 7 | 3b (8S,9R) | H | 69 (9b) |
| 8 | 7 | 3c (8S,9R) | OCH3 | 48 (9c) |
| 9 | 7 | 3d (8R,9S) | OCH3 | 63 (9d) |
aIsolated yield. bTHF/H2O solvent mixture.
Synthesis of disubstituted CD derivatives
To open the way for the preparation of more versatile types of enantioselective organocatalysts containing a CD skeleton and cinchona alkaloids, a method for the synthesis of a disubstituted derivative of cinchona alkaloid–non-methylated CD was developed. The prepared derivative was subsequently tested in an AAA reaction. In contrast to the monosubstituted derivatives, disubstituted CDs should be considered as possible mixtures of regioisomers and pseudoenantiomers [31,32]. Because of the results published by our group previously [33], we chose an AD regioisomer (as a pure regioisomer) on an α-CD skeleton for the preparation of the catalyst.
Initially, we synthesized the starting material, 6A,6D-diazido-6A,6D-dideoxy-α-CD (10) [34-36] and reacted it with propargylated cinchona alkaloid 3c. The disubstituted product 11 with a quinine moiety (3c) at position 1,4 on the primary rim of the α-CD skeleton was isolated in 76% yield (Scheme 3).
![[1860-5397-15-80-i3]](/bjoc/content/inline/1860-5397-15-80-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of difunctionalized α-CD 11 with quinine moieties.
Scheme 3: Synthesis of difunctionalized α-CD 11 with quinine moieties.
NMR elucidation of the prepared cinchona–CD derivatives
The structures of mono- (4a–d, 5a–d) and disubstituted (11) non-methylated CDs and permethylated (8a–d, 9a–d) CD derivatives were unambiguously confirmed by NMR measurements. As representative example of the prepared CD derivatives, we chose the α-CD derivative substituted with quinidine 4d. The 1H NMR spectra of the non-methylated CD derivatives in DMSO-d6 are in accordance with monosubstituted derivatives at position 6 on the primary rim (Figure 3). Generally, we observed four different regions with the typical signals: the first, well-resolved aromatic region belongs to the quinoline part of the cinchona alkaloid (9.00–7.55 ppm) and to the hydrogen signal of the triazole (8.21 ppm), thus confirming the successful attachment of the cinchona alkaloid to the CD skeleton through the CuAAC click reaction. The second part of the 1H NMR spectrum comprises the resolved signal for the double bond on the quinuclidine skeleton of the cinchona alkaloid (5.93 ppm). The third part of the spectrum consists of the CD region (5.50–3.20 ppm) with H-1 atoms of unsubstituted glucose units (4.80 ppm) and H-1I (5.03 ppm) for the substituted glucose. The signals of the H-2, H-3, H-4 and H-6 atoms of unsubstituted units are observed at around 4.00–3.00 ppm; on the other hand, the signal for H-6I is separately visible around 4.75 ppm (especially in the HSQC and 1H,1H COSY spectra). This part of the spectrum also includes the primary rim OH groups (4.49–4.34 ppm) and secondary rim OH groups (5.91–5.53 ppm). Finally, the quinuclidine skeleton part of the cinchona alkaloid is identified in the region around 2.00–1.20 ppm.
![[1860-5397-15-80-3]](/bjoc/content/figures/1860-5397-15-80-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Representative 1H NMR spectrum of the non-methylated quinidine–α-CD derivative 4d.
Figure 3: Representative 1H NMR spectrum of the non-methylated quinidine–α-CD derivative 4d.
Subsequently, 13C NMR, DEPT-edited HSQC and HMBC spectra also confirmed the substitution on the primary rim of the CD skeleton (Figure 4). The C-6 atom of the substituted glucose unit is correlated with the hydrogen signal of the triazole ring (50.41 ppm of C-6I to 8.16 ppm of H-14' of triazole) and 126.13 ppm of C-14' triazole is correlated to the signal at 4.57 ppm of the quinidine part. Additional 2D NMR spectra (COSY, HSQC, HMBC, ROESY) are included in Supporting Information File S2 and Supporting Information File S3.
![[1860-5397-15-80-4]](/bjoc/content/figures/1860-5397-15-80-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Representative 13C NMR spectrum and parts of the HMBC spectrum of the non-methylated quinidine–α-CD derivative 4d.
Figure 4: Representative 13C NMR spectrum and parts of the HMBC spectrum of the non-methylated quinidine–α-CD...
Moreover, we also investigated a possible inclusion of the cinchona alkaloid substituent in the CD cavity in the case of cinchonine–β-CD 5a in D2O. The 2D ROESY spectrum showed cross-peaks between the substituent (hydrogen atoms of the double bond of the quinuclidine skeleton) and the inner H-3 atoms of the β-CD cavity. However, the low solubility of non-methylated β-CDs in H2O (1 mg/1 mL) did not allow us to further investigate the nature of the inclusion, e.g., by concentration dependency measurements. Thus, the observed cross-peaks could be caused by intermolecular interactions (inclusion of the part of the cinchonine moiety into the second CD cavity) or by intramolecular interactions. Nevertheless, the rotation of the substituted glucopyranoside unit as discussed by Hapiot and Monflier [37], leading to the formation of the in isomer, is not very probable in our case, due to the large steric demand of the cinchona substituent. Moreover, the CD inner hydrogens showed no cross-peaks with the triazole ring hydrogen as well as with no cinchona hydrogens which are close to the triazole. The results of these NMR measurements in D2O are collected in Supporting Information File 2.
In conclusion, we unambiguously confirmed the cinchona alkaloid attachment to the CD skeleton through the triazole by 2D NMR measurements. This thorough investigation revealed no triple bond and a new triazole hydrogen signal while correlating carbon C6 of the substituted glucose unit with the triazole. Therefore, the prepared CD derivatives are substituted on the primary side. Further characterization data are included in Supporting Information Files 1–3.
Catalytic activity of cinchona–CD derivatives
Lastly, the activity of all prepared CD derivatives was tested in asymmetric organocatalytic reactions. After unsuccessful application in Morita–Baylis–Hillman and aldol-type reactions, we focused on their application in the decarboxylative asymmetric allylic amination (AAA) [38] of MBH carbamate 12 affording the product 13 with enantiomeric excesses of up to 75% (Scheme 4).
![[1860-5397-15-80-i4]](/bjoc/content/inline/1860-5397-15-80-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: AAA reaction of MBH carbamate 12 catalyzed by the prepared CD derivatives 4a–d, 5a–d, 8a–d, 9a–d, 11.
Scheme 4: AAA reaction of MBH carbamate 12 catalyzed by the prepared CD derivatives 4a–d, 5a–d, 8a–d, 9a–d, 11...
However, compared with the published procedure [38] (up to 97% ee, aromatic solvent, 40 °C, and 168 hours), the solvent of the reaction had to be changed in the case of non-methylated CDs due to their lower solubility in organic solvents. The reaction conditions and results are summed up in Table 3.
Table 3: Catalytic activity of the CD derivatives in the AAA reaction of MBH carbamate 12.a
| Entry | Catalyst | Solvent | Yieldb (%) | ee (%) |
| 1 | DABCO | toluene | 89 | – |
| 2c | per-Me-α-CD | toluene | n.d. | – |
| 3c | per-Me-β-CD | toluene | n.d. | – |
| 4 | 8a | toluene | 62 | 13 |
| 5 | 8b | toluene | 42 | 74 |
| 6 | 8c | toluene | 76 | 25 |
| 7 | 8d | toluene | 47 | 27 |
| 8 | 9a | toluene | 37 | 15 |
| 9 | 9b | toluene | 55 | 69 |
| 10 | 9c | toluene | 12 | 15 |
| 11 | 9d | toluene | 44 | 25 |
| 12 | 9b | CHCl3 | 63 | 69 |
| 13 | 9b | MTBE | 63 | 69 |
| 14 | 9b | MeOH | 73 | 33 |
| 15d | 9b | toluene | 15 | 75 |
| 16c | α-CD | ACN/H2O | n.d. | – |
| 17c | β-CD | ACN/H2O | n.d. | – |
| 18 | 4a | ACN/H2O | 10 | 3 |
| 19c | 4b | DMF | n.d. | – |
| 20 | 4b | ACN/H2O | 5 | 0 |
| 21 | 4c | ACN/H2O | 12 | 0 |
| 22 | 4d | ACN/H2O | 18 | 21 |
| 23 | 5a | ACN/H2O | 26 | 5 |
| 24 | 5b | DMF | 9 | 25 |
| 25 | 5b | DMSO | 19 | 23 |
| 26 | 5b | ACN/H2O | 19 | 19 |
| 27 | 5c | ACN/H2O | 5 | 0 |
| 28 | 5d | ACN/H2O | 21 | 13 |
| 29c | 11 | ACN/H2O | n.d. | – |
| 30c,e | 11 | ACN/H2O | n.d. | – |
aStandard conditions: 10 mol % catalyst, 0.4 M solution, solvent, 40 °C, 168 hours. bIsolated yield. cn.d. = not detected, – = not measured. dTemperature 25 °C. eWith 5 mol % (1S)-CSA.
First, the racemic version of this reaction with DABCO gave 89% yield of the product (Table 3, entry 1). Pure permethylated α- and β-CDs without cinchona alkaloid modification were also tested (Table 3, entries 2 and 3) as blank catalysts but completely failed. Promising results were achieved with permethylated CD–cinchonidine derivatives 8b and 9b affording the product in 74 and 69% ee, respectively (Table 3, entries 5 and 9). Other permethylated CD derivatives 8a, 8c, 8d, 9a, 9c, 9d resulted in low ee (Table 3, entries 4, 6–8, 10 and 11). Based on these results, the promising catalyst 9b was selected and investigated under different conditions (solvents and temperature, Table 3, entries 12–15) with similar results. Lastly, native α- and β-CDs were also tested as blank catalysts to confirm that the CD molecule without any modification has no influence on the reaction (Table 3, entries 16 and 17). Furthermore, the non-methylated monosubstituted CD derivatives 4a–d, 5a–d afforded on one side lower enantiomer excesses (Table 3, entries 18–28), on the other hand, they showed some catalytic activity in the solvent mixture acetonitrile/H2O, which could be promising for future applications of these catalysts in water. The disubstituted CD derivative 11 was not active in this enantioselective reaction (Table 3, entry 29) and this derivative was also tested in the AAA reaction with (1S)-10-camphorsulfonic acid (CSA) according to the original procedure [38], in which the cocatalyst (1S)-CSA enhanced the efficiency of dimeric cinchona alkaloids (Table 3, entry 30). However, there was no difference observable under these conditions.
Conclusion
We prepared a series of new 6-monosubstituted α-CD and β-CD derivatives modified with four different cinchona alkaloids, i.e., cinchonine, cinchonidine, quinine, and quinidine. The products were obtained in high yields through the CuAAC reaction and subsequently applied as catalysts in enantioselective reactions. We fully characterized the series of new 16 cinchona–CD derivatives including non-methylated and permethylated CDs by 2D NMR, MS, IR spectroscopy and we optimized their preparation (less than 3 h and up to 95% isolated yield). We applied them in the decarboxylative asymmetric allylic amination of a Morita–Baylis–Hillman carbamate (10 mol % of catalyst, up to 75% ee, up to 76% isolated yield). We believe that these new CD derivatives comprising cinchona alkaloids will be suitable catalysts of other asymmetric reactions using them under green chemistry conditions.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, copies of NMR spectra and chiral HPLC analysis. | ||
| Format: PDF | Size: 3.8 MB | Download |
| Supporting Information File 2: 2D NMR spectra of compounds 4a–d and 5a–d. | ||
| Format: PDF | Size: 3.7 MB | Download |
| Supporting Information File 3: 2D NMR spectra of compounds 8a–d, 9a–d and 11. | ||
| Format: PDF | Size: 4.1 MB | Download |
References
-
Crini, G. Chem. Rev. 2014, 114, 10940–10975. doi:10.1021/cr500081p
Return to citation in text: [1] -
Szejtli, J. Chem. Rev. 1998, 98, 1743–1754. doi:10.1021/cr970022c
Return to citation in text: [1] [2] -
Breslow, R.; Dong, S. D. Chem. Rev. 1998, 98, 1997–2012. doi:10.1021/cr970011j
Return to citation in text: [1] -
Easton, C. J. Pure Appl. Chem. 2005, 77, 1865–1871. doi:10.1351/pac200577111865
Return to citation in text: [1] -
Hapiot, F.; Tilloy, S.; Monflier, E. Chem. Rev. 2006, 106, 767–781. doi:10.1021/cr050576c
Return to citation in text: [1] -
Bogliotti, N.; Dalko, P. I. Shape and Site-Selective Asymmetric Reactions. In Enantioselective organocatalysis: reactions and experimental procedures; Dalko, P. I., Ed.; Wiley-VCH: Weinheim, 2007.
Return to citation in text: [1] -
Pedersen, C. M.; Bols, M. Cyclodextrin-Based Artificial Enzymes: Synthesis and Function. In Organic Synthesis and Molecular Engineering; Nielsen, M. B., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp 305–332. doi:10.1002/9781118736449.ch11
Return to citation in text: [1] -
Wu, J.; Du, X.; Ma, J.; Zhang, Y.; Shi, Q.; Luo, L.; Song, B.; Yang, S.; Hu, D. Green Chem. 2014, 16, 3210–3217. doi:10.1039/c3gc42400f
Return to citation in text: [1] -
Tayade, Y. A.; Padvi, S. A.; Wagh, Y. B.; Dalal, D. S. Tetrahedron Lett. 2015, 56, 2441–2447. doi:10.1016/j.tetlet.2015.03.084
Return to citation in text: [1] -
Sim, J. H.; Song, C. E. Angew. Chem., Int. Ed. 2017, 56, 1835–1839. doi:10.1002/anie.201611466
Return to citation in text: [1] -
Macaev, F.; Boldescu, V. Symmetry 2015, 7, 1699–1720. doi:10.3390/sym7041699
Return to citation in text: [1] [2] -
Hapiot, F.; Menuel, S.; Ferreira, M.; Léger, B.; Bricout, H.; Tilloy, S.; Monflier, E. ACS Sustainable Chem. Eng. 2017, 5, 3598–3606. doi:10.1021/acssuschemeng.6b02886
Return to citation in text: [1] -
Jouffroy, M.; Gramage-Doria, R.; Armspach, D.; Sémeril, D.; Oberhauser, W.; Matt, D.; Toupet, L. Angew. Chem., Int. Ed. 2014, 53, 3937–3940. doi:10.1002/anie.201311291
Return to citation in text: [1] -
Hapiot, F.; Ponchel, A.; Tilloy, S.; Monflier, E. C. R. Chim. 2011, 14, 149–166. doi:10.1016/j.crci.2010.04.003
Return to citation in text: [1] -
De Rosa, M.; La Manna, P.; Talotta, C.; Soriente, A.; Gaeta, C.; Neri, P. Front. Chem. (Lausanne, Switz.) 2018, 6. doi:10.3389/fchem.2018.00084
Return to citation in text: [1] -
Kanagaraj, K.; Suresh, P.; Pitchumani, K. Org. Lett. 2010, 12, 4070–4073. doi:10.1021/ol101658n
Return to citation in text: [1] -
Doyagüez, E. G.; Fernández-Mayoralas, A. Tetrahedron 2012, 68, 7345–7354. doi:10.1016/j.tet.2012.06.089
Return to citation in text: [1] -
Shen, H.-M.; Ji, H.-B. Tetrahedron Lett. 2012, 53, 3541–3545. doi:10.1016/j.tetlet.2012.04.140
Return to citation in text: [1] -
Liu, K.; Zhang, G. Tetrahedron Lett. 2015, 56, 243–246. doi:10.1016/j.tetlet.2014.11.084
Return to citation in text: [1] -
Kacprzak, K. M. Chemistry and Biology of Cinchona Alkaloids. In Natural Products; Ramawat, K. G.; Mérillon, J.-M., Eds.; Springer: Berlin, Heidelberg, 2013; pp 605–641. doi:10.1007/978-3-642-22144-6_22
Return to citation in text: [1] -
Ghosh, A. K.; Zhou, B. Tetrahedron Lett. 2013, 54, 3500–3502. doi:10.1016/j.tetlet.2013.04.080
Return to citation in text: [1] -
Nakayama, Y.; Gotanda, T.; Ito, K. Tetrahedron Lett. 2011, 52, 6234–6237. doi:10.1016/j.tetlet.2011.09.064
Return to citation in text: [1] -
Ogawa, S.; Shibata, N.; Inagaki, J.; Nakamura, S.; Toru, T.; Shiro, M. Angew. Chem., Int. Ed. 2007, 46, 8666–8669. doi:10.1002/anie.200703317
Return to citation in text: [1] -
Marcelli, T.; Hiemstra, H. Synthesis 2012, 44, 2114. doi:10.1055/s-0032-1316742
Return to citation in text: [1] -
Liu, Y.; Li, L.; Zhang, H.-Y.; Fan, Z.; Guan, X.-D. Bioorg. Chem. 2003, 31, 11–23. doi:10.1016/s0045-2068(02)00512-6
Return to citation in text: [1] -
Liu, Y.; Chen, G.-S.; Chen, Y.; Ding, F.; Chen, J. Org. Biomol. Chem. 2005, 3, 2519. doi:10.1039/b506053b
Return to citation in text: [1] -
Tang, W.; Ng, S.-C. Nat. Protoc. 2008, 3, 691–697. doi:10.1038/nprot.2008.37
Return to citation in text: [1] [2] -
Celewicz, L.; Kacprzak, K.; Ruszkowski, P. Application of Cinchona alkaloid derivatives as cytotoxic compounds. Canadian Pat. Appl. CA2891633A1, March 26, 2015.
Return to citation in text: [1] -
Bauer, M.; Fajolles, C.; Charitat, T.; Wacklin, H.; Daillant, J. J. Phys. Chem. B 2011, 115, 15263–15270. doi:10.1021/jp205917q
Return to citation in text: [1] -
Al Temimi, A. H. K.; Boltje, T. J.; Zollinger, D.; Rutjes, F. P. J. T.; Feiters, M. C. Bioconjugate Chem. 2017, 28, 2160–2166. doi:10.1021/acs.bioconjchem.7b00319
Return to citation in text: [1] -
Guieu, S.; Zaborova, E.; Blériot, Y.; Poli, G.; Jutand, A.; Madec, D.; Prestat, G.; Sollogoub, M. Angew. Chem., Int. Ed. 2010, 49, 2314–2318. doi:10.1002/anie.200907156
Return to citation in text: [1] -
Guieu, S.; Sollogoub, M. Advances in Cyclodextrin Chemistry. In Modern Synthetic Methods in Carbohydrate Chemistry; Werz, D.; Vidal, S., Eds.; Wiley-VCH: Weinheim, Germany, 2013; pp 241–283. doi:10.1002/9783527658947.ch9
Return to citation in text: [1] -
Tichá, I.; Benkovics, G.; Malanga, M.; Jindřich, J. Beilstein J. Org. Chem. 2018, 14, 2829–2837. doi:10.3762/bjoc.14.261
Return to citation in text: [1] -
Pearce, A. J.; Sinaÿ, P. Angew. Chem., Int. Ed. 2000, 39, 3610–3612. doi:10.1002/1521-3773(20001016)39:20<3610::aid-anie3610>3.0.co;2-v
Return to citation in text: [1] -
Kumprecht, L.; Buděšínský, M.; Vondrášek, J.; Vymětal, J.; Černý, J.; Císařová, I.; Brynda, J.; Herzig, V.; Koutník, P.; Závada, J.; Kraus, T. J. Org. Chem. 2009, 74, 1082–1092. doi:10.1021/jo802139s
Return to citation in text: [1] -
Fredy, J. W.; Scelle, J.; Guenet, A.; Morel, E.; Adam de Beaumais, S.; Ménand, M.; Marvaud, V.; Bonnet, C. S.; Tóth, E.; Sollogoub, M.; Vives, G.; Hasenknopf, B. Chem. – Eur. J. 2014, 20, 10915–10920. doi:10.1002/chem.201403635
Return to citation in text: [1] -
Menuel, S.; Azaroual, N.; Landy, D.; Six, N.; Hapiot, F.; Monflier, E. Chem. – Eur. J. 2011, 17, 3949–3955. doi:10.1002/chem.201003221
Return to citation in text: [1] -
Dočekal, V.; Šimek, M.; Dračínský, M.; Veselý, J. Chem. – Eur. J. 2018, 24, 13441–13445. doi:10.1002/chem.201803677
Return to citation in text: [1] [2] [3]
| 29. | Bauer, M.; Fajolles, C.; Charitat, T.; Wacklin, H.; Daillant, J. J. Phys. Chem. B 2011, 115, 15263–15270. doi:10.1021/jp205917q |
| 30. | Al Temimi, A. H. K.; Boltje, T. J.; Zollinger, D.; Rutjes, F. P. J. T.; Feiters, M. C. Bioconjugate Chem. 2017, 28, 2160–2166. doi:10.1021/acs.bioconjchem.7b00319 |
| 31. | Guieu, S.; Zaborova, E.; Blériot, Y.; Poli, G.; Jutand, A.; Madec, D.; Prestat, G.; Sollogoub, M. Angew. Chem., Int. Ed. 2010, 49, 2314–2318. doi:10.1002/anie.200907156 |
| 32. | Guieu, S.; Sollogoub, M. Advances in Cyclodextrin Chemistry. In Modern Synthetic Methods in Carbohydrate Chemistry; Werz, D.; Vidal, S., Eds.; Wiley-VCH: Weinheim, Germany, 2013; pp 241–283. doi:10.1002/9783527658947.ch9 |
| 6. | Bogliotti, N.; Dalko, P. I. Shape and Site-Selective Asymmetric Reactions. In Enantioselective organocatalysis: reactions and experimental procedures; Dalko, P. I., Ed.; Wiley-VCH: Weinheim, 2007. |
| 17. | Doyagüez, E. G.; Fernández-Mayoralas, A. Tetrahedron 2012, 68, 7345–7354. doi:10.1016/j.tet.2012.06.089 |
| 5. | Hapiot, F.; Tilloy, S.; Monflier, E. Chem. Rev. 2006, 106, 767–781. doi:10.1021/cr050576c |
| 18. | Shen, H.-M.; Ji, H.-B. Tetrahedron Lett. 2012, 53, 3541–3545. doi:10.1016/j.tetlet.2012.04.140 |
| 3. | Breslow, R.; Dong, S. D. Chem. Rev. 1998, 98, 1997–2012. doi:10.1021/cr970011j |
| 4. | Easton, C. J. Pure Appl. Chem. 2005, 77, 1865–1871. doi:10.1351/pac200577111865 |
| 38. | Dočekal, V.; Šimek, M.; Dračínský, M.; Veselý, J. Chem. – Eur. J. 2018, 24, 13441–13445. doi:10.1002/chem.201803677 |
| 16. | Kanagaraj, K.; Suresh, P.; Pitchumani, K. Org. Lett. 2010, 12, 4070–4073. doi:10.1021/ol101658n |
| 38. | Dočekal, V.; Šimek, M.; Dračínský, M.; Veselý, J. Chem. – Eur. J. 2018, 24, 13441–13445. doi:10.1002/chem.201803677 |
| 12. | Hapiot, F.; Menuel, S.; Ferreira, M.; Léger, B.; Bricout, H.; Tilloy, S.; Monflier, E. ACS Sustainable Chem. Eng. 2017, 5, 3598–3606. doi:10.1021/acssuschemeng.6b02886 |
| 14. | Hapiot, F.; Ponchel, A.; Tilloy, S.; Monflier, E. C. R. Chim. 2011, 14, 149–166. doi:10.1016/j.crci.2010.04.003 |
| 15. | De Rosa, M.; La Manna, P.; Talotta, C.; Soriente, A.; Gaeta, C.; Neri, P. Front. Chem. (Lausanne, Switz.) 2018, 6. doi:10.3389/fchem.2018.00084 |
| 37. | Menuel, S.; Azaroual, N.; Landy, D.; Six, N.; Hapiot, F.; Monflier, E. Chem. – Eur. J. 2011, 17, 3949–3955. doi:10.1002/chem.201003221 |
| 38. | Dočekal, V.; Šimek, M.; Dračínský, M.; Veselý, J. Chem. – Eur. J. 2018, 24, 13441–13445. doi:10.1002/chem.201803677 |
| 8. | Wu, J.; Du, X.; Ma, J.; Zhang, Y.; Shi, Q.; Luo, L.; Song, B.; Yang, S.; Hu, D. Green Chem. 2014, 16, 3210–3217. doi:10.1039/c3gc42400f |
| 9. | Tayade, Y. A.; Padvi, S. A.; Wagh, Y. B.; Dalal, D. S. Tetrahedron Lett. 2015, 56, 2441–2447. doi:10.1016/j.tetlet.2015.03.084 |
| 10. | Sim, J. H.; Song, C. E. Angew. Chem., Int. Ed. 2017, 56, 1835–1839. doi:10.1002/anie.201611466 |
| 33. | Tichá, I.; Benkovics, G.; Malanga, M.; Jindřich, J. Beilstein J. Org. Chem. 2018, 14, 2829–2837. doi:10.3762/bjoc.14.261 |
| 7. | Pedersen, C. M.; Bols, M. Cyclodextrin-Based Artificial Enzymes: Synthesis and Function. In Organic Synthesis and Molecular Engineering; Nielsen, M. B., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp 305–332. doi:10.1002/9781118736449.ch11 |
| 13. | Jouffroy, M.; Gramage-Doria, R.; Armspach, D.; Sémeril, D.; Oberhauser, W.; Matt, D.; Toupet, L. Angew. Chem., Int. Ed. 2014, 53, 3937–3940. doi:10.1002/anie.201311291 |
| 34. | Pearce, A. J.; Sinaÿ, P. Angew. Chem., Int. Ed. 2000, 39, 3610–3612. doi:10.1002/1521-3773(20001016)39:20<3610::aid-anie3610>3.0.co;2-v |
| 35. | Kumprecht, L.; Buděšínský, M.; Vondrášek, J.; Vymětal, J.; Černý, J.; Císařová, I.; Brynda, J.; Herzig, V.; Koutník, P.; Závada, J.; Kraus, T. J. Org. Chem. 2009, 74, 1082–1092. doi:10.1021/jo802139s |
| 36. | Fredy, J. W.; Scelle, J.; Guenet, A.; Morel, E.; Adam de Beaumais, S.; Ménand, M.; Marvaud, V.; Bonnet, C. S.; Tóth, E.; Sollogoub, M.; Vives, G.; Hasenknopf, B. Chem. – Eur. J. 2014, 20, 10915–10920. doi:10.1002/chem.201403635 |
| 21. | Ghosh, A. K.; Zhou, B. Tetrahedron Lett. 2013, 54, 3500–3502. doi:10.1016/j.tetlet.2013.04.080 |
| 19. | Liu, K.; Zhang, G. Tetrahedron Lett. 2015, 56, 243–246. doi:10.1016/j.tetlet.2014.11.084 |
| 20. | Kacprzak, K. M. Chemistry and Biology of Cinchona Alkaloids. In Natural Products; Ramawat, K. G.; Mérillon, J.-M., Eds.; Springer: Berlin, Heidelberg, 2013; pp 605–641. doi:10.1007/978-3-642-22144-6_22 |
| 28. | Celewicz, L.; Kacprzak, K.; Ruszkowski, P. Application of Cinchona alkaloid derivatives as cytotoxic compounds. Canadian Pat. Appl. CA2891633A1, March 26, 2015. |
| 26. | Liu, Y.; Chen, G.-S.; Chen, Y.; Ding, F.; Chen, J. Org. Biomol. Chem. 2005, 3, 2519. doi:10.1039/b506053b |
| 24. | Marcelli, T.; Hiemstra, H. Synthesis 2012, 44, 2114. doi:10.1055/s-0032-1316742 |
| 25. | Liu, Y.; Li, L.; Zhang, H.-Y.; Fan, Z.; Guan, X.-D. Bioorg. Chem. 2003, 31, 11–23. doi:10.1016/s0045-2068(02)00512-6 |
| 22. | Nakayama, Y.; Gotanda, T.; Ito, K. Tetrahedron Lett. 2011, 52, 6234–6237. doi:10.1016/j.tetlet.2011.09.064 |
| 23. | Ogawa, S.; Shibata, N.; Inagaki, J.; Nakamura, S.; Toru, T.; Shiro, M. Angew. Chem., Int. Ed. 2007, 46, 8666–8669. doi:10.1002/anie.200703317 |
© 2019 Tichá et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)